Review
Review

Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
ORCID: https://orcid.org/0009-0006-2656-7429
Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
Affiliation:
State Key Laboratory Base of Cell Differentiation and Regulation, College of Life Sciences, Henan Normal University, Xinxiang 453007, Henan, China
Email: 041125@htu.edu.cn
Explor Dig Dis. 2023;2:282–296 DOI: https://doi.org/10.37349/edd.2023.00031
Received: July 17, 2023 Accepted: August 26, 2023 Published: December 13, 2023
Academic Editor: Ming Yang, University of Missouri, USA
Metabolic dysfunction-associated steatotic liver disease (MASLD) is one of the most common chronic liver diseases. Over time, there has been a significant increase in the prevalence of MASLD. It has become one of the leading causes of hepatocellular carcinoma (HCC) in the United States, France, and the United Kingdom. Globally, the incidence of HCC related to MASLD may further increase with the growing prevalence of obesity. Non-alcoholic steatohepatitis (NASH) is an important stage of MASLD, which is more likely to cause cirrhosis and even HCC. And patients with NASH cirrhosis have a much higher incidence of hepatocellular cancer than patients with non-cirrhotic MASLD. As a result, it is critical to investigate the targets of MASLD therapy in HCC. This article reviews therapeutic targets of MASLD, such as farnesoid X receptor (FXR), peroxisome proliferator activated receptor (PPAR), fibroblast growth factor-21 (FGF-21), etc., and introduces the drugs related to these targets and their mechanisms of action in HCC. In addition, the developmental process and pathogenesis of MASLD, as well as risk factors for HCC development, are discussed. These are of great significance for the prevention and treatment of HCC.
Metabolic dysfunction-associated steatotic liver disease (MASLD) is one of the common chronic metabolic diseases characterized by fatty deposits in liver cells and liver damage, which in severe cases can develop into non-alcoholic steatohepatitis (NASH). A higher proportion (between 35% and 50%) of hepatocellular carcinomas (HCCs) develop in NASH patients before cirrhosis and routine cancer screening, compared to the incidence in other liver disorders [1]. According to estimates, 32.4% of people worldwide are estimated to have MASLD, and the prevalence has significantly increased over time [2]. Globally, the number of NASH patients is rising, with the incidence expected to reach 56% within the next ten years [3]. HCC is one of the five most frequent cancers in the world, and its incidence continues to rise globally [4]. In contrast to recorded declines in mortality from other prevalent cancers, HCC mortality continues to rise year after year, increasing from the third position to the second highest cancer mortality rate [5]. According to Global Cancer Statistics 2020 (GLOBOCAN 2020), there will be 906,000 instances of liver cancer worldwide in 2020, with 830,000 fatalities [6]. Surgery, local destructive therapy, and liver transplantation all have therapeutic potential for patients with early-stage HCC. HCC recurrence, however, continues to be a serious issue even after treatment, with a recurrence rate of 50–60% in just two years [7]. Alcohol and viral hepatitis are risk factors for HCC, but MASLD is now known to be a substantial contributor to the disease [8, 9]. Statistics show that as MASLD transformed into NASH, the annual incidence of HCC rose to 5.29 per 1,000 persons. In the United States, the United Kingdom, and France, MASLD is now the underlying cause of HCC which is increasing at the quickest rate [10]. Therefore, understanding the connection between MASLD and HCC as well as the therapeutic targets for MASLD and its role in HCC is crucial. In order to effectively lower the risk of HCC and enhance the therapeutic effect, we can think of the therapeutic target of MASLD as a new target for HCC treatment in the future.
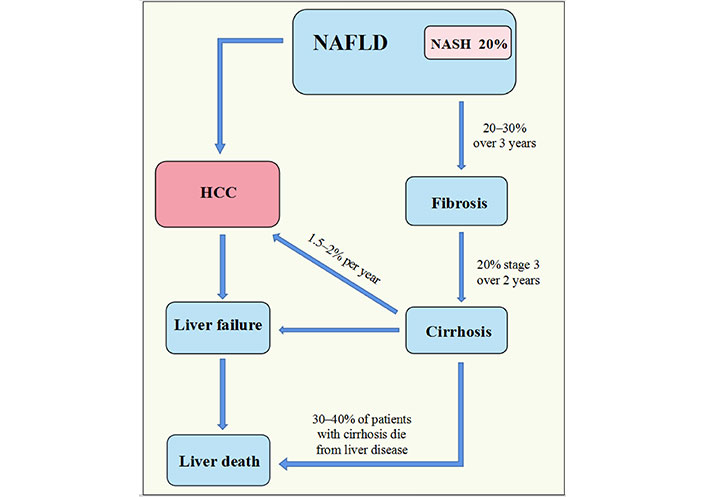
MASLD’s pathophysiology may typically be broken down into five stages: MASLD, NASH, cirrhosis, liver fibrosis, and even HCC. Approximately 20% of MASLD patients have NASH, and 20% to 30% will develop liver fibrosis within three years, and then approximately 15% of those who have liver fibrosis will develop cirrhosis, according to the data. In addition, individuals who have cirrhosis brought on by NASH have a 1.5% to 2% risk of developing HCC further [11]. We also discovered that liver failure, which can ultimately result in liver mortality, can be brought on by both cirrhosis and HCC (Figure 1).
The pathogenesis of MASLD involves complex multifactorial effects, leading to metabolic abnormalities that disrupt fatty acid metabolism in the body. This disruption causes the accumulation of large amounts of free fatty acids (FFAs) in the liver, resulting in liver fibrosis and damage to liver cells due to an inflammatory reaction. Dysregulation of the gut microbiota may also contribute to the development of MASLD, as it can lead to fatty deposits and inflammatory responses in the liver [12]. Furthermore, factors such as obesity, environment, and lifestyle are believed to play a role in the development of MASLD.
