
Abstract
Lipoprotein(a) [Lp(a)] is composed of a low-density lipoprotein (LDL) and glycoprotein (a)—apolipoprotein(a) [apo(a)]. The size and concentration of Lp(a) in serum can vary among individuals and is determined by genetic factors. The environmental factors, diet, and physical activity have a negligible effect on Lp(a) level. Observational, epidemiological, and genetic studies improved that high levels of Lp(a) > 50 mg/dL (> 125 nmol/L) have been associated with an increased risk of myocardial infarction (MI), stroke, and calcific aortic valve stenosis (CAVS). It is recommended to measure Lp(a) at least once in adults to identify individuals with a high cardiovascular risk. This screening is particularly important in certain populations, including: youth with a history of ischemic stroke or a family history of premature atherosclerotic cardiovascular disease (CVD; ASCVD) or high Lp(a), individuals with recurrent cardiovascular events despite optimal hypolipemic treatment and no other identifiable risk factors or patients with familial hypercholesterolemia (FH). Considering Lp(a) levels in the evaluation of cardiovascular risk can provide valuable information for risk stratification and management decisions. However, it’s important to note that the treatments of elevated level of Lp(a) are limited. In recent years, there has been ongoing research and development of new drugs targeting Lp(a): pelacarsen—antisense oligonucleotide (ASO), and olpasiran—a small interfering RNA (siRNA).
Keywords
Lipoprotein(a), olpasiran, pelacarsen, stroke, calcific aortic valve stenosis, myocardial infarction, diabetesIntroduction
Lp(a) is a molecule similar to low-density lipoprotein (LDL), consisting of a lipid core (cholesterol esters and triacylglycerols) which is surrounded by a shell composed of proteins, phospholipids, and free cholesterol. Apolipoprotein B (apoB)-100 is a large protein that plays a crucial role in lipid metabolism and transport. It acts as a ligand for the LDL receptor (LDLR), allowing the uptake of Lp(a) particles into cells. Moreover, the Lp(a) molecule contains glycoprotein—apolipoprotein(a) [apo(a)] with a similar structure to plasminogen. Apo(a) consists of two three-loop structures known as kringle domains: kringle IV (KIV) and kringle V (KV). Among these domains, KIV1, KIV3-10, and KV occur in one copy, while KIV2 may occur in varying numbers, ranging from 1 to more than 35 copies. The number of KIV2 repeats is determined by genetic factors. KIV10, one of the kringle domains, serves as the site where oxidized phospholipids (OxPL), which are highly atherogenic substances, can covalently bind (Figure 1) [1–4].
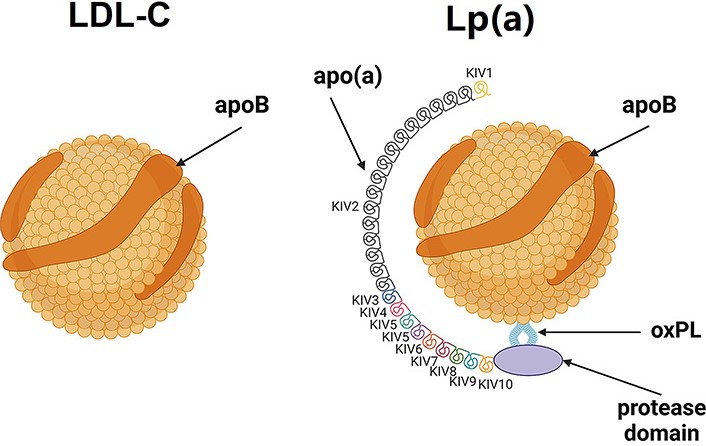
Apo(a) is synthesized in the liver [1]. The gene responsible for apo(a) production is the lipoprotein(a) [Lp(a)] gene (LPA). The size of apo(a) can vary among individuals due to genetic polymorphisms, resulting in different isoforms of Lp(a) [1–4]. A larger number of KIV2 repeats (≥ 23) results in a larger apo(a) isoform. Large apo(a) isoforms are less secreted from hepatocytes, and are more susceptible to degradation by protease [5], resulting in lower Lp(a) serum levels. Studies on the kinetics of apo(a) production have shown that individuals with smaller apo(a) isoforms exhibit higher synthesis rates compared to those with larger apo(a) isoforms [6]. There is an inverse correlation between apo(a) isoform size and plasma Lp(a) concentration (Figure 2). Patients with smaller apo(a) isoform size have higher Lp(a) concentrations [1–4].
