Review
Review

Affiliation:
1Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
†These authors contributed equally to this work.
Affiliation:
1Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
†These authors contributed equally to this work.
Email: margherita.perlato@gmail.com
Affiliation:
1Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
2Allergology and Clinical Immunology Unit, San Donato Hospital, 52100 Arezzo, Italy
Affiliation:
1Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
Affiliation:
1Department of Experimental and Clinical Medicine, University of Florence, 50134 Florence, Italy
Affiliation:
3Department of Emergency and Organ Transplantation, School and Chair of Allergology and Clinical Immunology, University of Bari - Aldo Moro, 70100 Bari, Italy
Affiliation:
4Section of Dermatology and Venereology, Department of Precision and Regenerative Medicine and Ionian Area (DiMePRe-J), University of Bari - Aldo Moro, 70124 Bari, Italy
Affiliation:
5Dermatology Section, Department of Medicine and Surgery, University of Perugia, 06100 Perugia, Italy
Affiliation:
3Department of Emergency and Organ Transplantation, School and Chair of Allergology and Clinical Immunology, University of Bari - Aldo Moro, 70100 Bari, Italy
Affiliation:
6Section of Dermatology and Infectious Diseases, Department of Medical Sciences, University of Ferrara, 44121 Ferrara, Italy
Affiliation:
5Dermatology Section, Department of Medicine and Surgery, University of Perugia, 06100 Perugia, Italy
Affiliation:
7Department of Clinical and Experimental Medicine, Section of Dermatology, University of Messina, 98121 Messina, Italy
Affiliation:
8Department of Health Sciences, Magna Graecia University of Catanzaro, 88100 Catanzaro, Italy
Affiliation:
9Department of Medical Sciences and Public Health, and Unit of Internal Medicine, University of Cagliari, Policlinico Universitario - AOU di Cagliari Azienda Ospedaliero Universitaria, 09042 Monserrato, Italy
Affiliation:
4Section of Dermatology and Venereology, Department of Precision and Regenerative Medicine and Ionian Area (DiMePRe-J), University of Bari - Aldo Moro, 70124 Bari, Italy
Affiliation:
10Section of Dermatology, Department of Clinical Medicine and Surgery, University of Naples Federico II, 80131 Napoli, Italy
Affiliation:
11Unit of Dermatology, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy
Affiliation:
12Studio Medico Cugia15, 09100 Cagliari, Italy
Affiliation:
13Department of Translational Medical Sciences, Allergy and Clinical Immunology, Center for Basic and Clinical Immunology Research (CISI), WAO Center of Excellence, University of Naples Federico II, 80131 Naples, Italy
Affiliation:
14Section of Dermatology, Department of Health Sciences, University of Genoa, IRCCS Ospedale Policlinico San Martino, 16100 Genoa, Italy
Affiliation:
15Department of Laboratory Medicine, ASFO, Pordenone Hospital, 33170 Pordenone, Italy
Affiliation:
11Unit of Dermatology, Foundation IRCCS Ca’ Granda Ospedale Maggiore Policlinico, 20122 Milan, Italy
Affiliation:
14Section of Dermatology, Department of Health Sciences, University of Genoa, IRCCS Ospedale Policlinico San Martino, 16100 Genoa, Italy
Affiliation:
16Internal Medicine Unit, Ospedale dell’Angelo, 30100 Venice, Italy
Affiliation:
17Laboratorio Unico Metropolitano, Maggiore Hospital AUSL Bologna, 40133 Bologna, Italy
Affiliation:
18Allergy Unit, Fondazione Policlinico Gemelli IRCCS, 00042 Rome, Italy
Affiliation:
4Section of Dermatology and Venereology, Department of Precision and Regenerative Medicine and Ionian Area (DiMePRe-J), University of Bari - Aldo Moro, 70124 Bari, Italy
Affiliation:
19Dipartimento di Medicina e Scienze della Salute “Vincenzo Tiberio”, Rheumatology Unit, University of Molise, 86100 Campobasso, Italy
Affiliation:
20Department of Internal Medicine, Antonio Cardarelli Hospital, 80131 Naples, Italy
Affiliation:
21Division of Allergy, Civitanova Marche Hospital, 62012 Civitanova Marche, Italy
Affiliation:
7Department of Clinical and Experimental Medicine, Section of Dermatology, University of Messina, 98121 Messina, Italy
Affiliation:
8Department of Health Sciences, Magna Graecia University of Catanzaro, 88100 Catanzaro, Italy
Affiliation:
5Dermatology Section, Department of Medicine and Surgery, University of Perugia, 06100 Perugia, Italy
Affiliation:
22Clinical Unit of Occupational Medicine, University of Trieste, 34100 Trieste, Italy
Affiliation:
3Department of Emergency and Organ Transplantation, School and Chair of Allergology and Clinical Immunology, University of Bari - Aldo Moro, 70100 Bari, Italy
Affiliation:
23Allergy and Clinical Immunology Unit, Azienda Sanitaria di Firenze, 50100 Florence, Italy
Affiliation:
24School and Operative Unit of Allergy and Clinical Immunology, Department of Clinical and Experimental Medicine, University of Messina, 98125 Messina, Italy
Affiliation:
4Section of Dermatology and Venereology, Department of Precision and Regenerative Medicine and Ionian Area (DiMePRe-J), University of Bari - Aldo Moro, 70124 Bari, Italy
Affiliation:
5Dermatology Section, Department of Medicine and Surgery, University of Perugia, 06100 Perugia, Italy
Affiliation:
3Department of Emergency and Organ Transplantation, School and Chair of Allergology and Clinical Immunology, University of Bari - Aldo Moro, 70100 Bari, Italy
Affiliation:
6Section of Dermatology and Infectious Diseases, Department of Medical Sciences, University of Ferrara, 44121 Ferrara, Italy
Affiliation:
25Immunoallergology Unit, Careggi University Hospital, 50100 Florence, Italy
Explor Asthma Allergy. 2024;2:421–440 DOI: https://doi.org/10.37349/eaa.2024.00055
Received: May 16, 2023 Accepted: May 13, 2024 Published: August 26, 2024
Academic Editor: Umit Murat Sahiner, Hacettepe University Faculty of Medicine, Turkey
Allergic and immunologic skin diseases are becoming increasingly common and this requires clinicians to be able to recognize and diagnose them. A joint meeting (GET TOGETHER 2022) of the Italian Society of Allergy, Asthma and Clinical Immunology (SIAAIC) and the Italian Society of Allergological, Occupational and Environmental Dermatology (SIDAPA) aimed to review the current knowledge on the differential diagnosis of contact dermatitis, atopic dermatitis, hereditary angioedema, urticaria, and cutaneous mastocytosis. The most important aspects to take into consideration when faced with a new cutaneous manifestation are the clinical features of the lesions, their distribution, age of onset, and comorbidities/aggravating factors. The document does not aim to provide an exhaustive and comprehensive description of all allergic and immunologic skin diseases. Instead, it should be a reference tool for the clinician who is faced with the onset of a new skin manifestation and its differential diagnosis.