It is generally acknowledged that diet and environmental factors can elevate cholesterol (CH) levels and serum-free FFA, which in turn affect the function of adipocytes [13]. Insulin resistance, which is thought to be the first strike of the “two-hit” hypothesis, is an important role in the development of MASLD [14]. When the body’s energy intake and expenditure are out of balance, insulin resistance will change, leading to steatosis, which in turn raises the level of FFA and increases inflammation and cancer [15]. When the level of FFA in the liver grows, it might cause lipid poisoning. On one hand, cathepsin B is released, altering lysosome permeability and causing liver injury. Increased FFA levels, on the other hand, produce a number of oxidative byproducts and trigger the production of adipokines and pro-inflammatory cytokines from the liver, such as tumor necrosis factor alpha (TNF-α) and interleukin 6 (IL-6), which can lead to mitochondrial dysfunction and, finally, the development of NASH [16]. At present, the mechanism of the occurrence of MASLD has gradually transited from the “two-hit” hypothesis to the “multiple hit” hypothesis, which is generally accepted by people. Oxidative stress is also one of the factors in the “multiple hit” hypothesis and is thought to be the main cause of liver damage [1, 17]. Oxidative stress reflects that the production and consumption of reactive oxygen species (ROS) cannot maintain a balance. A high concentration of reactive ROS will cause oxidative modification of macromolecules like DNA and proteins in cells, which will lead to the accumulation of macromolecules with abnormal structures and ultimately cause liver damage [18]. On the other hand, it can result in nuclear factor-kappa B (NF-κB) activation that lasts for a long time and the production of plenty of inflammatory cells, which might result in inflammation of the liver. Furthermore, it has been demonstrated that the activation of hepatic stellate cells (HSCs) by ROS and oxidized low-density lipoprotein (LDL) might result in liver fibrosis. To summarize, insulin resistance, oxidative stress, and inflammation all play critical roles in the development of MASLD, and a better knowledge of the etiology of MASLD is required to create new therapeutic options (Figure 2) [19].
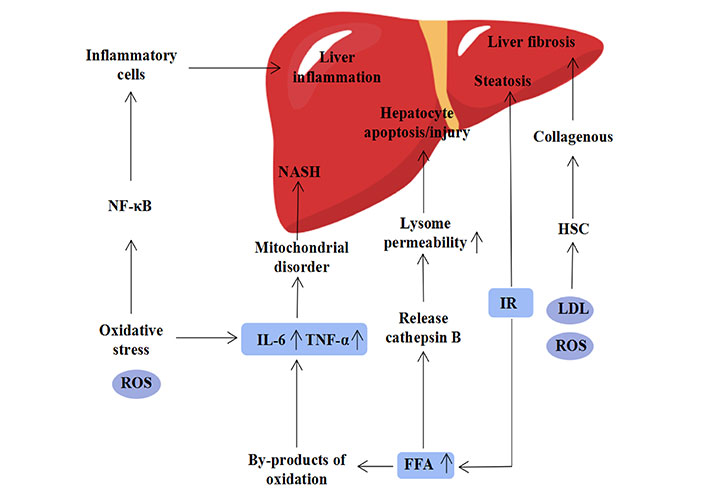
The pathogenesis of NAFLD, its causative factors include elevated FFAs, insulin resistance, oxidative stress, metabolism and inflammation [19]. IR: insulin resistance
Farnesoid X receptor (FXR) is a nuclear receptor, which is abundant in the liver, and it is thought to be a major regulator of lipid metabolism. In controlling lipid metabolism and cholesterol levels, liver X receptor (LXR), sterol regulatory element binding protein (SREBP), and FXR all play significant roles [20]. Bile acids have a crucial role in the liver, and their production is strictly regulated by negative feedback, in which FXR plays a key part. Bile acids have the ability to function as ligands for FXR. These ligands are secreted into the intestine and bind to FXR to activate fibroblast growth factor-15 (FGF-15). FGF-15 that has been activated can then inhibit the activity of hepatic cholesterol 7-alpha-hydroxylase (CYP7A1), which in turn inhibits the synthesis of bile acids [21]. Additionally, bile acids and FXR together can increase the expression of the small heterodimer partner (SHP), decrease the level of liver X receptor transcription, and decrease the synthesis of liver fat [22]. FXR agonists are crucial medications for the treatment of NASH because decreased levels of FXR expression in the liver may worsen the severity of liver disease in people with NASH. Among them, obeticholic acid, which was first created by Pellicciari et al. [23] in 2002, has been demonstrated in numerous trials to improve the hepatic histological characteristics of patients with MASLD as an FXR agonist. Obeticholic acid has been demonstrated in studies to be an effective treatment for metabolic problems and liver lesions brought on by ethanol while also safeguarding liver cells from oxidative stress [14].
In addition to obeticholic acid, there are many other potential FXR agonists, such as cilofexor, tropifexor, EDP-305, LY-2562175, etc. (Table 1) [24]. These agonists are rarely studied in humans and are mostly still studied in animal models. Following the discovery of GW4064, both GS-9674 and LY-2562175 were produced on its foundation, and their chemical structure formulas are quite similar. Unfortunately, due to its potential toxicity and extremely slow drug metabolism ability, GW4064 has not been studied in clinical trials [25]. Other drugs also have some potential risks and side effects in clinical practice, among which obeticholic acid, cilofexor and EDP-305 are most commonly associated with pruritus [26, 27]. Consequently, creating FXR agonists that work well without producing negative side effects is still a problem for pharmaceutical chemists.
