Review
Review

Affiliation:
Department of Medical Sciences and Public Health, University of Cagliari, 09100 Cagliari, Italy
Email: giuliacostanzo14@gmail.com
ORCID: https://orcid.org/0000-0003-2269-0439
Affiliation:
Department of Medical Sciences and Public Health, University of Cagliari, 09100 Cagliari, Italy
ORCID: https://orcid.org/0009-0003-5456-2359
Explor Asthma Allergy. 2024;2:410–420 DOI: https://doi.org/10.37349/eaa.2024.00054
Received: January 22, 2023 Accepted: May 21, 2024 Published: August 20, 2024
Academic Editor: Mario Di Gioacchino, Italian Society of Allergy and Clinical Immunology, Italy G. d’Annunzio University, Italy
The article belongs to the special issue The Era of Biologics in Allergy
A granulomatous vasculitis of the medium and large vessels, giant cell arteritis (GCA) is a persistent, idiopathic condition. The overlapping phenotypes of this condition include conventional cranial arteritis and extra-cranial GCA, also known as large-vessel GCA. Vascular problems linked with considerable vessel involvement may partly be caused by delayed diagnosis, emphasizing the necessity of early detection and the fast beginning of appropriate therapy. The cornerstone of treatment for GCA is glucocorticoids, but using them for an extended period has numerous, often severe, side effects. We aim to explore the most recent literature on GCA therapies to investigate the current and potential therapeutic options for induction and maintaining treatment in GCA. By now, only tocilizumab is approved for GCA treatment, but several other biological drugs may be efficient and safe for GCA patients, like abatacept, baricitinib and upadacitinib, mavrilimumab, secukinumab, ustekinumab, and anakinra.
Giant cell arteritis (GCA) is the most common form of primary systemic vasculitis in people over 50 years of age (it rarely occurs under 50, and the peak is around 80). This pathology causes granulomatous inflammation of the vessels, large and medium caliber. Initially thought to be a type of vasculitis primarily involving the carotid and vertebral artery branches, autopsy studies have revealed histological evidence of large-vessel involvement in 80% of cases, and imaging studies of patients affected by GCA have highlighted extensive radiographic large-vessel involvement (e.g., aorta and its major branches) in up to 83% of patients [1–3]. The risk of developing the disease is 1% in women and 0.5% in men. The Scandinavian-descent population has a higher frequency of the disease than the Mediterranean one, and GCA rarely occurs in patients of African or Asian descent: The disease has been linked to genetic predisposition and is associated with the HLA-DRB1*04 allele [4].
GCA and polymyalgia rheumatica are closely related. Indeed, polymyalgia rheumatica occurs alone, but it could be found in 40–50% of patients affected by GCA. Moreover, among patients who complained only of symptoms of isolated polymyalgia rheumatica (shoulder, hips, and thighs muscle stiffness), 10–20% of those later developed GCA. This substantial clinical relationship, along with findings from pathophysiologic research, has increasingly supported the idea that GCA and polymyalgia rheumatica constitute different clinical spectrums of the same disease process. Histopathologically, classic histology is characterized by inflammation of the arterial walls, lamina interna fragmentation, and intima thickening. Although the name of the pathology derives from the presence of multinucleated giant cells, they are documented only in about one-third of temporal artery biopsies, usually associated with a granulomatous inflammatory infiltrate composed of CD4+ T-cells located in the junction between the intima and average. Thickening of the intima can lead to partial or complete lumen occlusion with ischemic complications such as ischemic anterior optic neuropathy. Pathogenesis is related to inappropriate activation, maturation, and retention of adventitious antigen-presenting dendritic cells. These cells provide a favorable environment for viral and bacterial pathogens through the action of toll-like receptors (TLRs). Particular profiles of TLR are, in fact, specific for vessels. Mouse models show activation of wall-embedded dendritic cells that release chemokines that recruit CD4+ T-cells and macrophages. The pattern of arterial inflammation corresponds to TLR4 stimulation that induces panarteritis and TLR5 that stimulates perivasculitis. The onset of GCA tends to be insidious and lasts for weeks or months before manifesting with a symptom spectrum attributable to the localized effects of vascular or systemic inflammation: new-onset headache, scalp tightness, jaw claudication, fever, fatigue, malaise, anorexia, weight loss, polymyalgia, painless, and irreversible vision loss (bilateral or unilateral). The headache is usually constant, sudden onset, and typically located in the temporal region where it is severe enough to prevent nighttime sleep. It may also present alternately or worsen without treatment. Scalp tension, increased prominence, and decreased temporal artery pulsation raise suspicion of GCA, although one-third of patients have negative biopsies of temporal artery specimens. The diagnosis could be challenging, and many other conditions could mimic GCA (Table 1) [5].