“People wear their health on their skin”, said Lauren Gravitz [1], and we have the responsibility to understand what it is telling us. To guide us through this process, the Italian Society of Allergy, Asthma and Clinical Immunology (SIAAIC) and Italian Society of Allergological, Occupational and Environmental Dermatology (SIDAPA) organized the GET TOGETHER 2022 initiative. It consisted of virtual meetings held by specialists in allergic and immunologic skin diseases in Italy between September and December 2022. Its primary aim was to discuss and review the current knowledge on the differential diagnosis of contact dermatitis (CD), atopic dermatitis (AD), hereditary angioedema (HAE), urticaria, and cutaneous mastocytosis (CM). This document aims at providing a reference tool for the clinician who is faced with the onset of a new skin manifestation, considering the increasing prevalence of allergic and immunologic skin disorders.
CD is an inflammatory skin disorder that includes irritant CD (ICD) and allergic CD (ACD) [2, 3]. Up to 80% of CD is irritant and is commonly related to occupation, especially wet work. ICD is a nonimmunologic inflammation of the skin caused by contact with chemical, physical, or biological agents. Clinically, it presents as an acute monomorphic reaction that includes scaling, low-grade erythema, vesicles, or erosions; when it affects the hands, it is usually localized on the dorsum and fingers. It can resolve or progress to chronic ICD [2]. ACD is a cutaneous inflammatory delayed reaction caused by contact with a specific exogenous allergen, occupational and non-occupational, to which a person has developed allergic sensitization [3]. It commonly manifests as eczematous dermatitis, even if non eczematous clinical forms are also described (lichenoid, bullous, lymphomatoid). The acute phase is characterized by pruritus, erythema, edema, and vesicles usually confined to the area of direct exposure. Recurrent contact with the allergen causes chronic disease, characterized by lichenified erythematous plaques with variable hyperkeratosis and fissuring that may spread beyond the areas of direct exposure. Itching and swelling are key elements of the history. The hands, feet, and face (including the eyelids) are some of the common sites for ACD (Figure 1) [3]. Diagnosis is mainly clinical, while histological features are not specific. ICD and ACD can be clinically indistinguishable; however, in ICD, burning exceeds itching and it does not spread beyond the area of contact after continued exposure [2]. Patch testing is fundamental for the identification of causal allergens in ACD [3] and to exclude ACD in cases with suspected chronic irritant dermatitis [2]. Metals, especially nickel sulfate, fragrance mix, isothiazolinones, and para-phenylenediamine are the most commonly positive allergens. Herein we will focus on the most common differentials of CD: AD, dermatophytosis, seborrheic dermatitis (SD), psoriasis (PSO), and dyshidrosis.
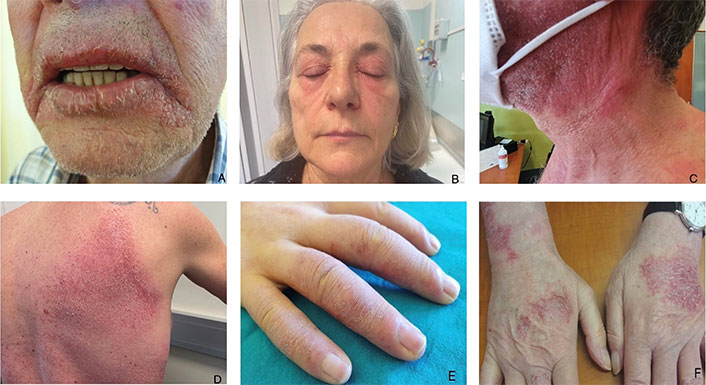
Contact dermatitis. (A) Allergic cheilitis from Peru balsam-containing lipstick; (B) allergic contact dermatitis (ACD) due to nickel sulfate; (C) ACD due to fragrance mix and Peru balsam; (D) ACD to cobalt-dichloride after wearing a tank top; (E) irritant contact dermatitis (ICD) in houseworker; (F) ACD to chrome thiuram mix in bricklayer. Used with permission from Dr. Stefano Francalanci, Allergological and Occupational Dermatology Unit, Department of Surgery and Translational Medicine, University of Florence
Differential diagnosis between atopic and contact eczema is based on several clinical characteristics. While AD affects patients with a personal and family history of atopy, patients who suffer from ICD or ACD generally lack a story of atopy and hardly ever experienced allergic diseases, such as rhinitis, conjunctivitis, or asthma. Age of onset is also important since AD usually starts in childhood while CD typically occurs in adulthood, often as an occupational disease. Another factor to consider is the localization of the lesions. CD distribution depends on the site of exposure and it often provides valuable information about the sensitizing agent. For example, ACD can manifest at the earlobe when patients wear nickel-containing earrings and occupational CD usually affects the hands, thus investigating “working gestures” can be important. Localization of AD changes overtime: in the adult patient, it shows on the face (eyelids and perioral region), neck, antecubital folds, and popliteal fossae. In AD, eczematous lesions can be accompanied by other signs such as Dennie-Morgan infraorbital fold, marked xerosis, pityriasis alba, cheilitis, keratosis pilaris, and palmar hyperlinearity [2, 4–6]. As for hand eczema, while the dorsal aspect of hands, digits, and interdigital spaces can be involved both in CD and AD, palmar involvement suggests a CD (cumulative use of detergents, repeated microtrauma). While CD resolves after removal of the noxa, AD has a chronic relapsing course that can show seasonal changes (generally worsening in winter and improving in summer) and worsen with physical stimuli such as sweating, wool fabrics, or soaps. Though there are no laboratory tests specific for these conditions, AD can be associated with high levels of IgE. Furthermore, prolonged and frequent exposure to potential allergens found in personal care and topical products used to treat chronic AD may predispose AD patients to developing ACD [7, 8]. This should be suspected in the presence of lesions with atypical morphology despite appropriate treatment, immediate rebound after discontinuation, and failure to improve or worsen after initiation of topical therapy [9].