Related drugs targeting FXR
| FXR agonists | Alias | Molecular formula | Chemical structure | R & D stage | References |
|---|---|---|---|---|---|
| Obeticholic acid | 6-Ethyl-chenodeoxycholic-acid, OCA | C26H44O4 | 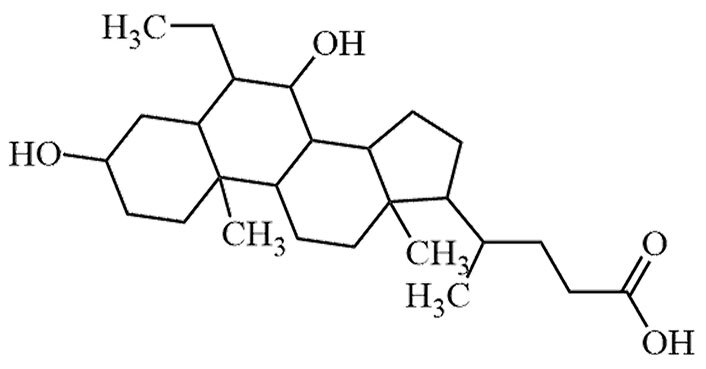 | Approval to market | [24, 25] |
| GW4064 | GW-4064 | C28H22Cl3NO4 | 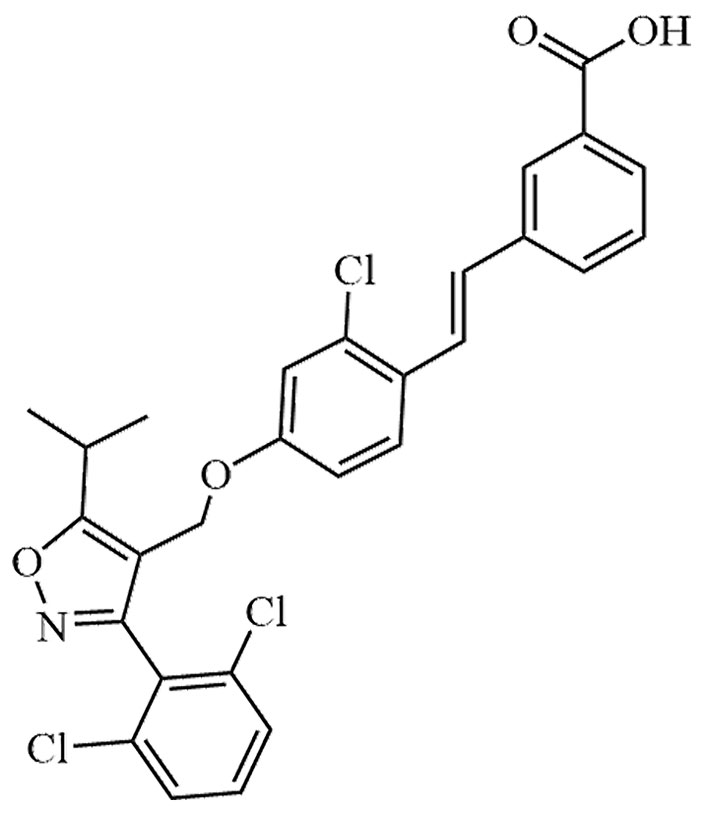 | Preclinical | [24, 25] |
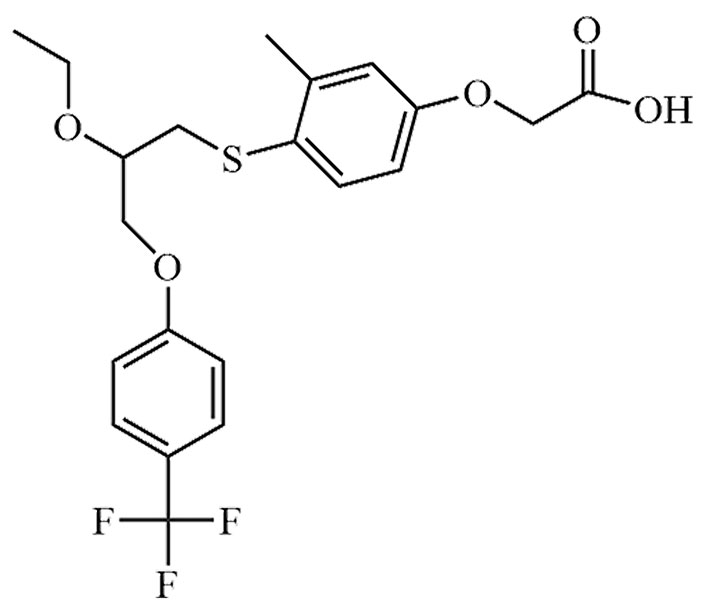
| Cilofexor | CILO, GS-9674 | C32H33Cl3N4O8 | 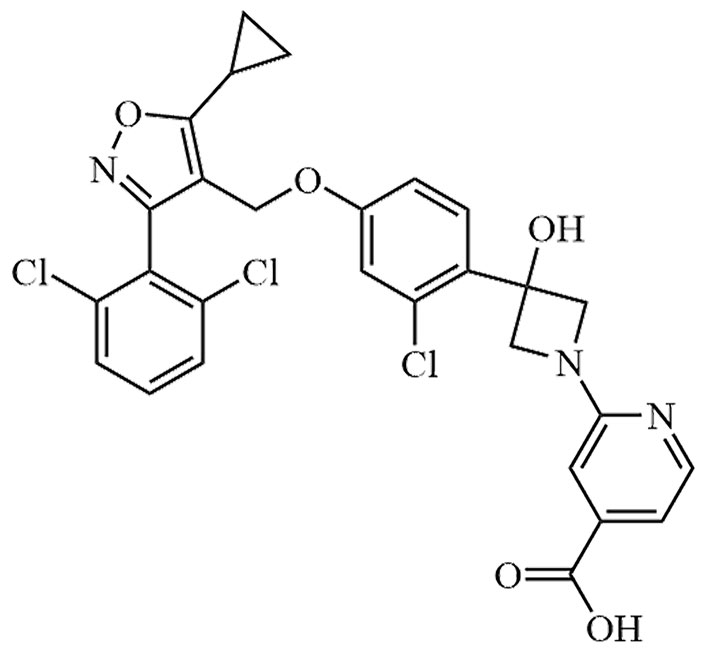 | Phase III | [24, 25] |
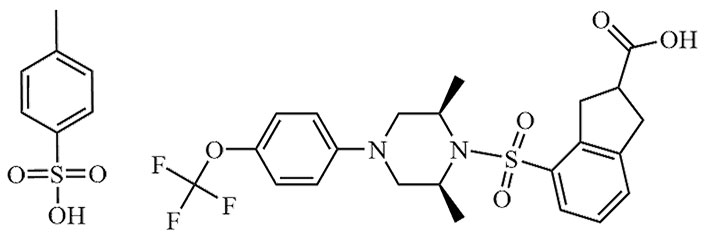
| Tropifexor | LJN-452, NVP-LJN452-NXA | C29H25F4N3O5S | 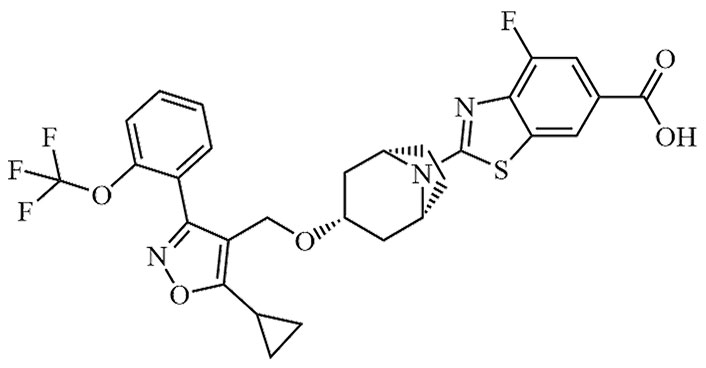 | Phase II | [24, 25] |
| EDP-305 | EDP 305 | C36H58N2O5S | 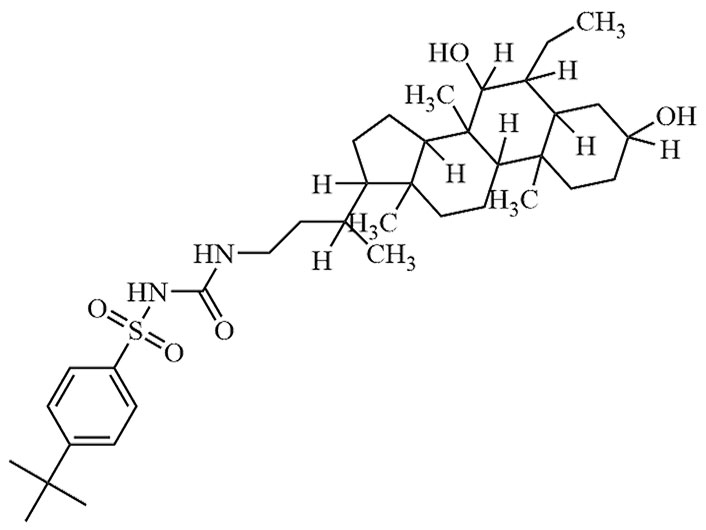 | Phase II | [27] |
| LY-2562175 | None | C28H27Cl2N3O4 | 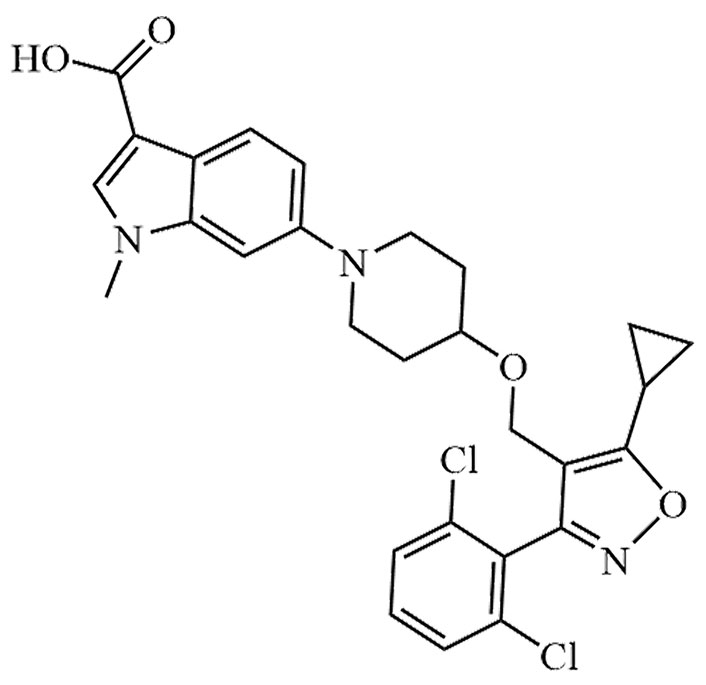 | Phase II | [24, 25] |
Peroxisome proliferator activated receptor (PPAR) is a class of ligand-activated transcription factors that regulate energy homeostasis and belongs to the nuclear receptor superfamily, which was first discovered in 1990 by the British scientist Issemann et al. [28]. Because it can be activated by a class of substances called peroxisome proliferators, PPAR was given its name [28]. Humans have three distinct PPAR isoforms: PPARα, PPARβ/δ, and PPARγ. The various isoforms are found in various tissues and each one has a particular regulatory function [29]. Despite having similar target gene sequences, they have different ligand selectivity. PPAR is a molecule that regulates hepatic metabolism and hence plays a significant part in glucose and lipid metabolism, as well as lipogenesis regulation and hepatic energy homeostasis maintenance [30].