Summary of the giant cell arteritis (GCA) mimickers [6]
| Conditions | Differential diagnosis |
|---|---|
| Takayasu arteritis | Age < 50, renal artery stenosis is common, and anterior ischemic optic neuropathic or polimyalgia reumatica is unusual. |
| Small and medium vessels arteritis | Different vascular distribution than GCA, other organ involvement, distinctive histopathology |
| Primary angiitis of the central nervous system | Inflammatory involvement of the intracranial vessels and stroke are more common than in GCA. |
| Idiopathic aortitis | Similar to hystopatology, the absence of the clinical expression of GCA |
| VEXAS syndrome | Neutrophilic dermatosis, hematologic abnormalities, characteristic vacuoles in myeloid precursor cells, UBA1 mutations |
| Nonarteritic anterior ischemic optic neuropathy | Clinical features and lab markers of GCA are generally absent. |
| Infection | Distinctive lab markers |
The classification is based on three or more criteria: age greater than 50 years, newly localized headache, decreased temporal artery tension or pulse, erythrocyte sedimentation rate (ESR) greater than 55 mm/h, and abnormal biopsy. The definitive diagnosis is obtained with the biopsy. From a laboratory point of view, the acute phase markers are often significantly elevated. Normocytic normochromic anemia is associated with thrombocytosis, and transaminases may be elevated with reduced albumin. In particular, the combination of increased C reactive protein (CRP) and positive biopsy has high sensitivity and specificity for GCA. A valid, non-invasive, and inexpensive test is ultrasonography of the temporal arteries, which renders the inflammation picture of the inflamed temporal arteries characterized by oedematous wall swelling. With the advent of glucocorticoids, survival is similar to age reference [5].
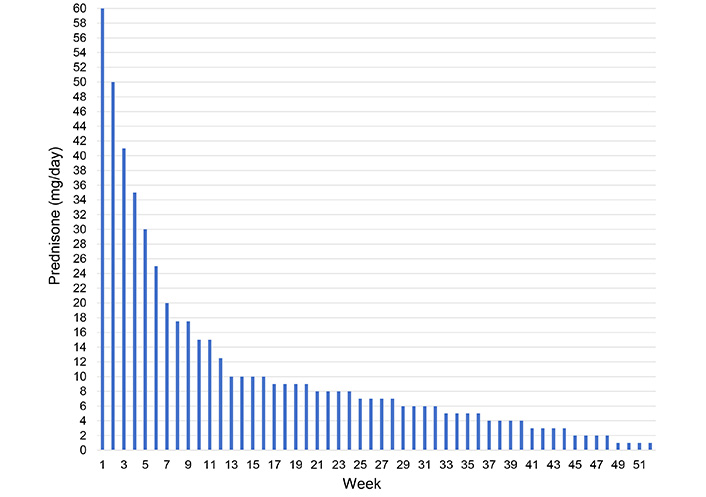
The first line of therapy for GCA has been systemic glucocorticoids (CSs) since their development in the 1950s. Although there are no randomized trials, most experts recommend starting immediately with high doses of corticosteroids (40 mg to 60 mg of prednisone per day) until clinical and laboratory values regress. Treatment every other day, even if combined with drugs such as methotrexate (MTX), does not seem to have greater efficacy. The response to CSs is so rapid that it is a diagnostic criterion of GCA: Most symptoms improve after 2 or 3 days of therapy, while symptoms related to blood flow, such as jaw claudication and visual impairment, can take longer to resolve. Visual loss may not be reversible, but CS therapy minimizes the risk of further deterioration. CSs can be gradually tapered as early as the first month after the symptoms’ resolution, reducing CRP or ESR values by at least 50%. The treatment should then be continued for at least 2 years, with some patients requiring 4–5 years of therapy [5]. Unfortunately, as known, the use of oral corticosteroids (OCS) is linked to substantial side effects, so between 50% and 100% of patients experience at least one adverse event (AE) from OCS. Furthermore, in case of discontinuation or tapering of OCS (Figure 1), the risk of exacerbation is 43–79% [7]. For these reasons, the need arises to find other drugs that can replace or integrate CSs in treating GCA.