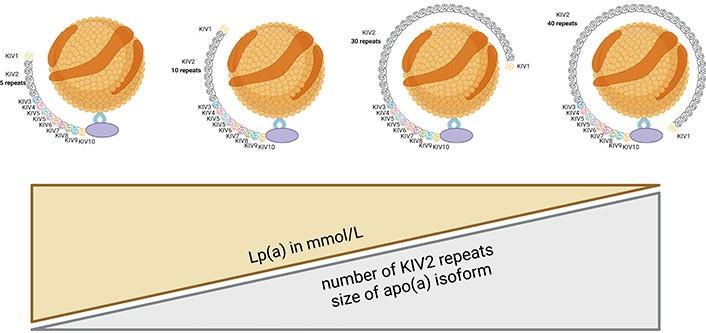
The concentration of Lp(a) in plasma is inversely proportional to the number of KIV2 repeats
Lp(a) particles are primarily cleared from the blood by the liver. The liver expresses specific receptors, such as the LDLR-related protein 1 (LRP1) and the scavenger receptor class B type I (SR-BI), which are involved in the uptake of Lp(a). Once taken up by the liver cells, Lp(a) undergoes intracellular degradation, leading to the breakdown of apo(a) and the release of LDL particles [7, 8]. The LDL particles released after Lp(a) degradation can be further metabolized through different pathways. They can be taken up by the liver via the LDLR or cleared by extrahepatic tissues that express LDLRs, contributing to overall LDL clearance.
Factors modifying the concentration of Lp(a)
Lp(a) concentrations are determined by the LPA gene, located on chromosome 6q26-q27. The variations in Lp(a) levels are predominantly influenced by the number of KIV2 repeats, which account for approximately 70% of the variation [1–4]. Most individuals have two different apo(a) isoform sizes (a small and a large one). In addition to the varying number of KIV2 repeats, the size and level of the Lp(a) isoform are determined by more than 200 single nucleotide polymorphisms (SNPs) in the LPA gene. These SNPs are variations in a single nucleotide within the DNA sequence and can impact gene expression and protein function. Among the SNPs, rs10455872 and rs3798220, rs 9457951 have the greatest impact on Lp(a) levels [9]. Furthermore, other genetic loci have been associated with Lp(a) levels, for example, the APOE, cholesterol ester transport protein (CETP), and APOH [8–10].
The concentration of Lp(a) rises very rapidly after birth and reaches a steady level in infancy. The circulating Lp(a) remains relatively stable throughout a lifetime [1–4]. Dietary, environmental factors, physical activity, and menopause have minimal effects on plasma Lp(a) levels [11, 12]. The non-genetic factors that can affect the Lp(a) concentrations are shown in Table 1 [4]. Many studies suggest that women have 5–10% higher concentrations of Lp(a) than men regardless of race [13, 14]. Higher concentrations of Lp(a) have been reported in Black than in Hispanics, Caucasians, and/or Asians [13–15]. However, all races are equally at risk for myocardial infarction (MI), stroke as a result of Lp(a) concentrations [16]. The Atherosclerosis Risk In Communities (ARIC) study [14], which had a 20-year follow-up period, found that Lp(a) levels were positively associated with cardiovascular events in both black and white subjects. Interestingly, despite black individuals having a larger range of Lp(a) distribution, the association with cardiovascular events was similar between the two racial groups. Among European populations, Finnish individuals were found to have the lowest concentrations of Lp(a) [17]. The Dallas Heart Study investigated specific genetic variations (SNPs) related to Lp(a) in different racial and ethnic groups. The study identified that the LPA SNP rs3798220 was most prevalent in Hispanics, rs10455872 in Whites, and rs9457951 in Blacks [18].