The first member of the PPAR family to be identified is PPARα [28]. It is mainly expressed in the liver, except for the kidney and muscle tissue where it is also widely expressed. It is widely believed that the activity of PPARα is localized in the nucleus, but in fact, it has been reported that PPARα is most often localized in the cytoplasm [31]. PPARα is a transcription factor that regulates the transcription of genes involved in fatty acid metabolism, including lipoprotein metabolism, gluconeogenesis, and bile acid metabolism [32]. It has certain anti-inflammatory properties in addition to its ability to control fat metabolism. In addition, studies have shown that the expression of PPARα is reduced in patients with MASLD. Therefore, increasing the expression level of PPARα is an important approach to the treatment of MASLD [33]. There are numerous PPARα agonists in preclinical development at the moment.
One of the agonists of PPARα, which can either directly or indirectly impact the expression of several genes, is fenofibrate. Fenofibrate can induce the expression of PPARα, thereby activating genes involved in fatty acid β-oxidation and oxidative phosphorylation [34]. It was indicated that fenofibrate may have positive effects on hepatocytes in some studies using specific NASH models [35]. However, long-term use of fenofibrate can cause intestinal discomfort, so many drug developers are constantly optimizing it [34]. There are numerous PPARα agonists that are licensed or in clinical trials in addition to fenofibrate, including gemfibrozil, bezafibrate, pemafibrate, etc. (Table 2). But unfortunately, fibrates (such as fenofibrate and bezafibrate) can cause side effects such as liver dysfunction, while pemafibrate is relatively safe [36].
