 Review
Review

Affiliation:
1Christopher S. Bond Life Sciences Center, University of Missouri, Columbia, MO 65211, USA
ORCID: https://orcid.org/0000-0003-2567-029X
Affiliation:
2School of Life Science, Shanxi Normal University, Linfen 041004, Shanxi Province, China
ORCID: https://orcid.org/0009-0002-9305-9310
Affiliation:
3The First Affiliated Hospital, Zhejiang University, Hangzhou 310006, Zhejiang Province, China
ORCID: https://orcid.org/0000-0001-9695-2492
Affiliation:
4Department of Surgery, University of Missouri, Columbia, MO 65211, USA
Email: yangmin@health.missouri.edu
ORCID: https://orcid.org/0000-0002-4895-5864
Explor Dig Dis. 2023;2:246–275 DOI: https://doi.org/10.37349/edd.2023.00029
Received: June 04, 2023 Accepted: August 05, 2023 Published: October 25, 2023
Academic Editor: Amedeo Lonardo, Azienda Ospedaliero-Universitaria di Modena, Italy
Non-alcoholic fatty liver disease (NAFLD) is the leading chronic liver disease worldwide, with a progressive form of non-alcoholic steatohepatitis (NASH). It may progress to advanced liver diseases, including liver fibrosis, cirrhosis, and hepatocellular carcinoma. NAFLD/NASH is a comorbidity of many metabolic disorders such as obesity, insulin resistance, type 2 diabetes, cardiovascular disease, and chronic kidney disease. These metabolic diseases are often accompanied by systemic or extrahepatic inflammation, which plays an important role in the pathogenesis and treatment of NAFLD or NASH. Metabolites, such as short-chain fatty acids, impact the function, inflammation, and death of hepatocytes, the primary parenchymal cells in the liver tissue. Cholangiocytes, the epithelial cells that line the bile ducts, can differentiate into proliferative hepatocytes in chronic liver injury. In addition, hepatic non-parenchymal cells, including liver sinusoidal endothelial cells, hepatic stellate cells, and innate and adaptive immune cells, are involved in liver inflammation. Proteins such as fibroblast growth factors, acetyl-coenzyme A carboxylases, and nuclear factor erythroid 2-related factor 2 are involved in liver metabolism and inflammation, which are potential targets for NASH treatment. This review focuses on the effects of metabolic disease-induced extrahepatic inflammation, liver inflammation, and the cellular and molecular mechanisms of liver metabolism on the development and progression of NAFLD and NASH, as well as the associated treatments.
Non-alcoholic fatty liver disease (NAFLD) is a complex and multifactorial disease with clinical manifestations ranging from hepatic steatosis to an advanced form of non-alcoholic steatohepatitis (NASH), which can progress to cirrhosis and hepatocellular carcinoma (HCC) [1]. NAFLD is the most common chronic liver disease. It is commonly associated with the development and progression of many chronic metabolic diseases, including type 2 diabetes mellitus (T2DM) [2], cardiovascular disease (CVD) [3], and chronic kidney disease (CKD) [4]. Multiple genetic, epigenetic, and environmental factors are involved in the pathogenesis of NAFLD [5, 6]. Hepatic steatosis is characterized by abnormal liver lipid accumulation, which is mainly caused by impaired fatty acid metabolism, continuously circulating fatty acids from adipose tissue lipolysis, and de novo lipogenesis (DNL) [3]. Dyslipidemia is also often accompanied by liver inflammation and metabolic disorders, such as insulin resistance [7, 8]. A panel of experts has suggested a new name for NAFLD, metabolic dysfunction-associated fatty liver disease (MAFLD) that is defined by the evidence of hepatic steatosis with one of the following three criteria: overweight or obesity, presence of T2DM, or evidence of metabolic dysregulation [9]. In this review, the terminology of NAFLD will be used in the context.
Liver inflammation promotes the progression of hepatic steatosis to NASH and liver fibrosis. Both innate and adaptive immune cells are involved in liver inflammation during NAFLD progression, including monocytes [10], macrophages [11], neutrophils, myeloid-derived suppressor cells (MDSCs) [12], natural killer (NK) cells [13], natural killer T (NKT) cells [14], and B and T lymphocytes [15, 16]. Chemokine receptors such as C-C motif chemokine receptor 2 (CCR2) and C-X3-C motif chemokine receptor 1 (CX3CR1) play important roles in the recruitment of these cells [17]. Pro-inflammatory cytokines and growth factors secreted from activated immune cells can promote the progression of NAFLD/NASH, such as interferon-γ (IFN-γ), interleukin (IL)-1β, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [13]. For example, metabolically activated macrophages in NASH livers can secrete proinflammatory cytokines and chemokines [e.g., IL-1β and C-C motif chemokine ligand 2 (CCL2)] to trigger the activation of hepatic stellate cells (HSCs) and infiltration of more inflammatory cells, resulting in the aggregation of liver inflammation and fibrosis [18]. Abnormal hepatic lipid accumulation, inflammation, and fibrosis, as well as the subsequent cell death, promote the progression of NAFLD to NASH and advanced liver disease, including cirrhosis and HCC [19]. Given the important roles of liver inflammation in liver diseases, treatment with anti-inflammatory drugs, either alone or in combination with metabolic signaling pathway regulators, is a potent strategy to prevent NAFLD progression [20].
In this review, we first summarize the role of extrahepatic and intrahepatic inflammation and inflammation-induced factors in the pathogenesis of NAFLD or NASH. Then, we discuss how systemic and local metabolites can regulate liver inflammation and hepatic cell responses and dig out the underlying molecular linkers or signaling pathways. Importantly, we explore the potential treatment options that can regulate abnormally metabolic and inflammatory pathways in NAFLD/NASH.
Extrahepatic inflammatory factors can contribute to the onset and progression of NAFLD, such as adipokines [21] and gut hormones [22]. For example, pro-inflammatory cytokines secreted from adipose tissues and intestinal epithelium cells, such as IL-1β and tumor necrosis factor (TNF)-α can transfer into the liver to induce immune cell activation [23, 24]. In this section, we discuss the roles of metabolic diseases and gut microbiota dysbiosis in extrahepatic and hepatic inflammation during the development and progression of NAFLD.