The non-genetic factors affecting Lp(a) levels [4]
| Non-genetic factors | Additional comments | |
|---|---|---|
| Factors increasing Lp(a) concentration | Hypothyroidism | 5–20% increase |
| Thyrostatic therapy, radioactive iodine therapy | 20–25% increase | |
| Acute and chronic inflammatory diseases | Increase related to interleukin-6 concentration | |
| Growth hormone | 2 times increase | |
| Pregnancy | 2 times increase | |
| Chronic kidney disease | Nephrotic syndrome: 3–5 times increase Peritoneal dialysis 2 times increase | |
| Protease inhibitors or antiretroviral therapy | Increase | |
| Air pollution (fine particulate) | Slight increase | |
| Factors decreasing Lp(a) concentration | Sepsis | Decrease |
| Postmenopausal hormonal replacement therapy | 25% decrease | |
| Tocilizumab | 30–40% decrease | |
| Low carbohydrate/high-fat diet | 10–15% decrease | |
| Hyperthyroidism | Decrease | |
| Thyroid hormone replacement therapy | 5–20% decrease | |
| Liver failure | Decrease | |
Threshold of Lp(a) concentration
Lp(a) concentrations in the general population range widely from < 0.1 mg/dL to > 200 mg/dL [19]. Cardiovascular risk increases linearly with increasing Lp(a) levels [19]. However, above the threshold value, the risk increases sharply [19]. The universal cut-off point for Lp(a) concentration is difficult to determine due to various measurements, methods, ethnic differences, and co-morbidities [4]. Currently, it seems that the most pragmatic position, extremely useful in clinical practice, has been adopted by the European Atherosclerosis Society (EAS) Experts Panel since 2022 [4]. According to the consensus, an Lp(a) level of less than 30 mg/dL or less than 75 nmol/L is suggested as a cut-off to “rule out” risk. This means that individuals with Lp(a) levels below these thresholds are considered to have a lower risk of cardiovascular diseases (CVDs). On the other hand, an Lp(a) level of greater than 50 mg/dL or greater than 125 nmol/L is proposed as a cut-off to “rule in” risk. This means that individuals with Lp(a) levels above these thresholds are considered to have a higher risk of CVD [4].
According to most guidelines, a concentration of Lp(a) ≥ 125 nmol/L (≥ 50 mg/dL) is considered a high risk (Table 2) [20–24]. In children, an Lp(a) >30mg/dl (> 75 nmol/L) is associated with a higher risk of recurrent ischaemic stroke [25]. According to the EAS is it noteworthy that individuals with extremely high Lp(a) concentrations [> 430 nmol/L (> 180 mg/dL)] may have high cardiovascular risk, similar to the risk in heterozygous familial hypercholesterolemia (FH) [26].