Tinea is a common superficial skin infection caused by dermatophytes belonging to three genera: Microsporum, Trichophyton, and Epidermophyton. Tinea may acquire various clinical aspects depending on inflammatory host response and damage to keratin. Tinea corporis is caused by the parasitization of dermatophytes on exposed glabrous skin. It is characterized by well-demarcated and sharply marginated patches with an erythematous and scaly surface. A raised border with vesicles, papules, or pustules is a distinguishing feature. The patches present a centrifugal spreading with central clearing that brings to an annular shape; coalescence of multiple lesions may result in bizarre configurations [10]. Hand involvement, tinea manuum (TM), is usually unilateral. It can be “dry”, with mild erythema and desquamation predominantly localized to palmar furrow, or “inflammatory” with vesicles and pustules that may mimic dyshidrotic eczema. It can be associated with foot involvement: tinea pedis. It usually involves the space between the fourth and fifth toes causing dryness, scaling, and fissures or wet and macerated aspects. Other possible forms are the dyshidrotic type, with erythema and vesicles that may leave a fine collarette after the rapture, and the moccasin type, with erythema, scaling, and occasionally hyperkeratotic lesions that may reach the lateral border of the foot where the typically raised edge of dermatophytosis can be observed [11]. In these cases, dermoscopic findings of scaling mainly located in the furrows can help us distinguish TM from hand eczema [12]. Most frequently, tinea can present as “two-foot, one hand syndrome”, mimicking CD. However, patch tests are negative, while direct microscopic examination of cutaneous scrapings may confirm the suspicion of fungal disorder. A definitive identification of the responsible agent requires culture in appropriate medium [11].
PSO is a common skin disorder characterized by erythematous plaques with characteristic adherent silvery-white scales. Lesions are typically localized at the knees, elbows, and scalp but other areas can be involved. Friction can cause the onset of psoriatic plaque (Koebner phenomenon). Its etiology is unclear: on a recognized genetic background, immune dysregulation, mainly Th1/Th17-driven, causes cytokine release with subsequent inflammation and keratinocyte proliferation [13–15]. Table 1 summarizes the similarities and differences between PSO and CD. While in most cases, differences are quite defined, PSO with palmoplantar localization can be hard to distinguish from CD due to overlapping characteristics [16]. Furthermore, palmoplantar PSO predisposes to contact sensitization [17, 18], thus patch testing should be performed to identify a possible sensitization that could benefit from preventive measures [19–21].
Characteristics of psoriasis and contact dermatitis for differential diagnosis
| Characteristics | Psoriasis | Contact dermatitis |
|---|---|---|
| Sites involved | Knees, elbows, scalps, palm and sole, all body | Sites in contact with irritants or allergens |
| Symptoms | Generally none | Burning in irritant form, itching in allergic form |
| Signs | Solitary or multiple erythematous plaques with characteristic adherent silvery white scales, Koebner phenomenon | Erythema, edema, papule and vesicles, fissures in irritant form; hyperkeratosis in chronic form |
| Histology | ||
| Parakeratosis | Moderate or strong, confluent; para-/ortho-keratosis | Mild and in multiple foci |
| Acanthosis | Regular | More irregular |
| Granular layer | Thinner | Normal |
| Capillaries in the upper dermis | Tortuous or dilatate | Horizontal |
| Lymphocytic exocytosis in epidermis | No (less likely) | Yes |
| Edema of papillary dermis | No (less likely) | Yes |
| Spongiosis | More frequent | Less frequent |
| Munro microabscesses | Yes | No |
| Prevention | Not available | Avoidance of contact with irritants or sensitizing agents |
SD is a common chronic dermatosis characterized by erythematous and desquamative lesions localized in those areas in which sebaceous glands are highly active (head, face, chest, and skin folds). It can affect infants and adults (M > F), as well as special patient groups such as immunocompromised (AIDS, organ transplant recipients) and neuropsychiatric disease (Parkinson’s and neuroleptic-induced parkinsonism, drug and alcohol addiction and subsequent malnourishment) [22]. Different factors can contribute to its development, most importantly seborrhea (an altered composition of cutaneous lipidic biofilm resulting in an oily-looking skin) and overgrowth of the yeast Malassezia spp., which may lead to inflammation [23]. Infantile SD is localized at skin folds, where lesions may be oozing and lack scale, the scalp, and the forehead [24]. The involvement of the diaper area is frequent (up to 10% of the newborn) during the first 6 to 9 months of life [25] and needs to be differentiated from CD. In fact, ICD is often caused by feces with worsening factors such as chemical irritants, increased pH, and excessively hydrated skin [24]. ACD can also occasionally cause diaper dermatitis, due to fragrances, preservatives, and emulsifiers, although much less common than ICD or SD [24]. In adulthood, SD is characterized by sharply demarcated, yellow to brown, greasy scaling patches and plaques affecting the scalp, ears, face, pre-sternal chest, and more rarely intertriginous areas. SD and CD usually differ not only in the typical distribution of the lesions but also in the type of desquamation, which is greasy in SD.
Dyshidrosis is a common chronic dermatitis of the hands and feet that may cause occupational impairment and significant physical and psychological distress [26]. Both the pathogenesis and the prevalence of dyshidrosis are not known, but it is more common in young adults and affects men and women equally. It appears suddenly, mainly during warm seasons [27, 28], and is very pruriginous. Clinically, it manifests as a recurrent vesicular—not erythematous—eruption affecting the hands and feet, particularly the lateral and dorsal aspects of the digits [27]. The lesions persist for several weeks and resolve with desquamation [29]. On the contrary, ACD manifests with different clinical phases. In the acute phase, it may present with a localized, well-demarcated skin eruption most commonly on the hands or face, but it can also be more widespread [30]. Chronic ACD usually shows lichenification, fissuring, and scale [30]. Dyshidrosis is usually a chronic condition and recurrence is possible, while ACD relapses on contact with the culprit allergen. Besides the clinical characteristics of the lesions, differential diagnoses can be guided by a combination of history and patch test results.
