 Review
Review

Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
†These authors share the first authorship.
Email: swin@usc.edu
ORCID: https://orcid.org/0000-0001-7694-5172
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0001-8818-9271
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
2Biological Sciences Collegiate Division, University of Chicago, Chicago, IL 60637, USA
Affiliation:
3Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
3Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
3Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
3Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
3Dornsife College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
4Bridge Undergraduate Science (BUGS) Program, Bridge Institute, College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
4Bridge Undergraduate Science (BUGS) Program, Bridge Institute, College of Letters, Arts and Sciences, University of Southern California, Los Angeles, CA 90089, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
1Department of Medicine, Division of Gastrointestinal and Liver Diseases, Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Affiliation:
5Program in Liberal Medical Education (PLME), Warren Alpert Medical School, Brown University, Providence, RI 02903, USA
ORCID: https://orcid.org/0000-0001-5680-8648
Explor Dig Dis. 2024;3:42–68 DOI: https://doi.org/10.37349/edd.2024.00039
Received: November 07, 2023 Accepted: January 03, 2024 Published: February 29, 2024
Academic Editor: Wen-Xing Ding, University of Kansas Medical Center, USA
The article belongs to the special issue Mitochondria and Lipid Signalling in Liver Diseases
Mitochondria are present in all mammalian cells except matured red blood cells. Mitochondria consist of several metabolic pathways for glucose, fatty acids, amino acids, and bioenergetic pathways for ATP synthesis, membrane potential, and reactive oxygen production. In the liver, hepatic mitochondria play a key role in hepatic steatosis because mitochondrial metabolism produces acetyl-CoA which is the building block for synthesis of lipids and cholesterol. Mitochondria inner membrane is impermeable of metabolites, reducing equivalents, and small molecules such as phosphate, and sulfate. Thus, mitochondrial shuttles and carriers function as the routes of influx and efflux of these metabolites and molecules across the inner membrane. The signal regulation of these shuttles and mitochondrial enzymes could play a key role in coordinating the mitochondrial metabolism to adapt the cytosolic part of metabolic pathways in liver metabolic stress. Intriguingly, the interaction of mitochondria protein SH3 domain-binding protein 5 (SAB/SH3BP5) and c-Jun N-terminal kinase (JNK) was found as a pivotal role in sustained activation of JNK and phosphorylated-JNK (P-JNK) mediated activation of lipogenic pathway in nutritional excess. Knockout or knockdown of SAB prevented or reversed the hepatic steatosis, inflammation, and fibrosis, and improved metabolic intolerance and energy expenditure. Moreover, blocking the SAB peptide prevents palmitic acid-induced P-JNK interaction with SAB and inhibition of mitochondrial bioenergetics, implying the P-JNK effect on mitochondrial metabolism. This review focuses on the flow of mitochondrial metabolites in metabolic stress conditions and the contribution of mitochondria and mitochondrial stress signals in hepatic steatosis.
Mitochondria occupy 22% of total cell volume, the second largest intracellular compartment of hepatocytes [1]. Mitochondria are very versatile and adaptive organelles, and the dynamics of mitochondrial shape, size, and number are congruent with the metabolic and energetic needs of a cell. Metabolic pathways such as the tricarboxylic acid (TCA) cycle, β-oxidation, ketogenesis, and energetic pathways including electron transport chain (ETC) and ATP synthesis are the unique components of mitochondria. Mitochondrial dysfunction is associated with metabolic dysregulation in liver [2]. Hepatic mitochondria provide a hub for liver metabolic homeostasis by uniquely integrating or coordinating multiple metabolic pathways of carbohydrate, lipid, cholesterol, bile acid, and amino acids for energy homeostasis and by keeping the balance of reducing equivalents and intermediary metabolites in the liver and whole body via mitochondria-cytoplasm shuttles. The TCA cycle is the core of metabolic pathways leading to production of energy for cells. Anaplerosis, the process of replenishing intermediates into the TCA cycle, is essential for energy metabolism. Anaplerosis is coupled with cataplerosis, which is the removal of TCA cycle intermediates. Anaplerosis occurs when intermediates are extracted (cataplerosis) for biosynthesis such as gluconeogenesis and lipogenesis. In gluconeogenesis, malate leaves the mitochondria, and a subsequent chain of reactions generates glucose. In lipogenesis, citrate leaves the mitochondria and is metabolized to form fatty acids. The main anaplerotic substrates are pyruvate, glutamine/glutamate, and fatty acyl-CoA. Together, anaplerosis and cataplerosis help regulate the biosynthesis and cellular energy status [3]. The overview of the biochemical pathways of mitochondria, followed by the mechanism of signal modulation on the mitochondrial metabolism, is the primary focus in this review. This review highlights the regulation of mitochondrial metabolic adaptation in hepatic steatosis.