The National Health and Nutrition Examination Survey (2003–2018) showed that the visceral adiposity index (VAI), which is calculated based on waist circumference (WC), body mass index (BMI), triglyceride (TG), and high-density lipoprotein (HDL) cholesterol levels, was increased in the U.S. adults with NAFLD [25]. In subjects with obesity, increased fat deposition and chronic low-grade inflammation are typical features of adipose tissue dysfunction, which play important roles in the pathogenesis of NAFLD, including hepatic steatosis, inflammation, and liver fibrosis [26]. Multiple mechanisms are implicated in obesity-induced NAFLD development, including insulin resistance, ectopic fat accumulation, the metabolism of free fatty acids (FFAs), and inflammatory adipokines secreted from adipose tissues. Adipose tissue insulin resistance contributes to the accumulation of intrahepatic TG associated with the upregulation in the production of FFAs [27]. The circulating FFAs are increased in subjects with obesity, which can induce insulin resistance in the liver and contribute to NAFLD development [28]. In addition, adipokines derived from obese tissues (e.g., brown adipose tissues) can be delivered into the liver to cause hepatic inflammation [26]. For example, an increase in circulating leptin levels and a decrease in adiponectin levels are associated with the increased severity of NAFLD [21]. Inflammatory cytokines and chemokines secreted from adipose tissue can impact systemic inflammation, including liver tissues. IL-23 expression in adipose tissues was increased in individuals with high low-density lipoprotein cholesterol (LDL-c) compared to subjects with low LDL-c. The increase of IL-23 expression was positively correlated with the expression levels of macrophage markers (e.g., CD11c, CD68, and CD86), pro-inflammatory cytokines (e.g., TNF-α, IL-12, IL-18), and chemokines [e.g., C-X-C motif chemokine ligand 8 (CXCL8), CCL5, and CCL20] [29]. The expression of IL-2 in adipose tissues was also found to be significantly increased in obese persons compared to lean subjects, as well as the levels of fasting blood glucose (FBG), hemoglobin A1c (HbA1c), TG, and C‑reactive protein (CRP). In addition, IL-2 expression was concomitant with the expression of cytokines IL-8 and IL-12a and chemokines and their receptors, such as CCL5, CCR2, and CCR5 [30]. Overall, adipose tissue metabolic disorder and inflammation play important roles in extrahepatic and hepatic inflammation.
The hormone insulin controls blood glucose levels. In the liver, insulin regulates glucose storage in the form of glycogen to avoid postprandial hyperglycemia. However, the loss of liver glycogen synthesis and aberrant lipid metabolites in metabolic disorders, such as obesity and NAFLD, can impair hepatic insulin action and cause insulin resistance [31, 32]. A study showed that BMI, fasting plasma glucose (FPG), TG, total cholesterol (TC), LDL-c, alanine aminotransferase (ALT), and the homeostasis model assessment of insulin resistance (HOMA-IR) index were significantly increased in NAFLD patients with T2DM compared to patients with T2DM alone [33]. Insulin resistance can downregulate the expression of oxysterol 7α-hydroxylase (CYP7B1) to increase toxic cholesterol accumulation in hepatocytes, resulting in liver inflammation [34]. Insulin resistance can directly contribute to NAFLD by increasing DNL and indirectly suppress lipolysis by increasing the delivery of FFAs to the liver [35, 36]. The function of insulin signaling pathways will be illustrated in the section of DNL.
T2DM is a chronic metabolic disease characterized by continual hyperglycemia. T2DM can also be induced by obesity and inflammation [37, 38], two contributing factors to NAFLD and NASH. Compared to patients with simple T2DM, T2DM patients with NAFLD had higher BMI and insulin resistance index and increased levels of TG [39]. Studies have shown that the prevalence of NAFLD in patients with T2DM is around 70% [40]. Insulin resistance is commonly a contributing factor for T2DM, promoting the development of NAFLD in patients with T2DM [41]. In addition, genetic factors such as patatin-like phospholipase domain-containing protein 3 (PNPLA3)-I148M variant [42, 43], gut microbial metabolites [44], and adipocyte dysfunction [45] in T2DM patients can promote NAFLD development, and vice versa.
Inflammation plays a pivotal role in the development and progression of CKD. An increase in the neutrophil-to-lymphocyte ratio (NLR), a systemic inflammation marker, contributes to the risk of CKD in overweight or obese women and men, but not in individuals with normal weight [46]. In addition to a higher NLR, patients with CKD at stages 1–2 have increased circulating levels of IL-6 and tumor necrosis factor receptor 2 (TNFR2) compared to controls [47]. Plasma biomarkers of tubular injury [e.g., kidney injury molecule-1 (KIM-1)] and inflammation (e.g., TNFR1 and TNFR2) are independently associated with CKD progression in children [48]. A higher prevalence of CKD has been shown in patients with NAFLD compared to that in subjects without NAFLD [49]. It has been shown that NAFLD is an independent risk factor for CKD [50]. However, the correlation between NAFLD and CKD may be interactive, as they share common causing factors such as unhealthy diets, dyslipidemia, gut microbiota dysbiosis, platelet activation, and aging [51, 52].
Pro-inflammatory cytokines such as IL-1β, IL-17, and TNF are commonly increased in the pathogenesis of CVD (e.g., coronary artery disease, myocardial infarction, and heart failure) and atherosclerosis [53–55]. CVD is the most common cause of mortality in patients with NAFLD, which is largely induced by abnormal lipid and lipoprotein metabolism [56]. Plasma hypertriglyceridemia and increased LDL-c, inflammatory cytokines, and extracellular vesicles are major contributing factors to CVD in patients with NAFLD [56, 57]. In addition, these two diseases share some risk factors, including obesity, insulin resistance, and T2DM. Several factors, including low-grade systemic inflammation, lipotoxicity, oxidative stress, adipokines, endoplasmic reticulum (ER) stress, microbiota dysbiosis, and other factors such as genetic and epigenetic variations, have been suggested to link CVD and NAFLD [58, 59]. However, the risk of CVD patients developing NAFLD and the associated mechanisms remain to be studied.
The liver is anatomically and functionally connected with the intestine. The gut-liver axis is defined as the bidirectional relationship between the gut, along with gut microbiota, with the liver [60]. This axis delivers the signals from bile acids (BAs), immunoglobulins, and gut-microbiota-derived products and metabolites to regulate intestinal homeostasis and liver function [60, 61]. Dysbiosis of gut microbiota and increased intestinal permeability result in NAFLD progression by increasing the transportation of gut-microbiota-derived components and metabolites into the liver [62]. For example, gut-microbiota-derived metabolite trimethylamine N-oxide (TMAO) from dietary choline, carnitine, and L-carnitine can aggravate hepatic steatosis in NAFLD by regulating BA metabolism through the regulation of farnesoid X receptor (FXR) signaling pathway [63]. In vitro treatment of TMAO together with pro-inflammatory cytokine TNF-α can increase the proliferation (detected by the cell-counting Kit-8 assay), migration (detected by the wound healing assay and the transwell assay), and invasion [detected by the expression levels of periostin, integrin-linked kinase (ILK)/RAC-α serine/threonine-protein kinase (AKT1), and the mammalian target of rapamycin (mTOR)] of mouse liver cancer cell line Hepa1–6 cells and human liver cancer cell line Huh7 cells [64]. In addition, TMAO-induced exosomes from hepatocytes can impair endothelial cell function and promote inflammation [65]. Gut microbiota can synthesize the secondary BAs (e.g., ursodeoxycholic acid) to regulate liver inflammation and hepatocyte apoptosis [66].