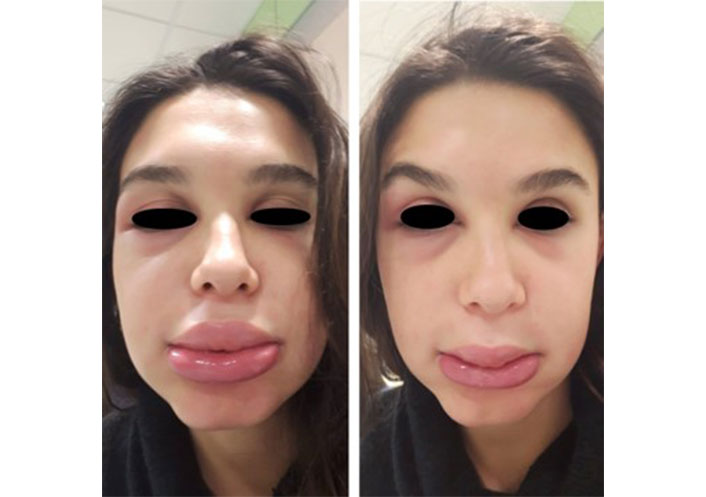
AD is a chronic and recurrent inflammatory skin disease caused by a complex interplay between the immune system, characterized by type 2 inflammation, the environment, and genetics, with mutations in genes encoding for structural proteins such as filaggrin (FLG), claudin 1 (CLDN1), and others [31]. The clinical characteristics of the lesions change over time and according to the age of onset. In infancy, we can find poorly defined erythematous patches or infiltrating erythematous-edematous plaques with papule microvesiculations and serous erosions, exacerbated by scratching, that progress from a wet to a crusty phase. These localize on the face, but the periorbital and perioral regions are normally uninvolved. In later childhood and adolescence, lesions primarily develop on the wrists, neck, antecubital, and popliteal fossae; eczematous cheilitis and eyelid dermatitis can also manifest. Acute flexural surface lesions present with painful fissures, erythematous patches or plaques, together with moist or crusty linear or punctiform erosions. On the other hand, lichenification, a thickening of the skin with accentuation of the pattern, is the main feature in the chronic stages. In eyelid dermatitis, signs of persistence are subpalpebral Dennie-Morgan folds and alopecia of the outer third of the eyebrows (Hertoghe sign). In adulthood, the lesions closely resemble that of an adolescent, but localization may narrow to the hands and eyelids (Figure 2). In the darker phototypes, AD might present as follicular eczema, which causes itchy follicular papules in the acute phase, and follicular lichenification, with punctiform desquamation in the chronic phase. Prurigo nodularis is common in older or extensively sensitized individuals, while others may show white dermographism and pityriasis alba [32]. An adult with newly developed dermatitis who has no history of childhood eczema or atopic disease should be evaluated for a variety of disorders. As a compass to guide us in the differential diagnosis of AD, we suggest evaluating the following items: time of onset, presence of itching, distribution of lesions, and skin features.
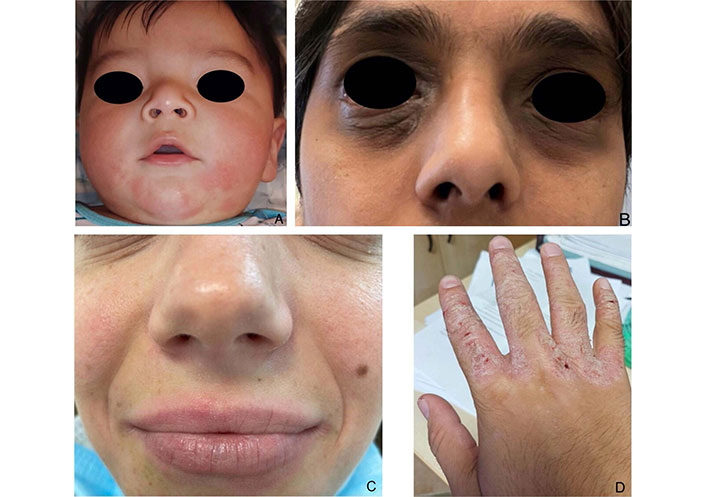
Atopic dermatitis (AD) manifestations stratified by age. (A) AD of the child; (B) eyelid dermatitis of the adult; (C) perioral cheilitis and Dennie-Morgan sign; (D) eczema of the hands
As previously seen, clinical manifestations of SD vary between infants and adults. In infancy, SD appears within the first months of life, affecting the frontal and parietal hairy scalp with an oily-looking, thick and fissured crust (cradle cap); intertriginous folds of the neck, axillae, groin, and anogenital area can also be involved with clearly outlined erythematous patches. The absence of pruritus, early onset, greasy instead of dry scaling of skin, salmon red skin, and the presence of cradle cap should lead us to the diagnosis of SD. On the other hand, moderate to severe pruritus, forearms and shins involvement, and a history of allergy, particularly to food allergens (milk, egg, and other geographically relevant allergens), should guide us towards AD. In adulthood, differential diagnosis is easier and should be based on localization and clinical features. In SD, lesions involve the scalp, the retroauricular area, the face with a T-shape distribution (eyebrows, glabella nose, and nasolabial folds), and the trunk with V-shaped areas; hands, arms, and legs, typical areas of AD, are rarely affected in SD. Xerosis, lichenification, and scratch wounds are typical in AD. White dandruff of the scalp, follicular and perifollicular redness, and flaky scales with mild redness that evolves to petaloid lesions characterized by oily thick scales and crusts on a mild to severe erythema, are characteristic of SD [33].
PSO can manifest in different forms. An acute form, such as guttate PSO, that presents as distinct salmon-pink drop-shaped papules; silvery scales, unusually loosely adhered to the skin surface, can reveal a pinpoint bleeding when removed (Auspitz sign). This form can be easily distinguished from AD, because of the characteristics of the lesions and their distribution. On the other hand, chronic forms manifest with erythematous purplish plaques with sharp margins, that tend to form polycyclic lesions, covered by silvery-white scales tenaciously adhered to the underlying skin (oyster-shell-like lesions) [34]. In these cases, differential diagnosis can be more difficult. It is important to note that these lesions are typically symmetric and localized at the scalp and extensor aspects of the extremities, especially knees and elbows, while in AD, other areas are involved. Furthermore, itching can be considered a prodromal symptom in AD, while patients with PSO manifest itching only after the development of skin plaques. Nail findings can also be a useful tool when PSO is suspected. Inflammation of the nail matrix can cause pitting depressions, leukonychia, red spots in lunula, and nail plate crumbling. Nail bed involvement can result in onycholysis, oil-drop discoloration, nail bed hyperkeratosis, and splinter hemorrhages [35]. As seen earlier, AD typically manifests in young children with other manifestations or a familiar history of atopy, while PSO is not associated with atopy and the mean age of onset is 20 years.