The TCA cycle (Figure 1), also known as the citric acid cycle or Krebs cycle, is a closed cycle of metabolic reactions. The TCA cycle constitutes the center of cell metabolism because multiple substrates such as pyruvate, fatty acids, and amino acids can feed into it. Pyruvate, an intermediate of glucose oxidation (glycolysis in the cytoplasm), is transported into mitochondria by mitochondrial pyruvate carrier (MPC). Pyruvate dehydrogenase (PDH) complex (PDHC) oxidizes pyruvate to acetyl-CoA. PDHC is subject to allosteric feedback inhibition by acetyl-CoA, NADH, and ATP. The TCA cycle begins with citrate synthase (CS) activated by NAD+ and its substrates acetyl-CoA and oxaloacetate (OAA). The combination of two-carbon acetyl-CoA and four-carbon OAA generates six-carbon citrate, which then is converted into cis-aconitate and isocitrate in the presence of aconitase (ACO). Two carbons from isocitrate are removed as CO2 by oxidative decarboxylation, releasing two NADH in two steps. First, isocitrate dehydrogenase (IDH) complex (IDHC) decarboxylates isocitrate to five-carbon α-ketoglutarate (α-KG). Second, α-KG is decarboxylated by oxoglutarate dehydrogenase (OGDH) complex (OGDHC) to four-carbon succinyl-CoA. Succinyl-CoA synthetase then converts succinyl-CoA into succinate, which is oxidized by succinate dehydrogenase (SDH) to fumarate and flavin adenine dinucleotide (reduced) (FADH2). Next fumarate is converted to malate by fumarase. Malate is oxidized to OAA by malate dehydrogenase (MDH). OAA combines with another acetyl-CoA to continue the TCA cycle [3]. Of note, three enzymes in particular, CS, IDH, and OGDH, catalyze rate-limiting steps in the TCA cycle. All three enzymes are allosterically inhibited by high levels of mitochondrial NADH generated from the TCA cycle [4–6]. Mitochondrial Ca2+ oscillation through endoplasmic reticulum (ER) calcium channels modulates the activity of PDHC, IDH, and OGDH [7]. ACO which contains a Fe-S cluster is susceptible to inactivation by superoxide radical (O2•–) [8].
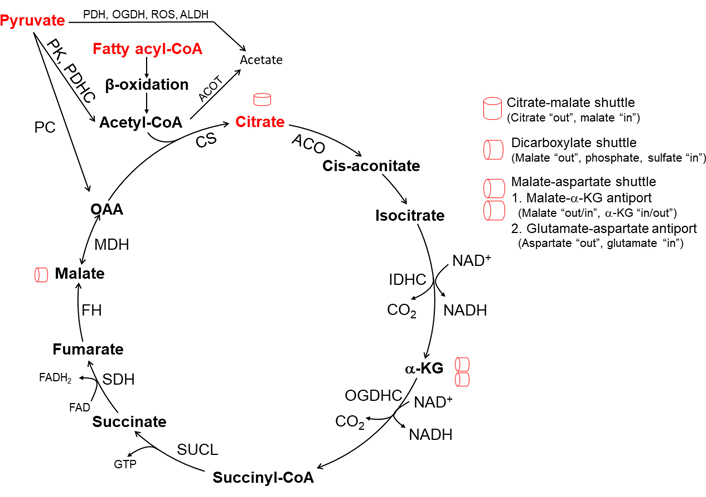
TCA cycle and mitochondrial intermediate metabolite shuttles. CS is the first and rate-limiting enzyme of the TCA cycle and is activated by its substrate acetyl-CoA generated from pyruvate metabolism and β-oxidation. Acetyl-CoA also activates PC in pyruvate to OAA to drive citrate synthesis. When pyruvate metabolism is suppressed by acetyl-CoA inhibition of PDH, mitochondrial β-oxidation mainly contributes necessary acetyl-CoA for citrate synthesis. IDH, and OGDH activity is suppressed and recycling of OAA is diminished due to an increased ratio of NADH/NAD+ state which could occur when consumption of NADH decreases due to inhibition of mitochondrial respiration. The depletion of OAA is rescued by the citrate-malate shuttle and malate-aspartate shuttle. Membrane permeable short-chain fatty acid (SCFA) acetate could be generated in mitochondria via the reaction of mitochondrial acyl-CoA thioesterase and PDH, OGDH, and ALDH. PC: pyruvate carboxylase; ROS: reactive oxygen species; ALDH: aldehyde dehydrogenase; ACOT: acyl-CoA thioesterases; SUCL: succinyl-CoA ligase; FAD: flavin adenine dinucleotide; FH: fumarate hydratase
One turn of the TCA cycle, starting with one acetyl-CoA derived from one pyruvate, generates one GTP (or ATP), three NADH, one FADH2, and two CO2. One NADH yields 10 H+ crossing the respiratory complexes (4 H+ through complex I, 4 H+ through complex III, and 2 H+ through complex IV), and one FADH2 yields 6 H+. Reentry of 4 H+ from inter-membrane space through ATP synthase generates one ATP. Therefore, one turn of the TCA cycle yields ten ATP [3].