Gut microbiota also plays important roles in obesity [67], insulin resistance [68], T2DM [69], CKD [70], and CVD [71]. The underlying cellular and molecular mechanisms of gut-microbiota-mediated functions in metabolic disorders are highly similar. These actions mainly include (1) the activation of innate and adaptive immune cells through bacterial components [e.g., CpG-rich oligonucleotides, lipopolysaccharides (LPSs), lipoteichoic acids (LTAs)], (2) energy metabolism [e.g., the production of short-chain fatty acids (SCFAs)], (3) the synthesis of secondary BAs, (4) other byproducts derived from potentially harmful bacteria (e.g., TMAO) [72–74].
Overall, systemic metabolic disorders and inflammation contribute to the development and progression of NAFLD (Figure 1). Some specific examples are listed in Table 1. The above-mentioned diseases and metabolic syndromes are commonly associated with NAFLD and liver inflammation.
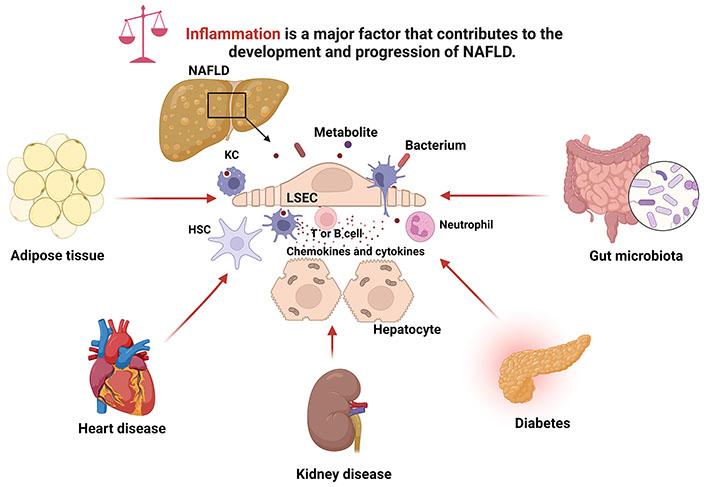
Inflammation contributes to the development and progression of NAFLD. Chronic metabolic diseases including obesity, type 2 diabetes, CKD, and CVD, as well as gut microbiota dysbiosis, can induce liver inflammation to promote the development and progression of NAFLD. LSEC: liver sinusoidal endothelial cell. Created with BioRender.com
Extrahepatic inflammation and metabolic disorders contribute to NAFLD development
| Metabolic diseases | Factors or regulators | Hepatic inflammation and steatosis | References |
|---|---|---|---|
| Obesity | Production of adipokines (e.g., increase of leptin levels and decrease of adiponectin levels) | Induce hepatic inflammation | [21] |
| Adipose tissue insulin resistance | Increase hepatic TG accumulation | [27] | |
| Production of FFAs | Increase hepatic steatosis | [28] | |
| Insulin resistance | High levels of FPG | Increase liver DNL | [33, 36] |
| Downregulation of CYP7B1 expression | Increase hepatic cholesterol accumulation | [34] | |
| T2DM | Continual hyperglycemia | Cause hepatic liver accumulation | [39, 41] |
| Insulin resistance | |||
| CKD | High circulating levels of IL-6 and TNFR2 | Cause liver inflammation | [47] |
| CVD | Increased levels of proinflammatory cytokines such as IL-1β and TNF | Cause liver inflammation | [53–55] |
| Dysregulation metabolism of lipids and lipoproteins | Increase hepatic steatosis | [58, 59] |
Hepatic injury, inflammation, and metabolism dysfunction play important roles in NAFLD development. Meanwhile, cell activation and death, liver inflammation, and fibrosis aggregate and accelerate NAFLD progression to NASH and HCC [75, 76]. In this section, we review some cellular processes that cause and promote liver inflammation and NAFLD progression.
Hepatic DNL contributes to fat accumulation in the fatty liver during the development and progression of NAFLD. Several transcriptional factors are involved in this process, such as sterol regulatory element-binding transcription factor-1c (SREBF-1c) [77] and peroxisome proliferator-activated receptor γ (PPARγ) [78], which can be regulated by non-coding RNAs (e.g., miR-615-5p and miR-130a). For example, in mice with fructose-induced NAFLD, hepatic SREBF-1c activation upregulated the expression of fatty acid synthase (FAS) an acetyl-coenzyme A carboxylase (ACC) to increase hepatic lipid accumulation [79]. In contrast, the expression levels of messenger RNAs (mRNAs) encoding enzymes of fatty acid and TG synthesis, such as ACC and FAS were decreased in the liver tissues of sterol regulatory element-binding protein-1c (SREBP-1c)-deficient mice with a normal diet [80]. The binding of SREBP-1c with sterol regulatory elements (SREs) of target lipogenic genes can be regulated by insulin and insulin-like growth factor signaling pathways [81]. Elevated hepatic DNL promotes NASH progression by inducing liver inflammation and fibrosis, which can be suppressed by inhibition of adenosine triphosphate-citrate lyase (ACLY), an enzyme in charge of generating acetyl-coenzyme A (CoA) and oxaloacetate from citrate [82].
Apoptosis is a form of programmed cell death. Hepatocyte apoptosis is often shown in cell or animal models and patients with NAFLD. Both the extrinsic (death-receptor-mediated) and intrinsic (organelle-initiated) pathways are activated during hepatocyte apoptosis [83]. For example, oleic acid (OA) can cause lipid accumulation in hepatocytes and their apoptosis by inducing mitochondrial membrane dysfunction and upregulating death receptor 5, the ligand of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) [84, 85]. Lysophosphatidylcholine (LPC), a metabolite derived from palmitic acid (PA), can directly cause hepatocyte cell rounding by reducing cellular extracellular matrix adhesion and cell-cell junction to cause hepatocyte apoptosis [86].
Pyroptosis is a programmed cell death accompanied by the activation of inflammasomes [87]. PA as a saturated FFA can induce pyroptosis in HepG2 cells by activating the expression of NOD-like receptor family pyrin domain-containing 3 (NLRP3) [88]. The release of extracellular NLRP3 inflammasome particles from injury hepatocytes can activate HSCs to express IL-1β and α-smooth muscle actin (α-SMA) proteins, leading to liver inflammation and fibrosis [89].
Ferroptosis, a non-apoptotic form of programmed cell death, has been exhibited in NAFLD and HCC [90]. An increase in iron accumulation and lipid peroxidation is shown in ferroptosis. Currently, the underlying mechanism of ferroptosis in NAFLD is not fully understood. Several genes have been shown to play important roles in ferroptosis in the case of NAFLD, such as glutathione peroxidase 4 (GPX4) [91], enolase 3 (ENO3) [92], tripartite motif-containing 59 (TRIM59) [93], and period circadian regulator 2 (PER2) [94].
Hepatocytes can also undergo necroptosis, a regulated process of necrotic cell death in NAFLD and NASH [95, 96]. The signaling pathway of receptor-interacting protein kinase (RIPK)/mixed lineage kinase domain-like protein (MLKL) is activated during hepatocyte necroptosis in NAFLD [95, 96]. The clearance of necroptotic hepatocytes by macrophages is impaired in NASH due to the upregulation of the CD47/signal regulatory protein α (SIRPα) axis [97].