Scabies (SC), the most common human ectoparasitosis, is caused by Sarcoptes scabiei. The pathognomonic lesions, burrows, are small serpiginous tunnels in the stratum corneum created by a mature female mite laying eggs and stools. In immunocompetent hosts, SC presents with pruritus exacerbating at night, which might be accompanied by crusted papules on erythematous patches. Burrows develop where the skin lacks adnexa and the epithelium is thin and soft, such as interdigital spaces, ulnar side of the hands, wrists, ankles, axillae, areolae (for women), waist, and groin [24, 36]. In inveterate SC, reddish/purplish papules or nodules, with burrows on top, might develop, typically on the genital and scrotal area in men and breasts in women. Both SC and AD cause an intense generalized itching that leads to sleep disturbance; in the first case, it manifests with nocturnal exacerbations, awakening patients, while AD patients often complain of difficulty in falling asleep. The presence of contact cases within the family and the different distribution of lesions should also aid differential diagnosis. In immunocompromised patients, Norwegian SC can develop. It presents with thick, hyperkeratotic, grayish plaques with characteristic sand-like scales that cover vivid erythema; itching is not always present and the head and neck may be involved [24, 36]. Norwegian SC is frequently misdiagnosed as eczema, SD, drug eruptions, Sezary syndrome, and PSO. Clinical confirmation of SC can only be made by detecting burrows with dermoscopy. Parasitological confirmation with a microscope is essential in cases of crusted SC or SC in healthcare settings, but failure to find mites is common and does not rule it out [24, 36].
AD and cutaneous T-cell lymphoma, particularly mycosis fungoides (MF), unlike B-cell lymphomas, share many analogies [37]. In childhood, the most frequent cutaneous lymphoma is MF, especially the pilotropic and the hypopigmented patterns, which should be distinguished from pityriasis alba [38, 39]. In patients with adult-onset AD, differential diagnosis with MF, especially in the early stages, might be difficult [40] because MF can manifest as variably erythematous, finely scaling lesions, hypopigmented or hyperpigmented patches [39, 41–45]. Skin lesions can become confluent, develop into erythroderma, or occur directly as erythroderma [41, 46, 47]. Partial loss of elastic fibers and atrophy of the epidermis (characteristic of a long-standing lesion) may confer a typical wrinkled appearance “cigarette paper-like” and represent a clue against an inflammatory dermatosis [39]. The fixed character of the lesions, their annular or figurate appearance due to partial spontaneous regression, and the localization of the lesions on photo-protected sites may help to distinguish MF from AD [46]. A histopathological examination is essential for diagnosis since epidermotropism of atypical lymphocytes is a distinguishing feature of MF and is not seen in AD [46]. However, in the early stages of MF, epidermotropism is not always evident, thus immunohistochemistry and research for T cell receptor (TCR) monoclonal rearrangement are essential.
Dermatomyositis (DM) belongs to the idiopathic inflammatory myopathies (IIMs), a diverse group of diseases that affect the skeletal muscles and frequently coexist with extramuscular involvements including, cancer, interstitial lung disease (ILD), and rheumatoid arthritis [48]. Up to 20% of patients with recognized DM skin features do not or only partially manifest muscle dysfunction. “Clinically amyopathic” DM (CADM) may be difficult to diagnose [49]. Skin findings include Gottron’s papules (violaceous erythematous papules over interphalangeal joints) and Gottron’s sign (symmetric macular violaceous erythema over interphalangeal joints, olecranon processes, patellas, medial malleoli), both of which have been proposed to be “pathognomonic”. “Characteristic” skin findings include heliotrope rash (periorbital violaceous erythema), periungual telangiectasias and dystrophic cuticles, macular violaceous erythema over dorsal hands, forearms, arms, shoulders (shawl sign), V area of neck and chest, and lateral surface of the thighs or hips (Holster sign). “Compatible” findings include poikiloderma atrophicans vascularis, subepidermal bullous lesions and superficial erosions, and calcinosis cutis [48]. Besides the identification of pathognomonic and characteristic lesions, DM can also be differentiated from AD with the aid of autoantibodies. Furthermore, DM can manifest in association with ILD and cancer.
HAE is a rare genetic disease characterized by recurrent episodes of cutaneous or submucosal edema, due to an increase of bradykinin, that leads to temporary enhanced vascular permeability. First described by Osler in 1888 as “hereditary angioneurotic oedema”, different forms of this disease have now been identified and genetically characterized [50]. The most common ones are associated with a defect of the C1-inhibitor (C1-INH) production (HAE type I, ~85% of cases) or function (HAE type II, ~15% of cases). In fact, over 450 mutations in the SERPING1 gene have been reported. More recently, mutations of other genes have been associated with HAE, they encode for proteins such as factor 12 (FXII), angiopoietin-1 (ANGPT1), plasminogen (PLG), kininogen 1 (KNG1), myoferlin (MYOF), or heparan sulfate-glucosamine 3-O-sulfotransferase 6 (HS3ST6) [51]. Attacks most frequently involve the upper respiratory tract, extremities, and abdomen, though genitals, muscles, joints, and bladder can also be affected (Figure 3). They have a self-limiting course, variable duration, and are characterized by intense pain and deformity of the involved sites [52]. Laryngeal attacks can be the first manifestation of disease and can occur in over 50% of patients; if left untreated, they can lead to death [53]. Swelling can be anticipated by prodromal symptoms and signs such as fatigue, tingling, and marginate erythema. Symptoms usually manifest early in childhood, worsen during puberty, and fluctuate during the patient’s lifetime. A positive family history can aid the diagnosis, though it may not be present in up to 25% of cases, and severity and localization may vary among the same genetic kindred [51]. Measurement of plasma C4 levels, C1-INH protein, and C1-INH function are required for the diagnosis of HAE type I and II. Currently, HAE with normal C1-INH can only be diagnosed by genetic testing as there are no laboratory tests available. The differential diagnoses of HAE should take into consideration bradykinin-mediated types of acquired AE, such as acquired AE due to C1-INH deficiency (AAE-C1-INH) and angiotensin converting enzyme inhibitor (ACE-I)-AE, and histaminergic AE [idiopathic histaminergic acquired AE (IH-AAE)] in patients with chronic spontaneous urticaria (CSU) without wheals or allergic AE, as well as idiopathic acquired AE and abdominal pain attacks [54].