The mitochondrial inner membrane is impermeable to intermediate metabolites generated in the mitochondria. In addition to carrier proteins such as pyruvate carrier and carnitine carrier, mitochondrial shuttles exchange mitochondrial metabolites in or out of mitochondria to replenish or dispose of metabolites. These transporters become major players in metabolic adaptation in mitochondria during hepatic metabolic stress. For instance, in the high-fat diet (HFD)-induced metabolic syndrome, liver mitochondria with an inefficient respiratory chain generate intermediate metabolites for gluconeogenesis. Antisense oligonucleotide (ASO) targeting PC reverses diet-induced gluconeogenesis [9–11]. Inhibition of citrate-malate transporter reverses the hepatic steatosis [12].
The malate-aspartate shuttle (Figure 2), one of the two redox balancing shuttles, ubiquitously expresses and generates NAD+ in cytoplasm and NADH in mitochondria. This cycle localized to the inner mitochondrial membrane (IMM) involves two antiporters (glutamate-aspartate and malate-α-KG antiporter), two oxidation or reduction MDHs (MDH1: cytosolic, MDH2: mitochondrial), and two transamination glutamate-OAA transaminases (GOTs, GOT1: cytosolic, GOT2: mitochondrial) [13]. Aspartate efflux is accompanied by the electro-gradient influx of glutamate and proton into the mitochondria. Therefore, this process is irreversible and the rate-limiting step of the shuttle [14, 15]. The exchange of malate and α-KG is bidirectional because it is driven by the concentration gradients of its substrates [16, 17]. Compartmentalization of NAD+/NADH through the shuttle is key in stress conditions of the liver in which cytosolic NAD+ promotes glycolysis and mitochondrial NADH for ATP production [18, 19]. Moreover, aspartate efflux through this shuttle increases the cytosolic NADPH/NADP+ ratio essential for glutathione synthesis [20]. In addition to redox regulation, the shuttle creates amino acid compartmentalization [21]. The primary action of this shuttle is to replenish substrates for the TCA cycle.
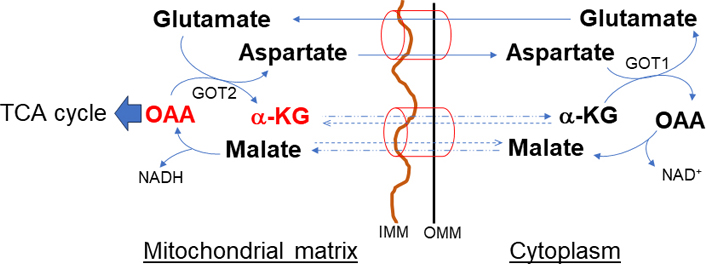
Malate-α-KG and glutamate-aspartate (malate-aspartate) shuttle. The shuttle has an important role in de novo amino acid synthesis. The primary action of this shuttle is to import glutamate and malate and convert to TCA intermediate metabolites necessary to sustain the OAA and α-KG and to maintain reducing equivalents. OMM: outer mitochondrial membrane
In contrast to the malate-aspartate shuttle, the citrate-malate shuttle [solute carrier family 25 member A1 (SLC25A1), Figure 3] equally distributes the reducing equivalents across the mitochondrial membrane. The shuttle also utilizes both MDH isoforms. However, MDH activity paired with CS, ATP-citrate lyase (ACLY), and the citrate-malate antiporter drives citric acid from mitochondria to the cytoplasm [12, 22]. Therefore, citric acid efflux through the citrate-malate shuttle promotes de novo fatty acid synthesis which is limited by citric acid compartmentalization [12]. Thus, the malate-aspartate and citrate-malate shuttles are not interchangeable, even though both have MDH activity to balance reducing equivalents. The citrate efflux activates ACLY generation of acetyl-CoA, resulting in post-translational modification of proteins through acetylation or deacetylation by acetyltransferases or sirtuins (SIRTs), respectively [23–25]. Importantly, enhanced activation of the citrate-malate shuttle could completely bypass the in-between intermediates reactions of the TCA cycle and thus could diminish mitochondrial NADH levels resulting in decreased mitochondrial bioenergetics.