Related drugs targeting PPARα
| PPARα agonists | Alias | Molecular formula | Chemical structure | R & D stage | References |
|---|---|---|---|---|---|
| Fenofibrate | FNF | C20H21ClO4 | 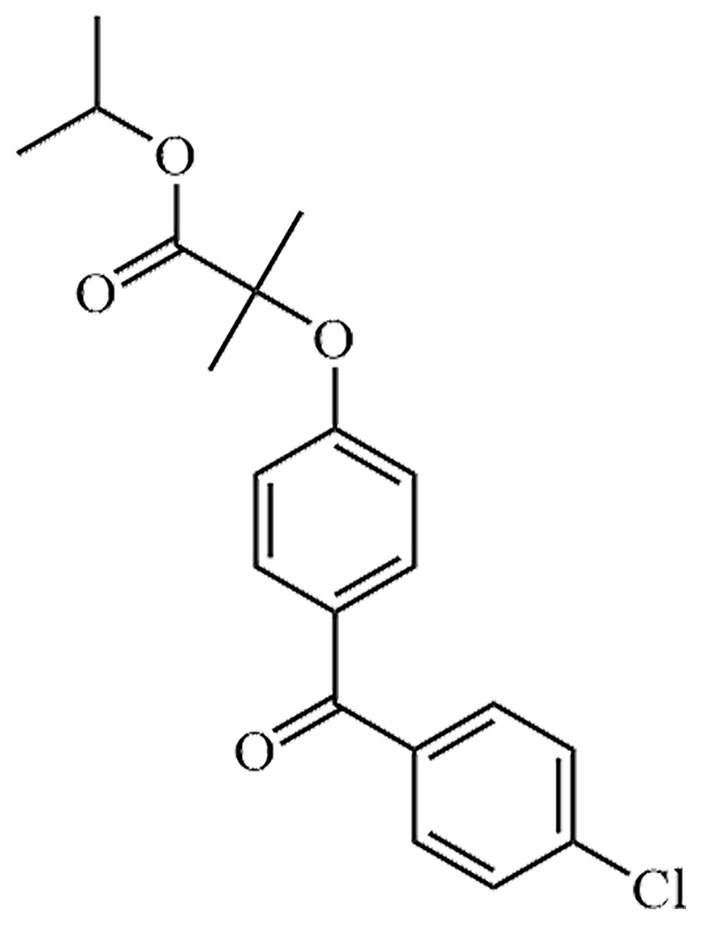 | Approval to market | [34] |
| Gemfibrozil | GEM, CI-719 | C15H22O3 | 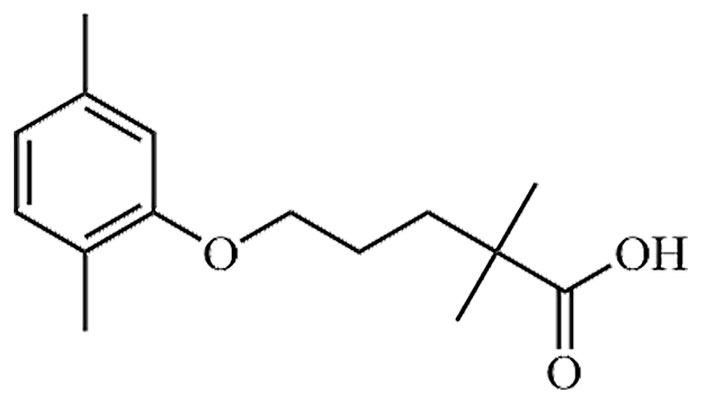 | Approval to market | [37] |
| Bezafibrate | LO-44 | C19H20ClNO4 | 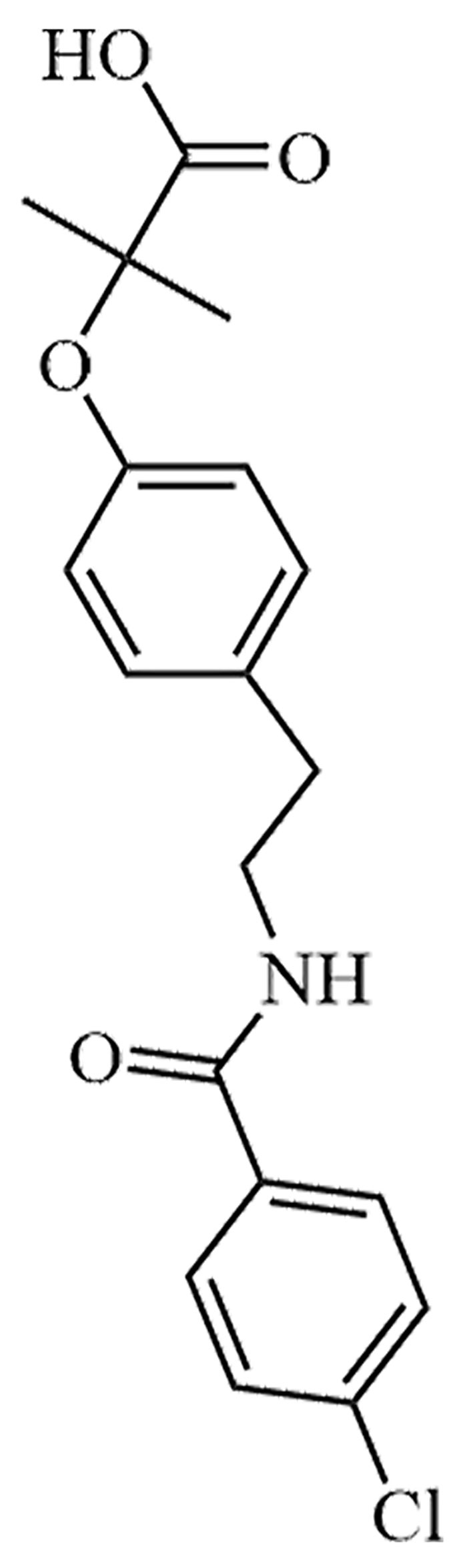 | Approval to market | [38] |
| Pemafibrate | JAN, K-877, K-877-FL, K-877-ER, K-877-BC | C28H30N2O6 | 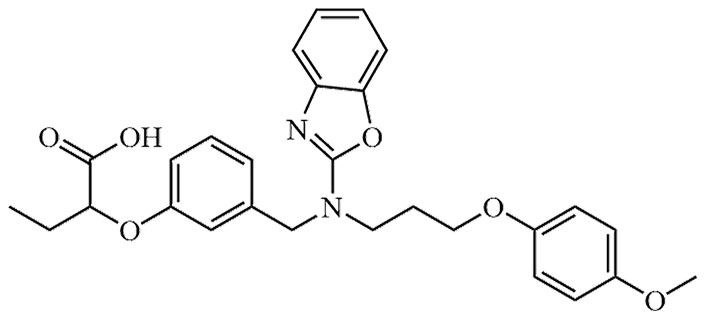 | Approval to market | [39] |
PPARδ, also known as PPARβ/δ, is the second subtype of the PPAR family. Essentially, it is a transcription factor involved in ligand-regulated gene expression. Upon activation by a ligand agonist, PPARδ binds to response elements within the promoter region of target genes, thereby regulating the transcription process of those genes. PPARδ possesses the ability to regulate cell growth, differentiation, metabolism, and survival [40]. PPARδ has a wide expression pattern. While PPARα and PPARγ are mainly present in the liver and adipose tissue, PPARδ is widely expressed in the digestive system, reproductive system, brain, bone marrow, muscle tissues, and others [41].