Thresholds of Lp(a) levels for estimation high cardiovascular risk related to the guidelines
| Statement/Guidelines | Cut-off |
|---|---|
| American College of Cardiology/American Heart Association Cholesterol Guidelines 2019 [20] | ≥ 125 nmol/L (≥ 50mg/dL) |
| National Lipid Association Scientific Statement 2019 [21] | ≥ 100 nmol/L (≥ 50mg/dL) |
| HEART UK Consensus Statement 2019 [22] | > 90 nmol |
| Endocrine Society Lipid Management Guidelines 2020 [23] | ≥ 125nmol/L (≥ 50mg/dL) |
| Canadian Guidelines for the Management of Dyslipidemia 2021 [24] | ≥ 100 nmol/L (≥ 50mg/dL) |
Lp(a) and CVD
The Copenhagen City Heart Study (CCHS) revealed that individuals with Lp(a) levels ≥ 120 mg/dL (≥ 95th percentile) had a significantly higher risk of MI compared to those with levels < 5 mg/dL (< 22nd percentile). The adjusted hazard ratios (HR) for incident MI were 3.6 in women and 3.7 in men [27]. The analysis of the UK Biobank database [19], which included a large number of participants (n = 460506), demonstrated a linear association between Lp(a) levels and the risk of atherosclerotic CVD (ASCVD) in both primary and secondary prevention. In primary prevention, the ASCVD risk increased for Lp(a) concentrations above 25 mg/dL [28], while in secondary prevention (recurrent events), the risk increased at values > 50 mg/dL [29]. Genetic studies have provided further evidence supporting the causal association between Lp(a) and coronary artery disease (CAD). Genetic variations in the LPA gene, which influence Lp(a) levels, have been found to be associated with CAD risk. This suggests that Lp(a) itself, rather than confounding factors, contributes to the development of CAD. The LPA variants rs1045587 and rs3798220 have been identified as being responsible for high levels of Lp(a) and reduced copy number of KIV2 repeats [30]. Studies have shown that these LPA variants and the resulting high Lp(a) levels are strongly associated with an increased risk of CAD. The association between these variants and CAD risk is considered to be stronger than the associations observed for LDL and proprotein convertase subtilisin/kexin 9 (PCSK9) gene loci, which are also known to be associated with CAD [31]. A meta-analysis of three large clinical trials involving patients with CAD revealed that individuals with Lp(a) levels in the highest quantile (top 25% of Lp(a) concentrations) had a 40% higher risk of major cardiovascular events (MACE) compared to those in the lowest quantile (bottom 25% of Lp(a) concentrations) [32].
Lp(a) and aortic stenosis
Calcific aortic valve stenosis (CAVS) is the most common acquired valve defect in the elderly. Currently CAVS remains a life-threatening condition with no proven disease-modifying pharmacotherapy. The genetic variation LPA rs10455872 has been associated with an increased risk of CAVS and the need for aortic valve replacement, independent of plasma Lp(a) levels [33]. This implies that this specific genetic variation plays a role in contributing to the risk of CAVS. Higher Lp(a) concentrations (> 58.5 mg/dL) were associated with a 1.5-time greater likelihood of progression to aortic stenosis and a 2-fold increase in the risk of valve replacement [34]. This indicates that elevated Lp(a) levels are associated with a higher risk of disease progression. Another study identified a cut-off value of 47.7 mg/dL for Lp(a) concentration as a marker for estimating the risk of CAVS [35]. Data from the SAFEHEART study involving patients with FH demonstrated that Lp(a), along with other risk factors such as older age, hypertension, and high levels of LDL, can predict the need for aortic valve replacement due to CAVS [2]. The EPIC-Norfolk prospective study involving a general population in the UK (17,553 adults) showed that individuals in the highest tertile of Lp(a) had a 57% higher risk for CAVS compared to those in the lowest tertile [36]. In a sub-analysis of the FOURIER study, increasing Lp(a) levels were associated with a higher risk of aortic stenosis progression or the need for aortic valve replacement [37]. The adjusted HR for CAVS was 1.55 per 1-SD increase in Lp(a) levels [37]. Notably, LDL-C levels were not found to be related to CAVS progression, suggesting that Lp(a) may play a more significant role than LDL-C in the pathogenesis of CAVS. These findings collectively suggest that Lp(a) is implicated in the development and progression of CAVS and may have a stronger association with CAVS than LDL-C. However, it’s important to note that further research is needed to fully understand the mechanisms underlying this relationship and to explore potential therapeutic interventions targeting Lp(a) for CAVS.