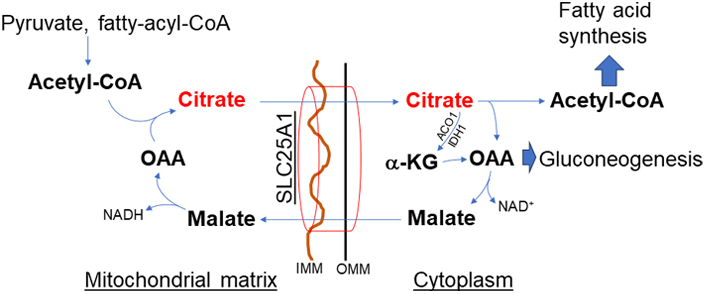
Citrate-malate shuttle (SLC25A1). Citric acid exists from mitochondria through this shuttle and has an important role in de novo fatty acid synthesis. This shuttle bypasses the TCA cycle, thus decreasing mitochondrial bioenergetics but increasing citrate
The α-glycerophosphate shuttle (Figure 4) is a unique redox-balancing shuttle without directly affecting mitochondrial NAD+/NADH. The shuttle has cytosolic and mitochondrial glycerol-3-phosphate dehydrogenase (cGPD and mGPD) enzymes (GPD1 and GPD2) which catalyze the reactions between dihydroxyacetone phosphate (DHAP) and glycerol-3-phosphate (G3P). cGPD1 utilizes NADH to reduce DHAP to G3P and generates cytosolic NAD+. G3P is subsequently oxidized to DHAP by the FAD-dependent mGPD2, which directly deposits electrons into the ETC. Of note, triose phosphate isomerase 1 (TPI1) rearranges DHAP into glyceraldehyde-3-phosphate (GA3P) which enters into glycolysis or gluconeogenesis. Thus α-glycerophosphate shuttle is tightly linked to glycolysis to regenerate cytosolic NADH while simultaneously sinking electrons into the ETC [26, 27]. G3P is also produced by the enzyme glycerol kinase from glycerol, the triose sugar backbone of triglycerides (TAG) and glycerophospholipids [28], to engage in thermogenesis, especially in brown adipose tissue. G3P can be synthesized from amino acids and citric acid cycle intermediates via the glyceroneogenesis pathway. G3P is a starting material for de novo TAG synthesis [29].