Abdominal pain occurs in 43% to 93% of patients with HAE. Approximately one third of patients with abdominal attacks undergo unnecessary surgery, such as appendectomy and exploratory laparotomy [55]. Pain localization is variable since the whole intestine, stomach, and colon can be affected. It may manifest as severe acute abdominal pain, often described as cramping or colicky, or as chronic recurrent abdominal pain of varying severity [56]. To date, there is no reliable marker to define abdominal AE upon presentation of symptoms. Blood tests are not decisive because they can show an elevation of leucocytes, which is present in many other abdominal conditions [55]. However radiologic imaging performed with the correct timing is extremely important: ultrasound or CT scan allows to identify the transitional presence of abundant free fluid in the abdominal cavity and intestinal swelling, which spontaneously disappears [57]. In conclusion, even in patients with a confirmed diagnosis of HAE, the diagnosis of an abdominal attack depends upon clinical judgment and is supported by imaging findings and the efficacy of specific treatments [55].
AAE-C1-INH is an extremely rare condition that can be caused by the presence of anti-C1-INH neutralizing antibodies and/or the consumption of C1-INH by neoplastic lymphoid cells. It is often associated with autoimmune diseases, lymphoproliferative diseases, or other malignancies; interestingly, AE attacks can precede several years of the onset of the hematological disease [58]. Symptoms of AAE-C1-INH are indistinguishable from those experienced by patients with HAE. Therefore, other clinical characteristics should be carefully taken into consideration when making the diagnosis. Besides the comorbidities associated with AAE-C1-INH, another distinguishing feature of the disease is its late age of onset, as it often manifests after the fourth decade of life. The lack of family history of recurrent AE attacks should also guide the diagnosis [59]. Laboratory test results show a reduction of the complement component C4, during and between attacks, and lower levels of the functional C1-INH test. This, however, is also seen in patients with HAE. A useful tool to aid the diagnosis is the measurement of the C1q level, as it is decreased only in AAE-C1-INH [54]. In conclusion, AAE-C1-INH should be suspected in all patients with a late onset of AE who have autoimmune or lymphoproliferative diseases; measurement of the C1q can help to discriminate between other forms of AE.
Drug-induced AE can be divided into three categories. The first one comprises IgE-mediated hypersensitivity reactions, with the drugs most commonly involved being penicillins, cephalosporins, iodinated organ contrast agents, neuromuscular blocking agents, pyrazolones, quinolones, and non-steroidal anti-inflammatory drugs (NSAIDs). In the case of NSAIDs, however, the most frequently involved mechanism is the non-allergic inhibition of cyclooxygenase, which leads to alterations in the metabolism of arachidonic acid and the overproduction of cysteinyl leukotriene [60]. The third type of drug-induced AE is a bradykinin-mediated type of acquired AE [61]; the drugs most frequently involved are ACE-Is. ACE is involved in the degradation of bradykinin, which can accumulate, thus leading to vasodilation and increased vascular permeability through the production of nitric oxide and prostaglandins [62]. The incidence of AE in patients taking ACE-Is ranges from 1 to 7 events per 1,000 patients [63]. Individuals with an intrinsic defect in bradykinin degradation have a propensity to develop AE with ACE-I. Furthermore, ACE also acts by degrading substance P, a peptide with vasodilatory properties. Angiotensin receptor blockers (ARBs) have also been associated with the development of AE, though the mechanism is not fully understood. In some cases, the appearance of IgG and IgA autoantibodies against ARBs, with cross-reactivity towards complement 1-esterase inhibitor, has been observed [64]. Fundamental in the differential diagnosis is the anamnesis, especially aimed at identifying drugs frequently associated with AE. Clinically, the lips, tongue, face, and upper airway are most commonly affected, though the intestine can also be involved. In these patients, C1-INH levels and function are within the normal range.
IH-AAE is one of the most common causes of AE without urticaria [65]. The etiology of IH-AAE is unknown, but the interruption of recurrences after administration of H1-histamine receptor blockers suggests an important role of mast cells and/or blood basophils in its pathogenesis [66]. IH-AAE typically spreads in minutes or a few hours, reaching its maximum within six hours, and resolves within 12–24 h [66, 67]. Nevertheless, Faisant et al. [68] reported a case series of 31 patients whose IH-AAE attacks lasted on average 28 h, suggesting that IH-AAE may last more than 24 h. IH-AAE involves the face and extremities, with a sparing of gastrointestinal mucosa [66, 67]. Some authors describe no involvement of laryngeal mucosa [66], while others report swelling of the upper respiratory tract in more than half of cases [67]. Despite these clinical manifestations, response to antihistamines makes the prognosis good, with no intubation or death reported [66, 68]. IH-AAE may present recurrences for months or years [67]. Diagnosis is based on prompt response to antihistamines and exclusion of other causes of AE, such as allergic triggers (drugs, foods, or other environmental allergens), infections, autoimmune diseases, and C1-INH deficiency. Differential diagnosis between IH-AAE and HAE is based on anamnestic, clinical, and laboratory features, as well as different responses to treatment. In IH-AAE patients, there is no family history of AE and the disease may present at any age, without predilection for childhood [66, 69]. Symptoms of IH-AAE usually last less than those of HAE and are not accompanied by prodromal symptoms [52, 67], and patients do not experience abdominal pain [66]. Plasma levels and activity of C1-INH are normal. Finally, antihistamines markedly improve IH-AAE but have no effect on hereditary forms of AE [52, 69].
Urticaria is a disorder characterized by the development of transient wheals (< 24 h), AE, or both (Figure 4) [70]. Urticaria is classified into different subtypes based on the duration of symptoms (i.e., acute ≤ 6 weeks or chronic > 6 weeks) and the relevance of the eliciting factor (i.e., spontaneous if no specific eliciting factor is involved, or inducible if one is identified) [70]. CSU is a highly heterogeneous condition that may hide potential underlying causes such as autoimmunity and infections [71]. In addition, a wide variety of skin and systemic disorders may mimic CSU, making the differential diagnosis a challenge in the clinical practice. Herein we will focus on the most common differential diagnosis of CSU including urticarial dermatitis (UD), dermatitis herpetiformis (DH), urticarial vasculitis (UV), and bullous pemphigoid (BP).