MTX is the most studied steroid-sparing therapy in GCA is MTX therapy. It can reduce the overall dose of CSs if used early in the course of the illness, but it has little clinical impact on the side effects of corticosteroid therapy [9]. In a very recent single-center real-life study, 49 GCA patients receiving MTX therapy showed a significant decrease in mean exacerbations per month but also an excellent steroid-sparing effect, as only half of these patients discontinued CSs therapy after a mean of 20.5 months of MTX therapy [10] according to previous randomized, double-blind studies [11, 12].
Mycophenolate mofetil (MMF) is an inosine monophosphate dehydrogenase inhibitor with an immunosuppressive effect used in lupus nephritis and various vasculitic disorders [9]. The available literature on this drug’s use in GCA is relatively sparse. A 2012 case series demonstrates how well-tolerated MMF can reduce disease activity and enable more rapid CSs dose reduction. The current report has demonstrated that the MMF therapy was well tolerated, controlled the GCA malady, and allowed for a quicker decline in prednisone dosage than had previously been suggested [13]. A 2020 retrospective cohort study that included 37 patients demonstrates how, starting with the diagnosis, the use of MMF on patients with GCA with interest in the large veins can reduce the cumulative dose in the first year by 5 g. It is well tolerated (only 9% of patients had to interrupt the therapy) [14].
Leflunomide is a pyrimidine synthesis inhibitor utilized in rheumatoid arthritis and psoriatic arthritis for its efficacy as a disease-modifying medication [9].
A recent comprehensive review and meta-analysis of cohort studies found that leflunomide has CSs-sparing abilities and a favorable clinical response in GCA and Takayasu arteritis (TAK). In GCA, there appears to be a good balance between efficacy and safety in preventing recurrences and reducing cumulative CSs dose over time. It could also be used as an alternative to MTX in cases of intolerance or contraindications [15].
According to a 2020 multicenter retrospective controlled trial, cyclophosphamide (CYC) appears beneficial in decreasing CSs dose within 6 months after commencing the medication. Furthermore, CYC was chosen for patients with ischemic problems and/or considerable artery involvement, which has recently been linked to decreased mortality in GCA and may support the use of deeper immunosuppression [16].
Table 2 resembles all biological options proposed for the GCA treatment. Today, only tocilizumab is fully approved for GCA therapy; the other drugs are currently under investigation at different points of the study phase.