Activated HSCs are the main cells that differentiate into myofibroblasts during liver fibrosis [98], and small parts of myofibroblasts are derived from portal fibroblasts and mesenchymal stem cells (PMSCs). Myeloid differentiation primary response 88 (MyD88) plays a pivotal role in HSC activation and the expression of extracellular matrix proteins, including α-SMA and collagen I [99]. Activation of MyD88/CXCL10 signaling pathway in HSCs can promote macrophage M1 polarization through CXCR3 by activating Janus kinase (JAK)/signal transducer and activator of transcription 1 (STAT1) signaling pathway. On the contrary, inhibition of CXCL10 secretion can reduce macrophage M1 polarization and decrease liver fibrosis [99]. Overexpression of some key genes such as ubiquitin-specific protease 33 (USP33) can regulate HSC activation and metabolic programming (e.g., glycolysis) to promote liver fibrosis [100]. Inflammation can further promote the activation of HSCs. For example, IL-18 is not only involved in the signaling pathway of NLRP3 inflammasome-mediated HSC activation, but also it can induce the trans-differentiation of HSCs into myofibroblasts by interacting with its receptor [101].
LSECs, hepatic gatekeeper cells, play multiple roles in chronic liver diseases [102], including NAFLD, NASH, and HCC. Gut-microbiota-derived components and metabolism (e.g., LPS and palmitate) can induce the capillarization of LSECs to promote NASH, liver fibrosis, and HCC development via the products such as mitogenic factor sphingosine-1-phosphate (S1P) and vascular cell adhesion molecule-1 (VCAM-1) [103, 104]. In addition, injury LSECs can secrete many proinflammatory cytokines (e.g., IL-6 and TNF-α) and chemokines (e.g., CCL2 and CXCL9) to mediate liver inflammation [102].
Hepatic ductular reaction (DR), a reactive bile duct hyperplasia, is involved in the proliferation and differentiation of cholangiocytes and hepatocytes or hepatic progenitor cells [105]. Feeding C57BL/6J mice a choline-deficient, amino acid-defined diet with 60% fat by calories for eight weeks can induce hepatic DR and advanced liver fibrosis [106]. The presence of centrilobular DR may predict the progression of liver fibrosis in patients with NASH [107]. The molecular signaling pathways in DR during NASH were reviewed in a literature report [105]. Angiogenic factors such as vascular endothelial growth factor and angiopoietin 2 play an important role in DR during NASH [108].
Biliary epithelial cells can differentiate into hepatocytes in chronic liver injury or severe liver disease [109, 110]. Cholangiocytes are a heterogenous population of epithelial cells that line bile ducts. Cholangiocytes can be activated to participate in hepatic inflammation and regulate liver fibrosis by interacting with myofibroblasts [111]. For example, cholecystokinin (CCK) released by duodenal enteroendocrine I-cells in response to dietary lipids and proteins can activate CCK receptors on cholangiocytes to promote NASH progression. Treatment with a CCK inhibitor proglumide can ameliorate choline-deficient, ethionine-supplemented (CDE) diet-induced NASH by activating FXR signaling pathway and altering gut microbiota profiles [112]. An accumulation of yes-associated protein (YAP)-positive reactive-appearing ductular cells (RDCs) in NAFLD/NASH has also been shown during liver fibrosis and hepatocyte injury [113]. In addition, senescent cholangiocytes can express pro-inflammatory cytokines, such as IL-1β, IL-6, and TNF-α, to promote liver inflammation and fibrosis [114]. Angiotensin-converting enzyme 2 (ACE2) is highly expressed in cholangiocytes within the liver, which may accelerate the impact of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection in NAFLD or NASH patients [115, 116].
The innate immune cells, including monocytes, liver macrophages or resident Kupffer cells (KCs), dendritic cells (DCs), neutrophils, NK cells, and NKT cells, play pivotal roles in liver inflammation during NAFLD and NASH [117, 118]. Accumulating evidence shows that liver macrophages or KCs play an essential role in liver metabolism and inflammation [119]. KC cells can be further differentiated into two populations including a major CD206lowendothelial cell-selective adhesion molecule negative (ESAM–) population (KC1) and a minor CD206highESAM+ population (KC2) [120]. The KC2 population is increased in fatty livers in obese mice with upregulation of the gene expression of carbohydrates and lipid metabolisms [120]. This study also found that KC2 cells function on hepatic lipid peroxidation and oxidative stress, with a relatively high expression of CD36 as fatty acid transporter. In contrast, KC1 cells express genes for the immune response and immune system process under the analysis of the Gene Ontology (GO) category [120]. Pro-inflammatory cytokines (e.g., TNF-α and IL-6) and M1 macrophages markers (e.g., CD11c) were significantly increased in the livers of hyperglycemic mice expressing hepatocyte-specific glycerol-3-phosphate phosphatase (G3PP) that hydrolyzes glycerol-3-phosphate (Gro3P) to glycerol [121]. Accumulating data reveal that an elevated NLR ratio is a risk factor for NAFLD and NAFLD-related HCC [122, 123]. However, a mouse study indicated that the depletion of neutrophils at the resolution phase of NASH can impair the repairing and remodeling processes due to the imbalance in the ratio of pro- and anti-inflammatory cytokines and macrophage phenotypic switching [124]. In addition, DCs [125, 126], NK cells [13], and NKT cells [127] play diverse roles in the pathogenesis of NAFLD and NASH.
In addition to innate immunity, adaptive immune cells also play critical roles in liver inflammation in NAFLD and NASH-related progression of HCC [128, 129]. T cells can secrete proinflammatory cytokines and profibrotic mediators to promote liver inflammation and fibrosis in NAFLD/NASH, including CD4, CD8, and γδ T cells [130]. For example, the ratios of regulatory T cells (Tregs) with T helper 1 (Th1) cells, Th17, and CD8 T cells are increased in the pathogenesis of NAFLD, NASH, fibrosis, and NASH-associated HCC [16, 131]. Recent studies have also demonstrated that CD4‒CD8‒ double negative T cells make an important contribution to NAFLD and NASH [132, 133]. B cells play dual roles in diet-induced NAFLD by secreting cytokines and antibodies, a phenomenon that was reviewed in a recent literature report [134]. Overall, both innate and adaptive immunities are implicated in NAFLD/NASH-related inflammation.
Interactions between diet and gut microbiota play an essential role in NAFLD pathogenesis, including the resulted metabolites of amino acids, glucose, and lipids, as well as gut-microbiota-associated products [135, 136]. The alteration of gut microbial metabolites such as SCFAs, secondary BAs, and choline metabolites can induce the development and progression of NAFLD [137]. Aberrant energy metabolism in the liver can also contribute to hepatic inflammation, fibrosis, NASH, and HCC [138]. This section discusses the metabolism of amino acids, BAs, glucose, lipids or lipoproteins, and SCFAs in the pathogenesis of NAFLD or NASH.