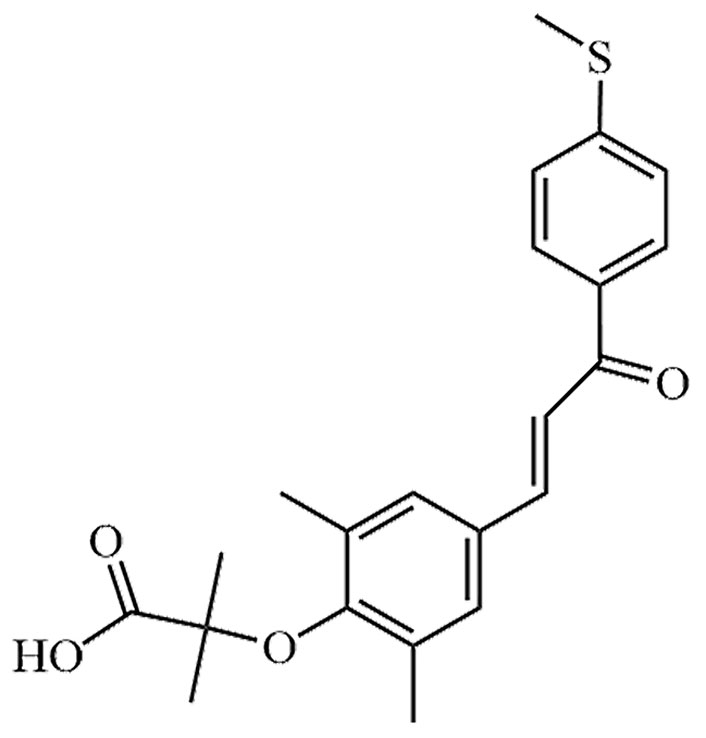
Seladelpar is a novel selective agonist of PPARδ. It has been tested in experiments aimed at alleviating lipotoxicity in diabetic obese mice, with the objective of reversing obesity, reducing blood lipid levels, and eliminating hepatic lipotoxicity in diabetic mice. The results demonstrated a decrease in hepatic lipotoxicity in the mice [42]. In addition to seladelpar, there are other PPARδ agonists that directly or indirectly act on MASLD, such as elafibranor, lanifibranor, KD-3010, etc. (Table 3). At the same time, potential risks of these drugs have been found clinically, among which, treatment with elafibranor can lead to increased serum creatinine [43].
Related drugs targeting PPARβ/δ
PPARγ is the third subtype of the PPAR family. Its main function is to regulate glucose and lipid metabolism in adipose tissue. Additionally, it plays a significant role in immune regulation in the human body. While PPARγ primarily plays a crucial role in adipose tissue by improving insulin sensitivity, increasing lipoprotein lipase expression, and inhibiting lipolysis, it is also found to be highly expressed in patients with MASLD [47]. In hepatocytes, PPARγ serves as a regulatory factor for lipid metabolism, specifically targeting the processes involved in fat synthesis.
The most widely used PPARγ agonists are the thiazolidinediones, which improve NASH, including rosiglitazone, and pioglitazone [19, 48]. There are some other PPARγ agonists, such as saroglitazar, AMG-131, etc. (Table 4). There are also potential risks associated with these drugs, including case reports that rosiglitazone may cause liver cell damage and liver failure in patients. Clinical use of pioglitazone has also been associated with some adverse effects, including weight gain, fluid retention, and possible bladder cancer [49].
Related drugs targeting PPARγ
| PPARγ agonists | Alias | Molecular formula | Chemical structure | R & D stage | References |
|---|---|---|---|---|---|
| Rosiglitazone | Avandia | C18H19N3O3S | 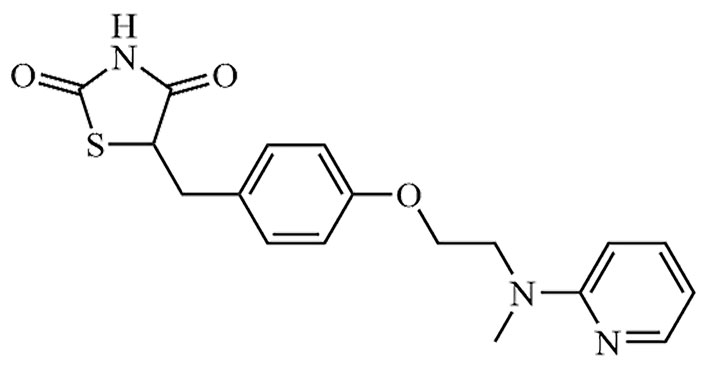 | Approval to market | [19] |
| Pioglitazone | Actos | C18H19N3O3S | 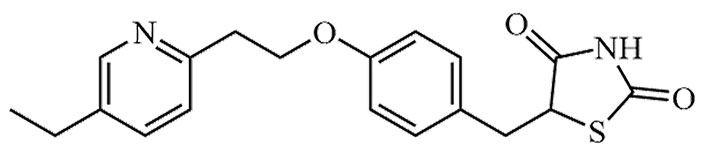 | Approval to market | [19] |
| Saroglitazar | [14C]-Saroglitazar, Bilypsa, Lipaglyn | C25H29NO4S | 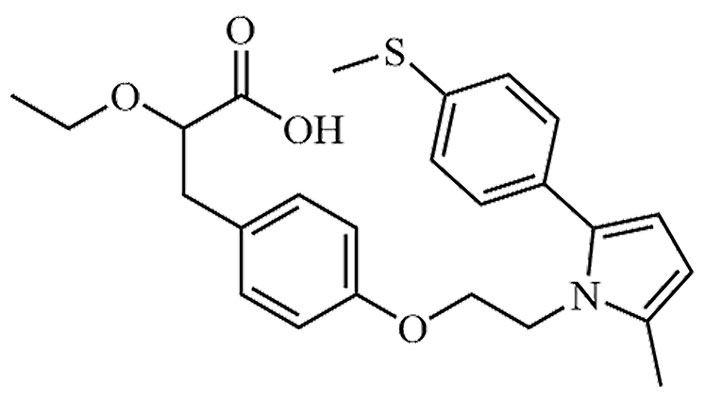 | Approval to market | [50] |
| AMG-131 | INT131 besylate, CHS-131, T-131, T-0903131, INT-131 | C21H12Cl4N2O3S | 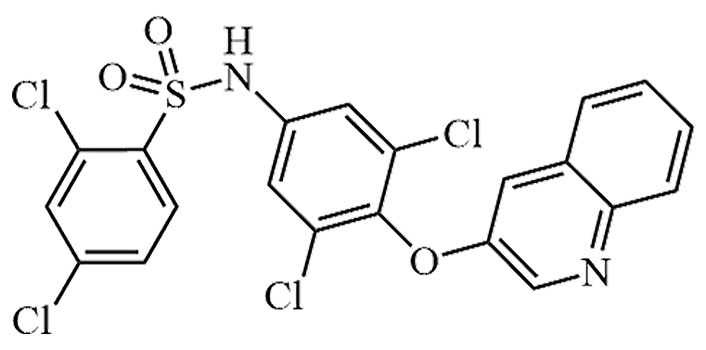 | Phase II | [51] |
FGF-21 is one member of the FGF protein family. In addition to FGF-21, this family also includes FGF-19 and FGF-23. They play crucial roles in maintaining overall body homeostasis. FGF-21 is a metabolic signal-regulating protein that enhances insulin sensitivity and improves insulin resistance in the liver, adipose tissue and other organs, thus exerting a metabolic regulatory effect that is beneficial to the health of the body [52]. Studies have shown that obesity, as well as MASLD/NASH, can increase the expression of FGF-21 in the liver. Furthermore, this research suggests that FGF-21 may serve as a novel biomarker for MASLD [53]. Contrary to initial beliefs that FGF-21 would increase fat breakdown, it actually inhibits fat synthesis through SREBPs, thereby lowering fat buildup in the liver [54]. In addition, FGF-21 exerts its metabolic effects only after binding to its receptors. It acts through the interaction with FGF receptor 1c (FGFR1c) and co-receptor β-Klotho. Both receptors are indispensable, and the absence of either receptor weakens its metabolic effects. However, the specific mechanism remains unclear [55]. FGF-21 can also alleviate endoplasmic reticulum stress in the liver, thereby inhibiting hepatic steatosis, reducing cellular apoptosis, and improving the severity of MASLD [56].