Summary of the current biological treatment used and experimented in the giant cell arteritis treatment [17–20]
| Treatment | Target | Dose |
|---|---|---|
| Abatacept | CD80/CD86 | 10 mg/kg iv every two weeks for 8 weeks then monthly |
| Baricitinib | JAK1/JAK2 | 4 mg/day |
| Upadacitinib | JAK1 | 15 mg/day |
| Tocilizumab | IL-6R | 162 mg sc every 1-2 weeks or 6 mg/kg iv every four weeks |
| Mavrilimumab | GM-CSFRα | 150 mg sc every 2 weeks |
| Secukinumab | IL-17 | 300 mg weekly for 5 doses, then 300 mg every 4 weeks |
| Ustekinumab | IL-12/IL-23 p40 | 90 mg sc at baseline then in week 4, then every 8 weeks |
| Anakinra | IL-1Ra | 100 mg/day for induction, then 100 mg three times a week |
IL-6R: interleukin 6 receptor; GM-CSFRα: granulocyte-macrophage colony-stimulating factor receptor alpha; JAK: Janus kinase
So far, only tocilizumab, an interleukin 6 receptor (IL-6R) inhibitor (Figure 2), has shown safety and efficacy in reducing exacerbations and decreasing the daily dose of CSs, and is the only drug to be cleared by the FDA and EDC for the treatment of GCA and this reason it was immediately incorporated into clinical practice [18]. From the Cochrane review, the authors conclude that tocilizumab has an essential role in increasing the percentage of patients who undergo sustained remission and the number of patients who do not require escape therapy. Tocilizumab also reduces the mean cumulative CS dose by lengthening the mean time to the first exacerbation after remission induction (Figure 1). Tocilizumab also improves the quality of life from a mental and general health perspective. The frequency of AEs in standardized studies appears to be similar between tocilizumab and placebo; most AEs for tocilizumab are infections [18]. Although tocilizumab has dramatically improved the treatment of this pathology, patients treated with tocilizumab have an exacerbation rate of 15–26%, and also, according to clinical trials and observational data in 30–47% of patients do not achieve a remission sustained clinic at 12 months. The length of treatment with tocilizumab also remains unknown at present. Therefore, further treatments are urgently needed, especially for patients who do not achieve remission or who, for various reasons, are not eligible for tocilizumab [18] (Figure 1).
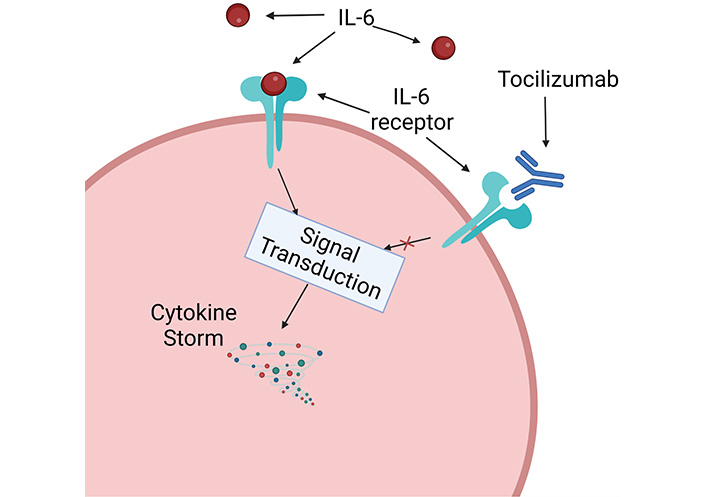
Graphically simplified mechanism of function of tocilizumab. Created with BioRender.com
Abatacept, a biologic used in rheumatoid arthritis, is a recombinant fusion protein composed of the extracellular domain of human CTLA-4 and the modified Fc region of human IgG1 that inhibits T-cell activation by explicitly binding to CD80 and CD86. Abatacept also modulates the activation of naïve T-cells. The critical concept beyond the idea of using abatacept as a treatment for GCA is that the pathogenesis is driven by antigen and the crosstalk between T, B lymphocytes, and dendritic cells. Abatacept blocks the signaling cascade mediated by CTLA-4. So T-cell activation is supposed to be one of the mechanisms behind the pathogenesis of GCA and was examined in a randomized trial of 49 patients with newly diagnosed or exacerbating GCA. Participants received abatacept every 14 days for 8 weeks until they were divided into two arms, one continuing the drug, the other switching to placebo associated with a progressive prednisone taper with total discontinuation at 28 weeks. The authors found that one-year survival in the absence of relapse was 48% in patients who continued abatacept vs. 31% in those who switched to placebo (p = 0.049) [17].