Both essential (e.g., histidine and threonine) and non-essential (e.g., alanine, glycine, and serine) amino acids play important roles in liver metabolism, such as lipid and nucleotide syntheses [139]. Circulating levels of amino acids impact both systemic and liver inflammation [140, 141]. Serum baseline levels of leucine, valine, and total branched-chain amino acids (BCAAs; including leucine, isoleucine, and valine) are significantly increased in patients with NAFLD compared to non-NAFLD controls [142]. In addition, serum leucine and total BCAAs are independent risk factors for the onset of NAFLD [142]. It has been shown that a decreased BCAA metabolism rate in the adipose tissue contributes to the increased levels of circulating BCAAs [143]. The catabolism of BCAAs is mainly regulated by PPARγ in inguinal white and brown adipose tissues in mice [144]. The impaired catabolism of BCAAs and downregulated BCAA metabolism gene sets in liver tissues have been shown in the pathogenesis of NAFLD and NASH [145, 146]. Studies also show that circulating BCAAs levels are negatively correlated with hepatic and peripheral insulin sensitivity and an increased valine level predicts an increase in hepatic fat [147]. Supplementation of BCAAs in a high-fat diet (HFD) by replacing carbohydrate calories not only can aggravate hepatic inflammation, fibrogenesis, and mitochondrial dysfunction but can decrease DNL [141]. Intake of a high-methionine diet (HMD) containing 2.58% of methionine can increase NAFLD development in mice by inhibiting hepatic H2S production. HMD treatment inhibits lipid catabolism and glycolysis metabolism and reduces adenosine triphosphate (ATP) production, resulting in mitochondrial dysfunction, oxidative stress, and inflammation in the liver [148]. Treatment of a high-protein (HP) diet significantly reduces HFD-induced hepatic steatosis in mice, causing a reduction in the plasma concentration of BCAAs and hepatic concentration of monomethyl branched-chain fatty acids (BCFAs) [149].
Glucose metabolism plays an important role in the pathogenesis of NAFLD [135, 150]. Insulin resistance can elevate blood glucose levels and increase DNL in the liver [151]. A study showed that about 40% of obese children had NAFLD with higher BMIs and fasting glucose, but lower insulin sensitivity indices compared to children without NAFLD [152]. In fatty liver, lipids such as diacylglycerols (DAGs) and ceramides can induce hepatic insulin resistance [153]. Hepatic levels of glycogen were decreased in HFD-fed mice, whereas mRNA expression levels of glycolysis rate-limiting enzymes hexokinase 2, phosphofructokinase, and pyruvate kinase were increased [154]. In addition, this study also showed that inhibiting glycolysis using 2-deoxy-D-glucose can reduce liver inflammation and fibrosis in liver-specific geranylgeranyl diphosphate synthase (GGPPS) knockout mice [154]. The levels of pyruvate are increased in the plasma and liver due to enhanced glycolysis of hepatocytes in the fatty liver. Pyruvate can be converted to oxaloacetate through anaplerosis to generate citrate through the tricarboxylic acid (TCA) cycle to enhance DNL [155]. Some key genes play important roles in hepatic glucose metabolism and steatosis. For example, one study showed that deletion of NOD-like receptor X1 (NLRX1) in mice can protect western diet-induced hepatic steatosis, fibrosis, obesity, insulin resistance, and glycosuria by decreasing glycolysis and increasing fatty acid oxidation in hepatocytes [156].
An excessive accumulation of lipids in hepatocytes is a typical feature of NAFLD. NAFLD is defined as > 5% of hepatocytes with fatty accumulation in patients without excessive alcohol consumption (< 20 g/day for women and < 30 g/day for men) [157, 158]. In NAFLD and NASH, lipid uptake and DNL in the liver are increased, whereas fatty acid oxidation is not sufficient to metabolize lipids, resulting in lipid accumulation and liver injury with oxidative stress and mitochondrial dysfunction [159]. Lipid-sensitive nuclear receptors, such as FXR, liver X receptor (LXR), and PPARs are involved in hepatic lipid metabolism and DNL [160, 161]. For example, FXR activation can decrease hepatic levels of monounsaturated fatty acids by repressing the expression of stearoyl-coenzyme A desaturase 1 (SCD1), diacylglycerol O-acyltransferase 2 (DGAT2), and lipin 1 (LPIN1), and reduce the liver polyunsaturated fatty acids via decreasing lipid absorption [162]. LXR plays an important role in hepatic lipogenesis by upregulating the expression of SREBP-1c [163] and FFA uptake transporter CD36 [164].
SCFAs are fatty acids produced by gut microbiota via the fermentation of polysaccharides. Acetate, propionate, and butyrate are three major SCFAs produced by the gut microbiota, which have immunomodulatory functions by regulating the expression of G protein-coupled receptors (GPCRs) [165] and can activate histone deacetylases and enzymes involved in post-translational modification [166]. A growing amount of evidence shows that SCFAs play important roles in health and disease, including NAFLD and NASH [167, 168]. For example, probiotics can increase the production of SCFAs (e.g., butyrate) to reduce systemic inflammation in NAFLD rats by activating GPCRs (e.g., GPR109a) [169]. Fermentation of dietary fiber can produce acetate, propionate, and butyrate [170]. Dietary fiber treatment can improve NAFLD and NASH pathogenesis by reducing liver inflammation, oxidative stress, lipid accumulation, and cell death [171, 172].
Treatment with inulin, a digestive fiber, can significantly reduce liver lipid accumulation and fibrosis in NAFLD/NASH by regulating the free fatty acid receptor 2 (FFAR2)-mediated signaling pathway [173]. In addition, inulin consumption increases the concentration of acetate with a concomitant enrichment of gut microbial genera Bacteroides and Blautia.
Proteolytic metabolites including amines, ammonia, indoles, phenolic compounds, hydrogen sulfide, and BCFAs play significant roles in metabolic diseases [174], including NAFLD. For example, the concentration of plasma iso-heptadecanoic acid (iso-C17:0), a monomethyl BCFA, has been found to decrease in the livers of children with steatosis [175]. Indole supplementation can decrease methionine- and choline-deficient-diet (MCD)-induced hepatic steatosis, inflammation, and fibrosis in mice by suppressing HSC activation and hepatocyte inflammation [176]. Indole-3-acetic acid (I3A), a gut-microbiota-derived metabolite from dietary tryptophan, can improve oxidative stress and hepatic steatosis by increasing mitochondrial oxidative phosphorylation in a PPARγ-coactivator-1α-dependent manner [177].
Many molecules are involved in the regulation of hepatic metabolism and inflammation. Here, we discuss some important proteins that can be used as targets for NAFLD treatment.
ACCs are important rate-limiting enzymes in DNL, which are in charge of the synthesis of malonyl-CoA from acetyl-CoA and control fatty acid β-oxidation in hepatocytes [178, 179]. Dual inhibitors of ACC1 and ACC2 can reduce liver fat accumulation, lipotoxicity, and TGF-β-induced activation of HSCs [179, 180]. In addition, the selective inhibition of ACC1 can inhibit malonyl-CoA content, hepatic TG content, and liver fibrosis [178].