Lp(a) and stroke
The previous observational studies examining the association between Lp(a) and stroke have yielded inconclusive results [38, 39]. The largest meta-analysis conducted by the Emerging Risk Factor Collaboration indicated that a 3.5-fold rise in Lp(a) levels was associated with a 10% increased relative risk (RR) for ischemic stroke. These findings suggested that Lp(a) is an independent risk factor for ischemic stroke [40]. In a prospective study involving individuals from the CCHS (n = 10,813) and Copenhagen General Population Study (CGPS; n = 49,699), it was found that patients with Lp(a) levels above 93 mg/dL (> 199 nmol/L) had a 1.6-fold higher HR for ischemic stroke compared to those with low levels of Lp(a) below 10 mg/dL (< 18 nmol/L) [41]. The study also observed a higher absolute 10-year risk of ischemic stroke in specific subgroups, such as active smoker women above the age of 70 suffering from arterial hypertension [41]. The high levels of Lp(a) and specific LPA genotypes, such as the variant rs10455872, are associated with an increased risk of ischemic stroke. However, the risk estimates for ischemic stroke associated with Lp(a) levels are reported to be less pronounced compared to those observed for MI and CAVS [41]. Studies examining different stroke subtypes have shown varying associations [42]. The ARIC study found that high Lp(a) levels increased the adjusted risk for non-lacunar stroke but not for lacunar or cardioembolic stroke [43]. A Korean study also observed higher Lp(a) levels in patients with large artery atherosclerotic stroke compared to other stroke types, with a significant correlation between Lp(a) levels and the degree of intracranial and extracranial carotid stenosis [44]. Current evidence is insufficient to establish a clear link between elevated Lp(a) levels and an increased risk of recurrent stroke [45]. However, a Chinese study indicated an increased risk of recurrent ischemic stroke events within the first three months after the initial event. Overall, these findings suggest that high Lp(a) levels are associated with an increased risk of ischemic stroke, particularly in certain subgroups and specific stroke subtypes. However, the exact relationship and the impact on recurrent stroke risk require further investigation.
Lp(a) and diabetes
Several studies suggest that lower levels of Lp(a) are associated with an increased risk of type 2 diabetes [46–48]. This association is particularly evident among individuals with very low Lp(a) concentrations. The highest risk of type 2 diabetes is observed when Lp(a) levels are below 1 mg/dL [49]. The exact mechanism underlying the inverse association between Lp(a) and diabetes is not fully understood. It is unclear whether the association is due to the effect of insulin resistance or hyperinsulinemia in suppressing Lp(a) levels, or if low levels of Lp(a) are causally related to the development of diabetes. Some studies suggest that high concentrations of insulin may suppress the synthesis of apo(a), the protein component of Lp(a) [48]. Additionally, glycation-related modifications of apo(a) in patients with type 2 diabetes may contribute to increased molecular weight apo(a) and lower Lp(a) levels [50]. While some studies have observed an inverse association between Lp(a) levels and type 2 diabetes, a Mendelian randomization analysis showed no relation between genetic variants associated with fasting insulin levels and Lp(a) concentrations [51]. Moreover, analyses of certain cohorts did not find an association between the LPA variant rs10455872 and incident type 2 diabetes [31, 39].
European consensus panel recommendations for Lp(a) testing
It is recommended to measure Lp(a) at least once in adults to identify individuals with a high cardiovascular risk [4]. This screening is particularly important in certain populations, including:
Youth with a history of ischemic stroke or a family history of premature ASCVD or high Lp(a).
Individuals with recurrent cardiovascular events despite optimal hypolipemic treatment and no other identifiable risk factors.
Individuals with FH, a family history of (very) high Lp(a), or personal/family history of ASCVD.
Repeated measurement of Lp(a) is generally not necessary, except in specific cases such as acute infections, acute and chronic kidney disease, or liver disease. Including Lp(a) levels in the assessment of cardiovascular risk allows for a more accurate estimation of risk, particularly for patients at borderline high and moderate risk categories [4]. Considering Lp(a) levels in the evaluation of cardiovascular risk can provide valuable information for risk stratification and management decisions.
Treatment to reduce Lp(a)
Among hypolipemic drugs, only a few have been shown to significantly reduce Lp(a) levels. These include nicotinic acid and estrogens, which can lead to a reduction of approximately 30% in Lp(a) levels. However, randomized trials with these drugs in cardiovascular prevention have not shown consistent benefits [52]. There are newer drugs that have shown some efficacy in reducing Lp(a) levels:
Mipomersen: an antisense oligonucleotide (ASO) designed to inhibit the synthesis of human apoB-100 in the liver. During weekly therapy with doses ranging from 100-300 mg per week, it has been shown to reduce Lp(a) levels by 15% to 25% [53].