Ustekinumab (UST) is a fully human IgG1κ monoclonal antibody binding the interleukin IL-12/23 p40 used in Chron’s disease and ulcerative colitis. The results in the literature are mixed. A prospective, single-center, open-label study evaluated the efficacy of UST in combination with prednisone in 13 patients with active GCA without demonstrating a significant rate of prednisone-free remission. However, adding UST to a 6-month prednisone taper did not result in a substantial rate of durable, prednisone-free remission in new onset or relapsing disease. Another prospective study in patients with refractory GCA who previously had difficulty tapering prednisone despite combination with another immunosuppressant showed that at 52 weeks, the daily prednisolone dose was reduced from 20 mg to 5 mg with 24% of patients were able to discontinue OCS and no patient experienced GCA relapse [17].
Secukinumab (SEC) was studied in a phase two randomized controlled trial of 52 patients with new-onset or exacerbation GCA who were biologic-naïve. Patients were randomized into two arms, one treated with SEC 300 mg, the other with a placebo administered weekly for five doses and then every 4 weeks until week 48, combined with a prednisolone taper at 26 weeks. The rate of patients in sustained remission at 28 weeks was 70.1% in patients with SEC compared to those on placebo. In addition, a higher sustained remission rate was associated with a longer time to first exacerbation in SEC patients [17].
Granulocyte-macrophage colony-stimulating factor (GM-CSF), is a multifunctional cytokine expressed by immune and endothelial cells in the temporal arteries that modulate dendritic cell, CD4+ T-cell, and macrophage biology. Preclinical data have suggested a role of GM-CSF in GCA: Blockade of GM-CSF receptors has been associated with a downregulation of the Th1 and Th17 response. In mouse models, inhibition of GM-CSF is associated with reduced arterial remodeling and inflammation. Mavrilimumab is a monoclonal antibody, specifically an IgG4, with demonstrated efficacy in phase 2 studies of rheumatoid arthritis, which blocks GM-CSF signaling by binding to the alpha chain of the receptor. In a phase 2 randomized double-blind placebo-controlled trial, the safety and efficacy of this drug in GCA were evaluated. The primary efficacy endpoint was the time to first flare (defined as an increase in ESR greater than or equal to 30 mm/h or in CRP more significant than 1 mg/dL together with the presence of sure cranial or extracranial signs or symptoms or the appearance on imaging of a new finding or signal of worsening abnormality suggestive of active vasculitis. The secondary efficacy endpoint was sustained remission rate, defined as the absence of flare from randomization to week 26. Of 70 randomized patients, 42 were randomly assigned mavrilimumab, and 28 were randomly assigned placebo. Twenty-one patients had a flare within 26 weeks, 8 of these patients received mavrilimumab, and the other 13 received a placebo. The median time to exacerbation was 25.1 weeks in the placebo, mavrilimumab was over 26 weeks. Mavrilimumab significantly reduced exacerbations versus placebo, and durable remission occurred in 83.2% of patients taking mavrilimumab while only 49.9% of patients taking placebo (p = 0.0038). The cumulative prednisone dose at 26 weeks was also higher in the placebo patients than in the mavrilimumab patients. Regarding safety, AEs were reported in 78.6% of patients on mavrilimumab compared to 89.3% of patients on placebo (severe AEs 4.8% vs. 10.7%). This trial shows that mavrilimumab with a prednisone taper at 26 weeks is superior to a placebo with a prednisone taper in reducing flares and maintaining sustained remission. Mavrilimumab is well tolerated and presents itself as a promising treatment option. However, it’s essential to recall that GM-CSF plays a critical role in pulmonary homeostasis by boosting alveolar macrophage-induced surfactant clearance. However, there were no differences between the treatment groups in this trial [21].