The AMP-activated protein kinase (AMPK) signaling pathway plays an essential role in the regulation of lipid metabolism in NAFLD, as well as in alcoholic fatty liver disease (AFLD). For example, AMPK activation can prevent the synthesis of fatty acids and cholesterol by upregulating the expression of genes involved in fatty acid oxidation and lipid decomposition, such as peroxisome proliferator-activated receptor γ coactivator 1 (Pgc1) and adipose triglyceride lipase (Atgl), and by down-regulating the expression of adipogenesis genes such as FAS and ACC [181]. Activation of the AMPK/ACC and AMPK/FAS signaling pathways can increase fatty acid oxidation and inhibit lipid synthesis to ameliorate steatosis in NAFLD pathogenesis [182]. Another study also shows that upregulation of the phosphorylation of AMPK or activation of AMPK/SIRT1 signaling pathway can significantly decrease hepatic TG content and ameliorate serum levels of LDL-c and ALT [183, 184].
Intestinal FXR activation can increase the production of ceramide in the ileum, which transfers into the liver and subsequently activates SREBP-1c to increase fatty acid production, resulting in hepatic steatosis [185]. In contrast, inhibiting intestinal FXR signaling can decrease diet-induced ileal ceramide production in cases of obesity, and ameliorate NAFLD. For example, the bioactive compound caffeic acid phenethyl ester works to inhibit FXR signaling by inhibiting bacterial bile salt hydrolase (BSH) to increase levels of tauro-β-muricholic acid (T-β-MCA) in the intestine [186]. In the liver, FXR activation can suppress hepatic lipogenesis by reducing the expression of SREBP-1c, while FXR can also increase the expression of PPARα to promote FFA catabolism via β-oxidation [187].
Fibroblast growth factors (FGFs) play essential roles in liver fibrosis and NASH progression. As a liver metabolic hormone secreted in response to various nutritional challenges, FGF21 plays a critical role in controlling liver fat and glucose metabolism, dietary protein intake, and body fat loss [188, 189]. The concentration of FGF21 can be regulated by the consumption of sugars regardless of their types, such as glucose, fructose, and sucrose [190]. The function of FGF21 is highly impacted by obesity-induced TNF-α in HFD-induced NAFLD by suppressing the expression of the FGF21 receptor, resulting in a decrease in FGF21 sensitivity [191]. Lysophosphatidic acid (LPA) produced from LPC by a liver enzyme autotaxin can suppress the PPARα/FGF21 axis to exacerbate NAFLD [192]. Secreted FGF21 can activate its receptor FGFR2 to suppress the expression of SREBP-2 to suppress cholesterol biosynthesis [193]. Treatment with curcumin increases the expression of FGF15 and suppresses HFD-induced insulin resistance, glucose intolerance, and hepatic TG accumulation by regulating gut microbiota [194].
As receptors for BAs and FFAs, GPCRs have been shown to play essential roles in metabolic disorders, including NAFLD and NASH [195]. The FFARs/GPCRs signaling pathways are involved in the pathogenesis of NAFLD and NASH. For example, GPR40-deficient mice with a low-fat diet (LFD) show metabolic abnormalities, including an increase in body weight, insulin resistance, and levels of cholesterol and ALT [196]. The hepatocyte-specific deletion of cannabinoid receptor 1 (CB1) can inhibit HFD-induced insulin resistance in mice [197]. Knockout of GPR40 in low-density lipoprotein-receptor (LDLR)-deficient mice increases HFD-induced plasma levels of cholesterol and FFAs, hepatic steatosis, inflammation, and fibrosis, potentially through activation of CD36-mediated signaling pathway [198].
Regulator of G protein signaling (RGS) proteins negatively regulate GPCR signaling. For example, RGS5 in hepatocytes can inhibit TAK1 phosphorylation and the subsequent activation of the c-Jun-N-terminal kinase (JNK)/p38 signaling pathway to reduce NAFLD [199].
Hypoxia-inducible factor-1α (HIF-1α) is ubiquitously implicated in the development of various chronic liver diseases, such as NAFLD and HCC [200, 201]. High trans-fat diet-induced weight gain, liver inflammation evidenced by an increased expression of TNF-α and IL-1β, and liver collagen production are decreased in mice with hepatocyte-specific deletion of gene Hif1a compared to wild-type mice [202]. On the contrary, silencing Hif1a promotes OA- and PA-induced lipid accumulation in HepG2 cells in vitro. Meanwhile, loss of HIF-1α increases the expression of pro-inflammatory cytokines IL-6 and TNF-α and lipid-metabolism-related proteins, such as apolipoprotein E (APOE) and SREBP-2 [203]. Exposure to intermittent hypoxia accelerates lipid accumulation in hepatocytes (human L02 cell line), which can be suppressed by silencing Hif2a or by treatment with a PPARα agonist. In contrast, hypoxia-induced overexpression of HIF-2α induces the suppression of fatty acid β-oxidation and promotes lipogenesis in the liver by suppressing PPARα expression [204].
The binding of insulin with its receptor (IR) can regulate the phosphoinositide-3-phosphate kinase (PI3K)/AKT pathway to induce glycogen synthesis by inhibiting the expression of glycogen synthase kinase 3 [205]. In addition, suppression of both AKT1 and AKT2 in the liver can cause insulin resistance and inhibition of lipid synthesis. In DNL, increased production of DAG can induce the translocation of protein kinase Cε (PKCε) to cell membranes to inhibit insulin/IR signaling [206].
In NAFLD, oxidative stress is commonly and significantly increased in liver inflammation, which is accompanied by the downregulation of nuclear factor erythroid 2-related factor 2 (Nrf2) expression [207]. Nrf2 is a transcription factor, which can induce the expression of antioxidant response element-dependent genes to display antioxidant activity [208]. The expression of Nrf2 in liver tissues of patients with NAFLD/NASH has been shown to correlate with the grade of liver inflammation but not the grade of steatosis [209]. Pharmacologic activation of Nrf2 using TBE-31 decreased hepatic inflammation, apoptosis, fibrosis, oxidative stress, and ER stress in mice with high-fat plus fructose, which was abrogated in Nrf2-deficient mice [210]. Another study showed that food-derived compound apigenin, a modulator of PPARγ, can increase Nrf2 nucleus translocation to inhibit the expression of lipid metabolism-related genes and increase the expression of oxidative stress-related genes, resulting in amelioration of HFD-induced NAFLD [211]. However, one study showed that, compared to Nrf2-LoxP mice, hepatocyte-specific Nrf2-knockout mice with HFD had significantly less liver size, inflammation, and steatosis, which was not shown in mice with macrophage-specific Nrf2-knockout [212]. A molecular mechanism study showed that knockout of Nrf2 can diminish the expression of PPARγ and its downstream lipogenic genes in primary hepatocytes [212]. Therefore, the role of Nrf2 in NAFLD may be cell dependent.