Anacetrapib: a CETP inhibitor. In a dose of 100 mg daily, it has been shown to reduce Lp(a) levels by 36% after 24 weeks of treatment [54].
PCSK9 inhibitors: these drugs, such as evolocumab and alirocumab, are members of the subtilisin/kexin family. They are primarily used for reducing LDL-C levels but have also been shown to lower Lp(a) levels by approximately 28–30% along with reductions in LDL-C and apoB [55].
Inclisiran: a small interfering RNA (siRNA) against PCSK9. It has also been shown to lower Lp(a) levels by 28–30% in addition to reducing LDL-C and apoB [56].
It’s important to note that the effectiveness of these drugs in reducing cardiovascular events and improving clinical outcomes may vary, and further research is needed to determine their long-term benefits and safety profiles.
Metformin treatment has been associated with a reduction in Lp(a) levels by 21% [57]. Similar reductions have been observed with other antidiabetic medications such as glimepiride, gliclazide, and rosiglitazone [57]. The effect of statins on Lp(a) remains unclear and controversial [58]. Some studies have suggested a modest reduction in Lp(a) levels with statin therapy, while others have not shown a significant effect [59]. A meta-analysis evaluating the efficacy of ezetimibe, either alone or in combination with statins, did not demonstrate a beneficial effect on Lp(a) levels [59].
Lipoprotein apheresis (LA) is a treatment option for individuals with extremely high Lp(a) levels. It involves the removal of Lp(a) from the blood, leading to transient reductions in Lp(a) concentrations ranging from 53% to a maximum of 73% from baseline values [60]. LA is approved in certain countries, such as Germany and Turkey, for patients with Lp(a) levels greater than 60 mg/dL and with progressive CVD despite maximal lipid-lowering therapy. However, LA is rarely used due to its inconvenience to patients, requiring weekly or biweekly sessions lasting 2 to 3 hours each for the rest of the patient’s life [61].
The ongoing study called “The effect of Lp(a) elimination by LA on cardiovascular outcomes (MultiSELECT)” aims to compare the clinical effects of weekly LA with maximally lipid-lowering therapy in patients with initial Lp(a) levels greater than 120 nmol/L and with progressive ASCVD [61]. The study will assess major adverse cardiovascular events (MI, percutaneous coronary intervention, coronary artery bypass graft surgery, ischemic stroke, transient ischemic attack, peripheral arterial revascularization, and cardiovascular death) in a population of 1,000 patients.
Novel therapies
There are several RNA-based therapeutics in clinical development that aim to reduce Lp(a) levels [62]. Pelacarsen is a hepatocyte-directed ASO that targets the mRNA of the LPA gene. By binding to hepatocyte apo(a) mRNA, pelacarsen prevents the translation of apo(a), leading to decreased apo(a) synthesis and lower circulating Lp(a) levels. Clinical trials with pelacarsen have shown promising results, with reductions in Lp(a) levels up to 80%, as well as reductions in OxPL by 70–88% and LDL-C up to 27%. and total apoB levels up to 16% at the highest doses used [62, 63]. Adverse effects observed in prior studies were mostly mild to moderate, with a small percentage of patients discontinuing treatment due to injection site reactions, myalgia, arthralgia, or malaise. The Lp(a) HORIZON trial is currently underway, evaluating the impact of pelacarsen on MACE in patients with established CVD and elevated Lp(a) levels [63].
Olpasiran is an siRNA that specifically targets LPA mRNA. The results of the phase 1 trial demonstrated that olpasiran effectively reduced mean Lp(a) levels in a dose-dependent manner compared to placebo, with maximal reductions greater than 90% from baseline. The Lp(a) reduction lasted for up to 150 days [64]. The trial included 32 participants with a mean age of 50 years, and 53% of the patients were female. The study intervention was well-tolerated, and all participants completed the trial. Only one patient experienced two serious adverse events: hospital admission for headache following SARS-CoV-2 vaccination and later for complications of cholecystitis. Both events were considered unrelated to the study drug.