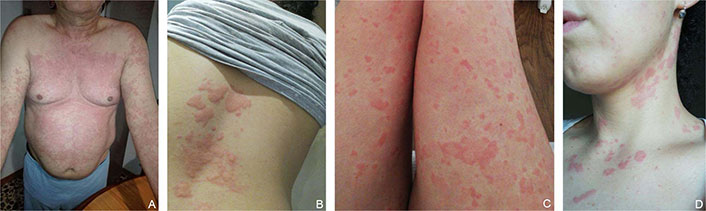
Different urticaria phenotypes. (A) Chronic spontaneous urticaria (CSU) of the trunk; (B) serpiginous and confluent CSU lesions of the back; (C) small punctiform CSU lesions of the limbs; (D) CSU of the neck in a young woman
UD is defined as a clinical entity presenting with itchy, erythematous plaques, and papules resembling urticaria, individually lasting longer than 24 h sometimes accompanied by eczematous lesions [72, 73]. The pathophysiology is thought to be a delayed type of hypersensitivity reaction [72, 74]. Clinical presentation may include the concomitance of plaques of urticated erythema and eczematous dermatitis-like areas or isolated urticarial papules and/or urticated patches [72, 74]. Clinical diagnosis may be challenging, especially when urticarial plaques rather than papules dominate the clinical presentation, as this condition may clinically mimic urticaria and other dermatological conditions such as UV, BP, DH, papular urticaria, drug reaction, viral exanthem, and herpes gestationis [72]. It is important to remember that the urticarial component of UD differs from conventional urticaria since the latter resolves in < 24 h [75]. Furthermore, UD primarily affects older patients (mean age of approximately 60 years) [72, 74], with a female predominance, and can sometimes be secondary to other underlying diseases [74]. A retrospective study showed that 15% of patients had a history of malignancies (e.g., myelodysplastic syndrome, pancreatic adenocarcinoma [76], prostate cancer, and vulval cancer) [74]. Association with autoimmune disorders and drug administration, such as rituximab and vaccines, has also been reported [77–79]. Cutaneous biopsies can be a useful diagnostic tool, usually revealing slight epidermal spongiosis with upper dermal perivascular lymphocytes and interstitial eosinophils with or without neutrophils and normal stratum corneum, in the absence of epidermal vesiculation or parakeratosis [72, 80].
DH is a rare cutaneous disease that can be considered the extraintestinal manifestation of celiac disease [81]. As celiac disease, it is strictly associated with human leukocyte antigen (HLA) DQ2 and DQ8 haplotypes [82] and it is caused by an autoimmune response to epidermal transglutaminase (TG3/eTG). It can occur at any age but affects primarily people between 30 to 40 years of age. The lesions are typically symmetric and usually visible in the extensor areas: lower limbs, elbows, dorsal forearms, buttocks, and sacral region. Scalp and neck are often affected, while mucosa oral lesions are spared [83]. The eruption is polymorphic with erythematous papules, urticarial plaques, and vesicles with serous or hemorrhagic content. Pruritus with burning sensation is an important clinical feature and many patients present erosions and crusted papules as a result of scratching [84]. Gastrointestinal symptoms occur only in 20% of patients [85]. As celiac disease, DH is associated with autoimmune diseases, such as thyroid disease, pernicious anemia, and type I diabetes, and a higher risk of non-Hodgkin lymphoma. Differential diagnoses with CSU should be guided primarily by the localization and duration of the lesions. The gold standard for the diagnoses is direct immunofluorescence (DIF) on skin biopsies, as it shows pathognomonic IgA deposits in a granular pattern in the dermal papillae and/or dermoepidermal junction [86]. Anti-TG3 antibodies are considered the primary diagnostic serology and are preferred over anti-TG2 antibodies for their superior specificity [87, 88].
BP is the most common subepidermal autoimmune blistering disease and mostly affects individuals aged > 60 years [89]. It is characterized by tissue-bound and circulating autoantibodies directed against components of hemidesmosomes involved in the dermal-epidermal cohesion: BP antigen 180, BP antigen 230, or both. Risk factors for BP include old age, neurologic diseases, and systemic therapies such as antibiotics, beta-blockers, NSAIDs, diuretics, anti-tumor necrosis factor (TNF)-α, dipeptidyl peptidase 4 inhibitors (DPP-4i), and immune checkpoint inhibitors [90]. Typically, it presents with tense blisters filled with serous and hemorrhagic fluid on erythematous skin. These blisters can persist for a few days before rupturing, leaving eroded and crusted areas. Predilection sites include the lower abdomen, the anterior and inner thighs, and the flexor region of the forearms. Mucous membranes are affected only in 10–35% of patients, with prevalent involvement of the oral mucosa, where blisters or erosions are generally non-scarring [91]. Up to 20% of BP patients have a “non-bullous prodromal phase” that lasts for weeks or months and, more rarely, some do not develop blisters at all (“non-bullous cutaneous pemphigoid”) [92]. Clinically this subtype of BP is characterized by intense pruritus which is associated with polymorphic lesions that are often urticarial in appearance, though in some cases lesions may be lacking. Diagnosis can be made by performing DIF of a biopsy from perilesional skin [90, 93] that reveals linear deposition of IgG and C3 along the dermo-epidermal junction. Serological tests that may confirm the diagnosis of BP include indirect immunofluorescence (IIF) on epithelial substrates (monkey esophagus; salt split skin) and ELISA [90].
UV is a leukocytoclastic small vessel vasculitis that frequently involves dermal capillaries and postcapillary venules [94]. It is a rare disease, mostly affecting middle-aged women. Different triggering factors have been identified, such as infections, medication reactions, autoimmune diseases, underlying malignancies, and others, though often the underlying cause remains unknown. All these factors can lead to complement activation and the generation of C3a and C5a, which act as anaphylatoxins, leading to mast cell degranulation. Based on complement levels, UV can be divided into normocomplementemic UV (NUV) and hypocomplementemic UV (HUV) [95]. Clinically, in UV urticarial plaques persist for more than 24 h leaving residual hyperpigmentation or ecchymoses, while in CSU wheals generally undergo spontaneous resolution in 2 h to 8 h (and always within 24 h). Furthermore, frank signs of vasculitis/purpura may be observed. While UV is generally associated with itchy or even painful lesions in up to one-third of patients, patients with CSU experience only itching. Response to antihistamines can also aid in the differential diagnoses, as patients with UV do not respond even after up-dosing [96]. Laboratory exams show low C1q levels and normal C1-INH levels, in association with anti-C1q antibodies in 55% of patients with HUV. Histologically, skin samples reveal dermal capillaries and postcapillary venules with leukocytoclastic vasculitis (fragmentation of leukocytes with nuclear debris), fibrinoid deposits, perivascular infiltrates composed of neutrophils, extravasation of red blood cells, and injury and swelling of the endothelial cells. DIF often reveals immunoglobulin and complement linear deposition in or around blood vessels of the upper dermis and/or at the dermal-epidermal junction [97].