There are three PPAR isoforms expressed in various tissues, including PPARα, PPARβ/δ, and PPARγ. PPARα is ubiquitously present in different tissues but highly expressed in the liver, while PPARβ/δ is mainly expressed in skeletal muscle and PPARγ is highly expressed in adipose tissue [213]. Pparα-deficient mice with a HFD have aggravated liver and adipose tissue inflammation compared to wild-type mice [214]. Both hepatic and whole-body deficiency of Pparα in mice can promote HFD-induced liver inflammation and NAFLD [215]. Hepatic Pparα-deficient mice with a standard diet can develop NAFLD during aging. In addition, PPARα can regulate hepatic and plasma FGF21 expression in mice with NASH [216]. Treatment with the PPARα agonist Wy-14643 can decrease hepatic steatosis, hepatocyte ballooning, and liver inflammation by suppressing nuclear factor-κB (NF-κB) and JNK signaling pathways and inhibiting the infiltration of macrophages and neutrophils in NASH livers [217]. The PPARα signaling pathway also contributes to the protective effects of torularhodin, a β-carotene-like compound from yeast Sporidiobolus pararoseus, on liver dyslipidemia and inflammation via up-regulating fatty acid β-oxidation, cholesterol excretion, and anti-inflammation gene expression [218]. The latest preclinical studies in diet-induced murine models also show that PPARα is a sexually dimorphic treatment target for NAFLD [219].
N-stearoylethanolamine (NSE), a bioactive lipid amine, can bind with PPARγ to inhibit the nuclear translocation of NF-κB in LPS-stimulated peritoneal macrophages and to reduce the expression of PPARγ-regulated genes solute carrier family 27 member 1 (SLC27A1) and interleukin-1 receptor antagonist (IL1RN) in insulin-resistant rats to suppress inflammation [220]. The roles of PPARβ/δ in the regulation of hepatic lipid and glucose metabolism and NAFLD development have been reviewed in another report, which is not discussed here [221].
The expression of sodium-glucose co-transporter 2 (SGLT2) is upregulated in liver samples from patients with steatosis and NASH compared to liver samples from individuals without NAFLD [222]. Treatment with SGLT2 inhibitor suppressed hepatic lipid accumulation, inflammation, and fibrosis in mice with diet-induced NASH by decreasing hepatocellular glucose update [222]. SGLT2 inhibitors are anti-hyperglycemic drugs that have been applied to treat diabetes [223], as well as other metabolic disorders such as CVD and renal dysfunction [224]. The effects of SGLT2 inhibitors on hepatic steatosis and fibrosis in patients with NASH and T2DM have been summarized in a review paper [225].
SREBP-1 consists of two isoforms, SREBP-1a and SREBP-1c. Specific SREBP-1a knockout in both hepatocytes and macrophages can exacerbate MCD-induced liver injury, while fatty liver disease is significantly worsened in mice with SREBP-1a knockout in hepatocytes compared to that in mice with SREBP-1a knockout in macrophages and wild-type mice [226]. Downregulation of SREBP-1c expression in HFD-fed rats with the treatment of Capparis spinosa can decrease hepatic steatosis and fibrosis by regulating the genes in DNL and β-oxidation signaling pathways [227].
In summary, the above-mentioned proteins play important roles in the regulation of hepatic inflammation and energy metabolism. A graphic figure summarizes the roles of some of these proteins in liver inflammation and lipid metabolism during NAFLD development and progression (Figure 2).
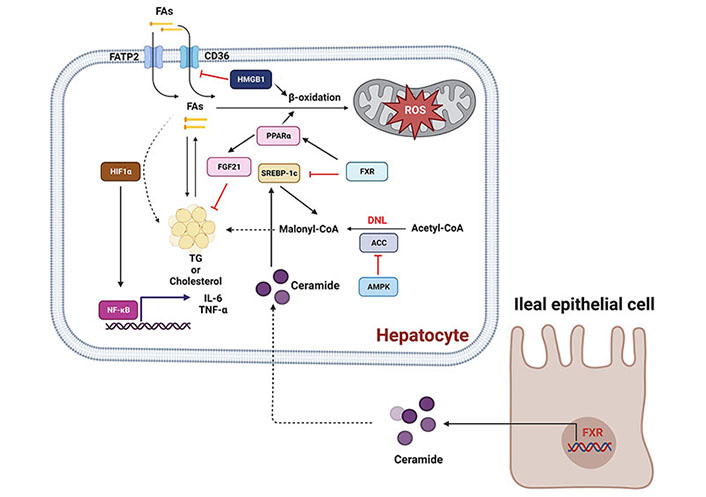
Molecular signaling pathways involved in NAFLD development and progression. Activation of AMPK/ACC signaling pathway can inhibit lipogenesis. The PPARα/FGF21 signaling pathway can be activated to increase fatty acid β-oxidation and suppress cholesterol biosynthesis. The activation of CD36 and fatty acid transport proteins (FATPs; e.g., FATP2) can promote the uptake of fatty acids (FAs), causing lipid steatosis. In contrast, HIF-1α can upregulate free-fatty-acid-induced lipid accumulation in hepatocytes and increase the expression of the pro-inflammatory cytokines IL-6 and TNF-α. In addition, FXR activation can increase the synthesis of ceramides in the intestine, which translocate into the liver to activate SREBP-1c, promoting lipogenesis or inducing hepatocyte death. Arrows and stop arrows indicate activation and repression, respectively. Created with BioRender.com
Appropriately inhibiting liver inflammation and regulating metabolic signaling pathways can decrease hepatic steatosis, fibrosis, and cell apoptosis to ameliorate NAFLD/NASH progression. Many molecular inhibitors and drugs, such as antidiabetic and anti-obesity drugs, antibiotics, pre/probiotics, caspase inhibitors, and CCR2/5 antagonists, are currently under clinical investigation for NAFLD or NASH treatment [228].
In this section, we review different classes of treatment agents undergoing clinical trials (Table 2), including vitamins [229, 230], FGF21 agonist antibodies or analogs [231, 232], diets [233], combined metabolic activators (CMAs) [234], anti-T2DM drugs [235], PPARγ agonist [236], regulation of lipogenesis [237], bacterial alteration [238], hormone therapy [239], and C-C chemokine receptors antagonist [240].