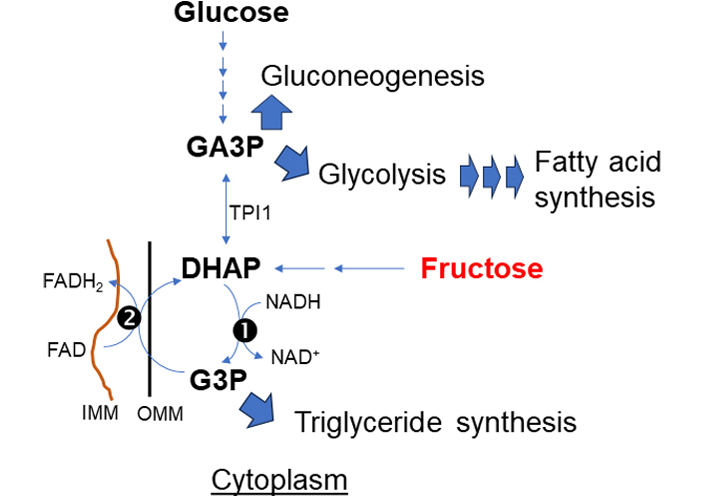
α-glycerophosphate shuttle. The primary action of this shuttle is to deliver electrons directly sink to the respiratory chain complex (II). This shuttle is at the crossroads of pathways for fructose, glucose metabolism, and synthesis of TAG and fatty acid for lipogenesis. (1) cytosolic and (2) mitochondrial glycerol-3-phosphate dehydrogenase: cGPD and mGPD enzyme (GPD1 and GPD2)
Malate in the cytosol is distributed into mitochondria through SLC25A1 (citrate-malate antiporter) in exchange for citrate in mitochondria. Cytoplasmic malate is replenished in two ways: (1) breakdown of citrate by ACLY to acetyl-CoA in the cytoplasm, and (2) exchange via dicarboxylate shuttles SLC25A10 (dicarboxylate-phosphate antiporter, Figure 5). SLC25A10 transports succinate and malate out of mitochondria in exchange for phosphate, sulfate, and other small molecules. Inhibition of SLC25A10 diminishes the citrate transport via SLC25A1 [30, 31]. In excess of mitochondrial acetyl-CoA, PC is activated but PDH is inhibited. PC catalyzes the ATP-dependent carboxylation of pyruvate to OAA which is then converted to malate by MDH. Malate is transported out of the mitochondria to the cytoplasm for gluconeogenesis.
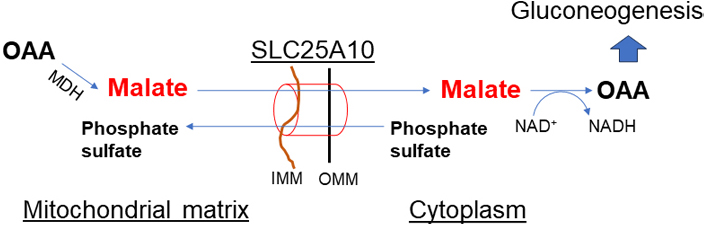
Dicarboxylate shuttle (SLC25A10). This shuttle is important in malate exchange out of mitochondria when pyruvate to OAA conversion is upregulated by activated PC in mitochondria due to increased mitochondrial acetyl-CoA. The activity of this shuttle has a key role in gluconeogenesis and TAG synthesis
Fatty acid β-oxidation (FAO) is the major pathway for the degradation of fatty acids and is essential for maintaining energy homeostasis. Free fatty acids (FFAs) are a crucial energy source when glucose supply is limited such as in the post-enteric phase and fasted states. Fatty acyl-CoA converted from FFA in the non-mitochondrial compartment is a substrate of β-oxidation in mitochondria. Based on length, FFA transfer to the mitochondria varies. SCFAs, fatty acids with aliphatic tails of fewer than 6 carbons are produced when bacteria in the intestines ferment fiber. The three main SCFAs are acetate (C2), propionate (C3), and butyrate (C4) [32–34]. Medium-chain fatty acids (MCFAs) are fatty acids with 6 to 12 carbons. MCFA includes caprylic acid (C8:0), capric acid (C10:0), and lauric acid (C12:0). Coconut oil, palm kernel oil, and bovine milk are rich in medium-chain TAGs [35, 36]. Long-chain fatty acids (LCFAs), fatty acids with 13 to 21 carbons, such as oleic, linoleic, palmitic, docosahexaenoic acid (DHA), and eicosapentaenoic acid (EPA) are transported into mitochondria for β-oxidation [37, 38]. Very LCFAs (VLCFAs) with 22 or more carbons such as peanuts are oxidized in peroxisomes [39].
Fatty acids taken up by hepatocytes and fatty acids accrued in the cytoplasm are converted to fatty acyl-CoA by acylation, catalyzed by acyl-CoA synthases (ACS). At this step acyl-CoA thioesterase hydrolyzes and deactivates fatty acyl-CoA generating FFA and CoA, thus limiting the availability of fatty acyl-CoA to transport to mitochondria [40]. Mitochondrial membranes are not permeable to LCFAs. Cytoplasmic long-chain fatty acyl-CoA is converted to fatty acyl-carnitine by carnitine acyl transferase [carnitine palmitoyl transferase-I (CPT-I)], located at the inner aspect of the OMM. CPT-I is sensitive to inhibition by malonyl-CoA, an intermediate of the TAG synthesis pathway. Malonyl-CoA level determines the switch between cytoplasmic fatty acid synthesis and mitochondrial β-oxidation [41]. Carnitine in mitochondria is converted by carnitine acetyltransferase (CrAT) to acetyl-carnitine which is transported back to the cytoplasm. The details of carnitine transport are available in previous reviews [42, 43]. MCFAs such as octanoic acid (caprylic acid, C8:0), enter the mitochondria by diffusion and rapidly undergo β-oxidation independently of the carnitine transport system [44–46]. SCFAs, such as acetate, enter mitochondria by diffusion and are converted by ACS short-chain family member 1 (ACSS1) to acetyl-CoA in mitochondria [47].