The term “mastocytosis” refers to a group of distinct disorders characterized by a clonal expansion of mast cells and their subsequent accumulation in different tissues and organs. It has been considered part of the myeloproliferative disorders for many years, but in 2016 it has been classified as an independent entity [98]. We can distinguish two forms of mastocytosis, the CM, where the skin is the only organ involved, and the systemic mastocytosis (SM) where many other organs, such as the bone marrow and bones, may be affected at the same time [98, 99].
Skin involvement can be considered the most characteristic feature of both CM and SM. Cutaneous lesions may be single (mastocytoma of the skin) or multiple macules or papules, often showing a pathognomonic positive Darier sign (lesion urtication after local scratching) [100]. Color may vary from yellowish to red and brown and commonly affected areas are the trunk and the extremities, especially in adults with diffuse forms [100]. Maculopapular lesions are common in both children and adults with mastocytosis. However, SM is an extremely rare entity during childhood, while if “urticaria pigmentosa-like” lesions (Figure 5) are present in adulthood, SM should always be investigated [101]. Furthermore, the persistence of these lesions in adulthood suggests a possible transition to SM. The differential diagnosis between CM and SM is based on the involvement of other organs other than the skin, in the latter. Clinically, this can be suspected in the presence of flushing, itching, urticaria, abdominal pain, diarrhea, osteoporosis, hematological disorders, and anaphylactic reactions to several triggers [99]. A definite diagnosis requires bone marrow or other organ biopsies, to identify the presence of mast cells in aggregates [98, 99].
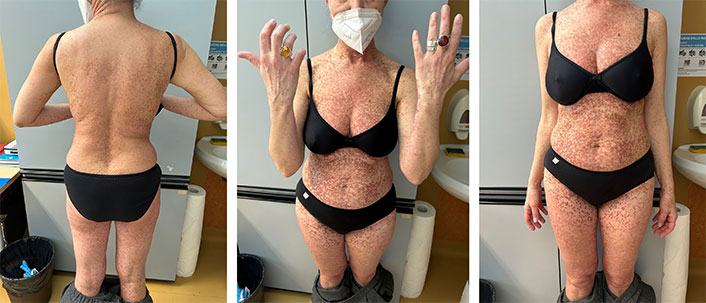
Cutaneous mastocytosis urticaria pigmentosa of the adult. Used with permission from Dr. Gabriella Perillo, Resident in Dermatology, Department of Health Sciences, Section of Dermatology, University of Florence
Hereditary alpha-tryptasemia (HαT) is an autosomal dominant genetic condition, caused by increased copy numbers of the α-tryptase encoding gene (TPSAB1) on chromosome 16p13.3, resulting in elevated basal serum tryptase (BST) levels and multisystem disorders [102]. The prevalence of this condition has been estimated as 4–6 % in the general population [103]. Affected patients may have high normal (> 8 ng/mL) or clearly high (> 20 ng/mL) BST [104]. Clinical manifestations of HαT may resemble symptoms described in clonal mast cell disorders, including flushing, pruritus, and urticaria. Other symptoms are autonomic dysfunction, functional gastrointestinal symptoms (irritable bowel syndrome, dysmotility, and chronic gastroesophageal reflux), anaphylaxis (food-mediated, insect stings, and idiopathic), pain (chronic arthralgia, headache, and body pain), neuropsychiatric manifestations, and musculoskeletal abnormalities (joint hypermobility and retained primary dentition) [105, 106]. Such as for clonal mast cell disorders, these symptoms can be chronic or episodic, sometimes induced by specific triggers, such as exercise, vibration, stress, food, heat, or physical trauma [107]. In contrast to SM, “urticaria pigmentosa-like” lesions are never present in HαT. However, recent studies have revealed a significantly higher HαT prevalence in patients with SM (12–17%) [103, 107, 108]. Therefore, the presence of mast cell associated disorders does not rule out the diagnosis of HαT. Furthermore, the coexistence of both disorders associates with a higher risk of life-threatening anaphylactic episodes and more severe mediator-related symptoms [106, 109]. Thus, TPSAB1 genotyping could be a novel biomarker for the risk of developing severe anaphylaxis in patients with mastocytosis.
The increasing prevalence of allergic and immunologic skin diseases requires clinicians to feel confident in distinguishing these common pathologies. This document does not aim to provide an exhaustive and comprehensive description of all these skin diseases. Instead, it focuses on critical clinical aspects that should be taken into consideration when navigating through the differential diagnosis of CD, AD, HAE, urticaria, and CM.
AAE-C1-INH: acquired angioedema due to C1-inhibitor deficiency
ACD: allergic contact dermatitis
ACE: angiotensin converting enzyme
ACE-I: angiotensin converting enzyme inhibitor
AD: atopic dermatitis
AE: angioedema
BP: bullous pemphigoid
C1-INH: C1-inhibitor
CD: contact dermatitis
CM: cutaneous mastocytosis
CSU: chronic spontaneous urticaria
DH: dermatitis herpetiformis
DIF: direct immunofluorescence
DM: dermatomyositis
HAE: hereditary angioedema
HαT: hereditary alpha-tryptasemia
ICD: irritant contact dermatitis
IH-AAE: idiopathic histaminergic acquired angioedema
MF: mycosis fungoides
NSAIDs: non-steroidal anti-inflammatory drugs
PSO: psoriasis
SC: scabies
SD: seborrheic dermatitis
SM: systemic mastocytosis
UD: urticarial dermatitis
UV: urticarial vasculitis
EC and M Perlato equally contributed to: Conceptualization, Data curation, Project administration, Supervision, Writing—original draft, Writing—review & editing. EN, MC, and OR: Conceptualization, Writing—original draft. L Salvati, EDA, AR, DP, FA, RM, SM, NS, MT, LDB, M Passante, MDP, ADM, LP, LA, RC, IM, IT, MEC, SF, RG, AZ, EB, AB, DB, MN, MB, TDP, FG, CP, KH, FLF, IZ, ST, SG, CF, and L Stingeni: Writing—original draft. All authors read and approved the final version of the manuscript.
The authors declare that they have no conflicts of interest.
Ethical approval was waived by the “AREA VASTA CENTRO” Ethics Committee since this is a scientific literature review.
The informed consent to participate in the study was obtained from relevant participants or the parents of relevant participants (under 16 years old).
The informed consent to publication was obtained from relevant participants or the parents of relevant participants (under 16 years old).
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 14328
Download: 91
Times Cited: 0