Clinical trials for NAFLD or NASH treatments
| Treatment | Class | Target | Trial number | References |
|---|---|---|---|---|
| δ-tocotrienol and α-tocopherol | Vitamin E | A combined treatment of two compounds improved hepatic steatosis, oxidative stress, and insulin resistance in patients with NAFLD. δ-tocotrienol was more effective than α-tocopherol in decreasing body weight, inflammation, and apoptosis | SLCTR/2019/038# | [229] |
| Fish oil plus vitamin D3 | Fish oil and vitamin D3 | The supplementation of two products reduced biomarkers of hepatocellular damage and plasma TAG levels in patients with NAFLD, which had additional benefits for insulin levels and inflammation compared to fish-oil-treated | ChiCTR1900024866* | [230] |
| Efruxifermin | A long-acting Fc-FGF21 fusion protein | Treatment with efruxifermin significantly decreased hepatic fat fraction measured by magnetic resonance imaging-proton density fat fraction in patients with NASH and fibrosis (F1–F3 stages) | NCT03976401 | [231] |
| Pegbelfermin | A PEGylated human FGF21 analog | The subcutaneous administration of pegbelfermin significantly reduced the hepatic fat fraction in patients with NASH | NCT02413372 | [232] |
| Mediterranean diet (MD) and LFD | Diets | A 12-week consumption of MD and LFD in adolescents with obesity and NAFLD reduced the BMI, fat mass, hepatic steatosis, and insulin resistance, decreased high transaminase levels, and improved inflammation and oxidative stress | NCT04845373 | [233] |
| L-carnitine tartrate, nicotinamide riboside, L-serine, and N-acetyl-l-cysteine | CMAs | CMA significantly reduced hepatic steatosis and levels of aspartate aminotransferase, ALT, uric acid, and creatinine | NCT04330326 | [234] |
| Tofogliflozin and glimepiride | An inhibitor of SGLT2 and anti-type 2 diabetes drug | Hepatic steatosis, hepatocyte ballooning, and lobular inflammation were decreased post-tofogliflozin treatment, whereas only hepatocellular ballooning was improved after the glimepiride treatment. In addition, the expression of genes related to energy metabolism, inflammation, and fibrosis was overturned after the tofogliflozin treatment | NCT02649465 | [235] |
| Lifestyle intervention (LSI) + pioglitazone (PGZ) | Lifestyle + PPAR-γ agonist | A combined PGZ and LSI treatment significantly decreased liver fat in both women and men compared to the LSI treatment alone, but it proved less effective in men than in women | NCT00633282 | [236] |
| Diacylglycerol acyltransferase 2 inhibitor (DGAT2i) and acetyl-coenzyme A carboxylase inhibitor (ACCi) | Inhibition of intrahepatic TG synthesis and blockade of DNL | A combined treatment of DGAT2i, PF-06865571, and ACCi (PF-05221304, clesacostat) was applied to treat NASH with liver fibrosis | NCT04321031 | [237] |
| Helicobacter (H.) pylori eradication treatment | Bacterial alteration | H. pylori eradication significantly decreased FBG, glycosylated hemoglobin, HOMA-IR, TGs, BMI, and inflammatory markers such as high-sensitivity CRP, and inflammatory cytokines such as IL-6 and TNF-α | ChiCTR2200061243* | [238] |
| Recombinant leptin therapy | Hormone therapy | Exogenous leptin treatment decreased hepatic steatosis and injury in patients with NASH who have relative leptin deficiency with partial lipodystrophy | NCT00596934 | [239] |
| Cenicriviroc | C-C chemokine receptors type 2 and 5 dual antagonist | In response to cenicriviroc treatment, patients with NASH achieved ≥ 1-stage fibrosis improvement at year 1 and maintained it at year 2 | NCT02217475 | [240] |
# Sri Lanka Clinical Trials Registry; * Chinese clinical trial registration
Furthermore, combinational therapies are potential therapeutic options for NAFLD or NASH treatment. Glucagon-like peptide-1 (GLP-1) is a gastrointestinal hormone (an incretin hormone) with numerous metabolic functions, including stimulation of insulin secretion, reduction of food intake, and inhibition of pancreatic β-cell apoptosis [241]. GLP-1 receptor agonists (e.g., short-acting agent lixisenatide and long-acting agent liraglutide) as glucose-lowering agents have been developed to treat T2DM and other metabolic or chronic diseases [242]. A dual agonist against GLP-1 and FGF21 improves the non-alcoholic fatty liver disease activity score (NAS) with improved efficacy compared to each single treatment alone [243]. Another clinical trial shows that a combination of Clostridium butyricum capsules with rosuvastatin [to lower bad cholesterol or low-density lipoprotein (LDL) and TG], which is used to decrease high cholesterol and TG levels, can effectively advance the intestinal flora balance, reduce blood lipid levels, and improve liver fibrosis and injury in NAFLD patients [244].
Abnormal liver metabolism and inflammation contribute to the progression of NAFLD to NASH, as well as the end stage of liver disease. The extrahepatic inflammation induced by metabolic disorders, including obesity, insulin resistance, T2DM, CKD, and CVD promotes the progression of NAFLD. The gut-microbiota-derived metabolites and their components can impact the function and inflammation of hepatocytes and liver non-parenchymal cells, such as LSECs and HSCs, to promote NAFLD and fibrosis. In addition, gut microbiota dysbiosis links different metabolic diseases. Proteins, including ACCs, AMPKs, FXRs, FGFs, GPCRs, HIF-1, Nrf2, and PPARs play pivotal roles in NAFLD pathogenesis; therefore, they are molecular targets for NAFLD or NASH therapy. Clinical trials have been undertaken to explore the inhibitors or agonists for molecular targets in the treatment of chronic liver disease and its comorbidities. However, their efficacy and safety must be further explored in the future.
ACC: acetyl-coenzyme A carboxylase
AKT1: RAC-α serine/threonine-protein kinase
ALT: alanine aminotransferase
AMPK: AMP-activated protein kinase
BAs: bile acids
BCAAs: branched-chain amino acids
BCFAs: branched-chain fatty acids
BMI: body mass index
CCK: cholecystokinin
CCL2: C-C motif chemokine ligand 2
CCR2: C-C motif chemokine receptor 2
CKD: chronic kidney disease
CMAs: combined metabolic activators
CoA: coenzyme A
CVD: cardiovascular disease
CXCL8: C-X-C motif chemokine ligand 8
DNL: de novo lipogenesis
DR: ductular reaction
FAS: fatty acid synthase
FFAs: free fatty acids
FGFs: fibroblast growth factors
FXR: farnesoid X receptor
GLP-1: glucagon-like peptide-1
GPCRs: G protein-coupled receptors
HCC: hepatocellular carcinoma
HFD: high-fat diet
HIF-1α: hypoxia-inducible factor-1α
HSCs: hepatic stellate cells
IL: interleukin
KCs: Kupffer cells
LDL-c: low-density lipoprotein cholesterol
LFD: low-fat diet
LPSs: lipopolysaccharides
LSEC: liver sinusoidal endothelial cell
NAFLD: non-alcoholic fatty liver disease
NASH: non-alcoholic steatohepatitis
NF-κB: nuclear factor-κB
NK: natural killer
NKT: natural killer T
NLR: neutrophil-to-lymphocyte ratio
NLRP3: NOD-like receptor family pyrin domain-containing 3
Nrf2: nuclear factor erythroid 2-related factor 2
PA: palmitic acid
PPARγ: peroxisome proliferator-activated receptor γ
SCFAs: short-chain fatty acids
SGLT2: sodium-glucose co-transporter 2
SREBP-1c: sterol regulatory element-binding protein-1c
T2DM: type 2 diabetes mellitus
TG: triglyceride
TMAO: trimethylamine N-oxide
TNF: tumor necrosis factor
TNFR2: tumor necrosis factor receptor 2
CZ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. YS and SL: Investigation, Writing—original draft, Writing—review & editing. MY: Conceptualization, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