LY2405319 is the first FGF-21 analogue to be evaluated in humans [57]. It is currently in phase I clinical trials, and the clinical results demonstrate that this drug can improve dyslipidemia, lower low-density lipoprotein cholesterol, increase high-density lipoprotein cholesterol, and also assist obese individuals and patients with type 2 diabetes in weight reduction [58]. In addition to LY2405319, analogues of FGF-21 include pegbelfermin, PF-05231023, and efruxifermin. Among them, pegbelfermin and efruxifermin are currently in phase II clinical trials, while PF-05231023 is still in phase I clinical trials. Among them, LY2405319 is currently performing well in clinical trials, while PF-05231023 patients will show a slight tendency to drop blood sugar during treatment, and in severe cases may develop diarrhea and nausea [59].
In addition to the previously mentioned FXR, PPAR, and FGF-21, sirtuins (SIRTs) and hepatocyte leukotriene B4 receptor 1 (Ltb4r1) are possible therapeutic targets for MASLD. SIRTs are thought to be involved in insulin signaling, redox signaling, and inflammation, while inhibition of hepatocyte Ltb4r1 reduces steatosis and insulin resistance [12, 60].
The course of MASLD and its pathogenesis were described previously, in which NASH may be the main cause of the development of cryptogenic cirrhosis, while patients with cryptogenic cirrhosis are more likely to develop HCC [61]. Currently, it has been determined that diabetes and obesity are risk factors for developing NASH and cryptogenic cirrhosis. According to studies, patients with type 2 diabetes mellitus (T2DM) had a five-fold higher prevalence of MASLD than those without T2DM. Consequently, persons with T2DM and obesity have a higher risk of developing HCC [62, 63]. Clinically, NAFLD co-exists with type 2 diabetes and obesity and plays a synergistic role. The low-grade chronic inflammatory characteristics of IR are more conducive to macrophage recruitment and pro-inflammatory cytokine release in the case of obesity. TNF-α and IL-6 contribute to the development of HCC through their effects on ikappaB kinase (IKK) and janus kinase (JNK) signaling pathways [63, 64].
FXR has a preventive effect on HCC, which was first discovered in mice with spontaneous HCC and FXR deficiency [65]. Researchers from all around the world have focused on HCC, but the precise pathogenic mechanisms behind its onset and development are still unknown. The role of FXR in HCC has progressively become a new research hotspot in recent years, following the clarification of its significance in various liver diseases. Furthermore, there is mounting evidence suggesting that patients with HCC exhibit impaired transduction signaling of FXR.
FXR expression is lower in HCC patients compared to normal liver [66]. Metabolic disorders, MASLD, and bile acid-induced liver injury are developing as the major causes of HCC, and FXR, as a metabolic regulator, plays a bridging role in HCC caused by metabolic disorders. In 2007, Kim et al. [65], conducted experiments on FXR-deficient mice and the results demonstrated that these mice spontaneously develop HCC as they age. Subsequently, Liu et al. [67], conducted further research and confirmed the existence of significant similarities between the mechanism of HCC development in FXR-deficient mice and humans. The experiments conducted by these researchers definitively established the correlation between FXR and HCC, highlighting the potentially crucial role FXR may play in the treatment of HCC. Hepatitis B and C viruses are known to induce chronic liver disease that can progress to HCC [68]. Several studies have demonstrated that FXR exhibits hepatoprotective effects by mitigating liver disease associated with these two hepatitis viruses and exerting anti-inflammatory properties. Bile acids have a key role in the onset and progression of HCC, with FXR playing an important regulatory role in bile acid metabolism and production. It has the potential to regulate the process of HCC occurrence by modifying bile acids, including inflammation, oxidative stress, and other factors, in order to minimize the occurrence and progression of HCC [69, 70].