Anakinra is an IL-1Ra that blocks both IL-1α and IL-1β activity and has previously been licensed in the USA, Canada, Europe, and Australia for use in rheumatoid arthritis and different forms of cryopyrin-associated periodic syndromes. A series of three clinical cases show its possible use in GCA. In an initial case report, an 80-year-old male patient with a fever of unknown origin, night sweats, weight loss, and arthralgias was treated with anakinra after the failure of other drugs such as MTX, azathioprine, and etanercept. The 100 mg/day dosage allowed remission of symptoms and laboratory abnormalities after one month, allowing a tapering of the OCS from 60 mg/day of prednisone up to 7.5 mg/day with the maintenance of disease control. A second case is that of a female patient diagnosed with GCA resistant to methylprednisolone therapy which was administered off-label anakinra, with prior informed consent, at a dosage of 100 mg/day with resolution of symptoms and normalization of laboratory values (CRP). Within 3 weeks allowing a tapering of both the OCS up to 5 mg/day of prednisone and of the same anakinra up to 100 mg 3 times a week and with maintenance of remission confirmed by the disappearance on PET/CT of the signs of mural arterial inflammation present at a first test performed before administration of anakinra.
The third case is that of an 80-year-old female patient suffering from symptoms of polymyalgia and ischemic pain in the upper and lower limbs with abolition of the radial and cubital pulse due to peripheral arterial occlusive disease with thrombosis of the right humeral artery resistant to OCS therapy. The patient underwent therapy with anakinra 100 mg every other day, associated with 20 mg/day of prednisone with disappearance after only three months of the signs of arterial inflammation on PET/CT. In this case, it was possible to taper the OCS up to 5 mg/day with maintenance of remission. These cases suggest a possible use of anakinra in forms of GCA refractory to other treatments [20]. Another case series of 6 patients, all female and with a mean age at diagnosis of 70.5, were involved. Each patient displayed corticosteroid resistance or dependence prior to the start of anakinra. Four patients presented large-vessel involvement with aortitis and four presented polymyalgia rheumatica. After an average disease duration of 13 months and a diagnosis of corticosteroid dependence or resistance at an average dosage of 14 mg/day, anakinra was administered subcutaneously at a dosage of 100 mg/day. All patients displayed a clinical and biological response of both GCA and polymyalgia rheumatica symptoms after receiving medication for a median of 19 months. Additionally, one patient with large-vessel involvement demonstrated the disappearance of aortitis under anakinra. Four individuals were able to discontinue oral steroid therapy after 56 months, another two reduced the daily dose to 5 mg/day. Regarding safety and tolerance, 3 individuals displayed injection site reactions, and 1 patient experienced a pneumonia episode that required hospitalization 6 months after beginning anakinra therapy and brief drug interruption. As a result, in this case, series anakinra appears to have a steroid sparing effect, with good tolerance even over an extended period of time, and may be effective on large-vessel involvement [22].
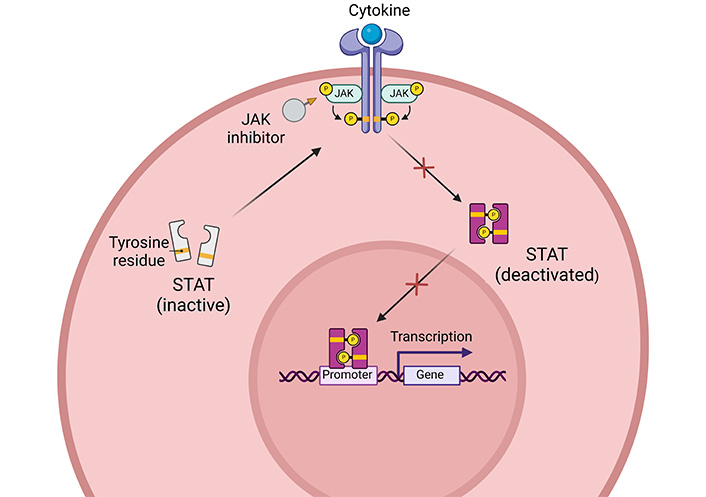
Baricitinib is a small Janus kinase (JAK) inhibitor (Figure 3) approved for the treatment of certain autoimmune diseases and autoinflammatory disorders. Initially licensed for the treatment of moderately to severe TNF-antagonist-resistant rheumatoid arthritis, its remarkable potential for blocking proinflammatory signaling and its selective effect on immune cells makes it a good candidate for resolving an exaggerated immune response in a large number of inflammatory disturbs—the Koster 2022 study [18] aimed to evaluate the safety and efficacy of this drug in exacerbating GCA. The study showed that the drug is effective and safe in treating forms characterized by exacerbations. The 4 mg/day dose appears to have sufficient control over both GC tapering and discontinuation (only 1 out of 14 patients had a flare) with the ability to discontinue cortisone 22 weeks after initiation of therapy, with 13 out of 14 patients achieved and maintained clinical remission at 52 weeks. Treatment should be carefully monitored in patients with renal dysfunction.