The phase 2 OCEAN(a)-DOSE trial (“Olpasiran Trials of Cardiovascular Events and Lipoprotein(a) Reduction-Dose Finding Study”) was specifically designed to assess the efficacy and safety of olpasiran in patients with established ASCVD and elevated Lp(a) levels more than 150 nmol/L) [65]. The study evaluated several different dosing regimens, including 10 mg every 12 weeks, 75 mg every 12 weeks, 225 mg every 12 weeks, and 225 mg every 24 weeks as well as a placebo group for comparison. At the 36-week observation, the Lp(a) concentration had increased by a mean of 3.6% in the placebo group. In contrast, olpasiran therapy led to a significant and dose-dependent reduction in Lp(a) concentrations. The placebo-adjusted mean percent changes were as follows: 10 mg dose: –70.5%; 75 mg dose: –97.4%; 225 mg every 12 weeks: –101.1%; 225 mg every 24 weeks: –100.5%.
The overall incidence of adverse events was similar across the trial groups, indicating that olpasiran was generally well-tolerated. The most common adverse events related to olpasiran were injection-site reactions, primarily pain. This trial has several limitations. The treatment period was limited to 48 weeks, which might not be sufficient to assess the long-term efficacy and safety of olpasiran. All the patients had elevated Lp(a) concentrations at baseline. To generalize the findings to a broader population, it is crucial to investigate whether similar pharmacodynamic effects are observed in patients with lower baseline Lp(a) concentrations. We are currently awaiting the completion of randomized controlled trial on the effect of Lp(a) lowering therapy (with olpasiran) on the prevention of CVD including MI, stroke, and CAVS.
Conclusions
Multiple investigations suggest that elevated Lp(a) levels play a causal role in the development of ASCVD. However, to establish a definitive link and demonstrate the clinical benefit of lowering Lp(a) large-scale clinical trials are needed. Despite recommendations from EAS to measure Lp(a) in all adults at least once, routine clinical practice often does not involve frequent Lp(a) measurements.
There is no consensus on the appropriate threshold for defining elevated Lp(a) levels as abnormal. Lp(a) concentration is primarily determined by genetics, leading to significant variation in its distribution across different patient populations, including variations based on race, ethnicity, and sex. Despite these differences, Lp(a) concentration consistently predicts coronary heart disease risk in a log-linear manner regardless of race or ethnic group.
Further research and large-scale clinical trials are necessary to establish effective treatments and management strategies for patients with elevated Lp(a) and ASCVD. In addition to potential future treatments (pelacarsen, olpasiran) managing Lp(a) levels typically involves lifestyle modifications and addressing other cardiovascular risk factors. This may include maintaining a healthy diet, regular exercise, quitting smoking, controlling blood pressure, and managing diabetes if present.
Abbreviations
| apo(a): |
apolipoprotein(a) |
| apoB: |
apolipoprotein B |
| ASCVD: |
atherosclerotic cardiovascular disease |
| CAD: |
coronary artery disease |
| CAVS: |
calcific aortic valve stenosis |
| CVD: |
cardiovascular disease |
| EAS: |
European Atherosclerosis Society |
| FH: |
familial hypercholesterolemia |
| HR: |
hazard ratios |
| KIV: |
kringle IV |
| LA: |
lipoprotein apheresis |
| LDL: |
low-density lipoprotein |
| LDL-C: |
low-density lipoprotein cholesterol |
| LDLR: |
low-density lipoprotein receptor |
| Lp(a): |
lipoprotein(a) |
| LPA: |
lipoprotein(a) gene |
| MI: |
myocardial infarction |
| PCSK9: |
proprotein convertase subtilisin/kexin 9 |
| SNPs: |
single nucleotide polymorphisms |
Declarations
Acknowledgments
The author deserves special thanks to Dr. Simona Fenizia for her valuable contribution to English-style editing.
Author contributions
Miłosz B: Conceptualization, Resources, Visualization, Writing—original draft. Marlena B: Conceptualization, Supervision, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.