PPAR is a distinct nuclear receptor that serves as a major regulator of lipid metabolism, as well as a link between fatty acid, cellular metabolism, and immune function, and has emerged as a critical regulator of immunological and lipid metabolic processes [71]. Overexpression of PPARα enhances cancer cell chemosensitivity, which may be due to the pro-apoptotic properties of PPARα. Zhang et al. [72], conducted experimental research to investigate the inhibitory mechanism of the NF-κB signaling pathway mediated by PPARα in hepatocarcinogenesis. They conducted experiments using three groups of mice: wild-type mice, PPARα knockout mice, and mice injected with diethylnitrosamine (DEN). The results showed that PPARα-deficient mice were more susceptible to DEN-induced tumorigenesis. It was observed that regulating the NF-κB signaling pathway could effectively achieve anti-tumor effects. Based on these findings, it can be concluded that PPARα functions as a tumor suppressor. In vivo studies have demonstrated the significance of host PPARα in immune-mediated tumor control, as the growth of transplantable tumors is significantly suppressed in PPARα knockout mice, irrespective of the PPARα status in cancer cells [73]. Similar to PPARα, PPARβ/δ inhibits NF-κB activity by binding to its subunit p65 and also possesses some anti-inflammatory effects. It has been shown that activating PPARβ/δ in the Kupffer cells of hepatitis B transgenic mice can inhibit the development of liver tumors [74]. GW501516, a compound commonly used in in vitro studies, also known as cardarine, was observed to increase cell proliferation rates in human HCC cell lines and to inhibit cell growth by RNA interference (RNAi) technology using PPARβ/δ. The outcomes also demonstrate that, in HCC cell lines, activation of PPARβ/δ upregulates the expression of cyclooxygenase (COX)-2, a rate-limiting enzyme in prostaglandin synthesis and tumor growth [75]. The literature suggests that PPARγ can exert anti-tumor effects by regulating metabolic pathways, increasing ROS levels, inducing apoptosis, and inhibiting the proliferation, migration, and infiltration abilities of tumor cells. Furthermore, activation of PPARγ can counteract proliferation by promoting cell differentiation. These findings imply that activating PPARγ may be beneficial in slowing down or halting the proliferation of undifferentiated tumor cells [76]. By searching for PPARγ agonists, scientists were able to uncover the multiple mechanisms of action that PPARγ plays in the inhibition of tumor cell growth. Research on the importance of this transcription factor in physiology and pathophysiology has attracted a lot of attention in the field [77]. According to experimental research, it has been found that the PPARγ agonist rosiglitazone (RGZ) can induce apoptosis in human hepatoma cells hepatoma G2 (HepG2) by activating PPARγ and inhibiting the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) signaling pathway [78].
FGF-21 is a powerful metabolic inflammation and obesity inhibitor. FGF-21 suppresses overload and lipid steatosis, preventing the onset of fatty liver disease and fibrotic injury, and eventually reduce the risk of the occurrence of HCC. It is highly induced in the liver in response to metabolic and pathological disturbances [79]. In addition, FGF-21, which is induced by liver injury and stress, can be used as a prognostic biomarker to monitor liver carcinogenesis and has been established as a biomarker for the early diagnosis of HCC [80]. P53, a human tumor suppressor gene, cooperates with signal transducer and activator of transcription 3 (STAT3) to regulate the expression of FGF-21. In the presence of endoplasmic reticulum stress and oxidative stress, FGF-21 is upregulated by P53 and STAT3, thereby promoting the development of liver tumors. Consequently, it is regarded as an early diagnostic biomarker [81, 82]. In another study, it was demonstrated that the overexpression of FGF-21 in transgenic mice resulted in a significant delay in the onset of diethylnitrosamine-induced liver tumors, indicating a substantial protective effect [83].
This article provides a comprehensive review of the pathogenesis of MASLD, therapeutic targets, and their roles in HCC. Firstly, the paper provides a thorough explanation of the pathogenesis of MASLD. Factors such as insulin resistance, oxidative stress, and inflammatory response are identified as the underlying causes of MASLD development. Furthermore, based on these etiologies, the paper presents a comprehensive list of corresponding therapeutic targets such as FXR, PPAR, and FGF-21. For each of these targets, the paper also provides a compilation of relevant drugs including FXR-targeted drugs such as obeticholic acid and cilofexor, PPAR-targeted drugs such as fenofibrate, seladelpar, and rosiglitazone, and FGF-21 analogs like LY2405319. These drugs exhibit significant potential in the treatment of MASLD.
It is well known that MASLD is one of the most common risk factors for liver cancer, and patients with MASLD have a higher risk of developing liver cancer, which has gradually become a major cause of HCC development. At the same time, compared to the declining mortality rates of other common cancers such as breast cancer and lung cancer, the mortality rate of HCC continues to rise every year. Therefore, using MASLD as an entrance point and then searching for new therapeutic techniques and tactics for HCC, this research focuses on the association between MASLD and HCC as well as the function of MASLD therapeutic targets in HCC.
Finally, we believe that investigating the role of therapeutic targets in MASLD treatment on HCC is of significant importance for both prevention and treatment of liver cancer. As this article points out, certain therapeutic targets in MASLD treatment, such as FXR and PPARα, have shown inhibitory effects on tumor initiation and progression in liver cancer. In conclusion, the investigation of therapeutic targets in MASLD treatment holds essential significance and promising prospects. It is hoped that in the future, more effective treatment methods can be developed, bringing more benefits to patients with MASLD and HCC.
FFAs: free fatty acids
FGF-15: fibroblast growth factor-15
FXR: farnesoid X receptor
HCC: hepatocellular carcinoma
MASLD: metabolic dysfunction-associated steatotic liver disease
NASH: non-alcoholic steatohepatitis
PPAR: peroxisome proliferator activated receptor
T2DM: type 2 diabetes mellitus
TNF-α: tumor necrosis factor alpha
CW and XY: Writing—review & editing. JW and CZ: Investigation. YZ and CL: Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the Science and Technology Research Project of Henan Province [222102310244]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.