Upadacitinib is a second-generation JAK inhibitor. The literature describes the clinical case of a 72-year-old patient with symptoms of fever, anorexia, headache, arthralgia, and jaw claudication who was diagnosed with GCA following a biopsy of the femoral artery. The patient also suffered from pustular psoriasis and type II diabetes mellitus. This patient was administered upadacitinib 15 mg/day associated with prednisolone (0.6 mg/kg) which induced a prompt remission of the disease. Following tapering of OCS for six months, upadacitinib alone therapy maintained remission for 7.5 months without predicting disease progression. This suggests that the update could be a further weapon against the GCA, although clinical trials are needed to determine the efficacy of specific JAK1 inhibition for large-vessel vasculitis (LVV) remission [19].
Some studies suggest selenium nanoparticles’ effect on inflammatory signaling [23]. Furthermore, revascularized microspheres could be used for vascular tissue engineering, saving the time required by angiogenic [24]. In the future, liposomes of 100 nm diameter with encapsulated drugs, like, for example, dexamethasone in the treatment of rheumatoid arthritis, could also be used in vasculitis [25].
Immense progress has been made in the therapeutic approach to giant cells therapy due to the novel findings on the pathogenetic pathways. Indeed, the massive use of OCS as induction and maintenance treatment gave researchers the pulse to find modern drugs to avoid the accrual organ damage due to chronic continuative use of OCS. Hence following the wave of the steroid-sparing effect of the other biological treatment, tocilizumab has been used firstly as an anchor drug with a well-recognized steroid-sparing effect. Among the proposed remedies for GCA, perhaps JAK inhibitors are the most promising therapies because of their pharmaceutical formulation (patients are often more compliant when taking their daily pills than injections as prescribed) and their efficacy. Mavrilimumab also seems to be a promising therapeutic option for induction and maintaining therapy for GCA; its efficacy underscores the importance of translational research to highlight novel pathogenic pathways and good potential therapeutic targets. Eventually, the goal of treatment in GCA is still the limitation of using OCS to avoid their harmful systemic damage.
AE: adverse event
CRP: C reactive protein
CSs: systemic glucocorticoids
CYC: cyclophosphamide
ESR: erythrocyte sedimentation rate
GCA: giant cell arteritis
GM-CSF: granulocyte-macrophage colony-stimulating factor
IL-6R: interleukin 6 receptor
JAK: Janus kinase
MMF: micofenolato mofetil
MTX: methotrexate
OCS: oral corticosteroids
SEC: secukinumab
TLRs: toll-like-receptors
UST: ustekinumab
GC and AGL: Conceptualization, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Alejandra Carrón-Herrero ... Giovanni Paoletti
Francesca Losa, Arianna Cingolani
Palma Carlucci ... Danilo Di Bona
Carlo Alberto Vignoli, Riccardo G. Borroni
Karl-Christian Bergmann ... Torsten Zuberbier
Diego Bagnasco ... Fulvio Braido
Shuichiro Matsumoto ... Hisatoshi Sugiura
Christian Paolo Ratti ... Silvia Mariel Ferrucci
Alexandru Corlateanu, Cristina Toma