Fatty acid oxidation occurs in four stages—dehydrogenation, hydration, oxidation, and thiolysis. These four stages repeat until all the carbons in the fatty acid are turned into acetyl-CoA. For every cycle, the acyl-CoA unit is shortened by two carbon atoms. Concomitantly, one molecule of FADH2, NADH, and acetyl-CoA are formed. The purpose of fatty acid oxidation in mitochondria is to generate acetyl-CoA for the citric acid cycle, and FADH2 and NADH for the ETC and ATP synthesis. The ATP yield for one oxidation cycle is theoretically a maximum yield of 14, as one NADH produces 2.5 ATP, one FADH2 produces 1.5 ATP and a full rotation of acetyl-CoA in the citric acid cycle produces 9 ATP and 1 GTP [48].
Ketogenesis is the formation of ketone bodies such as β-hydroxybutyrate (β-HB), acetoacetate (AcAc), and acetone. Ketogenesis primarily occurs in liver mitochondria when there is excess mitochondrial acetyl-CoA from β-oxidation in fasting and starvation during which glucagon increases peripheral lipolysis. Mitochondrial acetoacetyl-CoA thiolase [acetyl-CoA acetyltransferase 1 (ACAT1)] condenses two acetyl-CoA to form acetoacetyl-CoA. Next, the third acetyl-CoA delivers acetyl-SH to acetoacetyl-CoA. The reaction is catalyzed by mitochondrial hydroxymethylglutaryl (HMG)-CoA synthase 2 (HMGCS2) to form HMG-CoA that is transformed into the AcAc and acetyl-CoA by HMG-CoA lyase (HMGCL). The majority of newly formed AcAc is then reduced to β-HB by NADH-dependent β-HB dehydrogenase (BDH). β-HB is the most abundant ketone body delivered from the liver into circulation as an energy source to the brain, heart, and skeletal muscles. A small fraction of AcAc is spontaneously decarboxylated into volatile acetone. Ketogenesis depends on the acetyl-CoA pool coming from FAO. Chronic starvation, cancer cachexia or diabetic ketoacidosis, and amino acids catabolism such as lysine, phenylalanine, tyrosine, tryptophan, isoleucine, and leucine provide acetyl-CoA for ketogenesis. The rate of ketogenesis decreases when the concentration ratios of acetyl-CoA to acetoacetyl-CoA and acetyl-CoA to free CoA-SH are low. HMGCS2 is the rate-limiting enzyme of the ketogenesis pathway [49].
AcAc and β-HB are transported across the cell membrane via monocarboxylate transporters 1 (MCT1) and MCT 2 (also known as SLC16A1 and SLC16A7) to extrahepatic tissues for terminal oxidation. The transporter across the IMM is not known [50–52]. AcAc in the cytoplasm is converted to AcAc-CoA by acetoacetyl-CoA synthetase (AACS) which enters cholesterol biosynthesis or is converted to acetyl-CoA for de novo lipogenesis [49, 53].
The majority of lipids in hepatocytes are generated in ER, but fatty acid (lipoic acid) synthesis [mitochondrial fatty-acid synthesis type II (mtFASII) cycle] occurs in mitochondria. Mitochondrial fatty acid elongation, a minor pathway, occurs by a reversal of β-oxidation. In the mitochondria, fatty acid (lipoic acid) is synthesized de novo using intermediates from the mtFASII cycle, s-adenosylmethionine, and iron-sulfur clusters [54].
In the mtFASII cycle, malonyl-CoA synthetase [acyl-CoA synthetase 3 (ACSF3)] links malonate to CoA, producing malonyl-CoA in mitochondria. The malonyl moiety from malonyl-CoA is transferred by malonyl-CoA-acyl carrier protein transacylase (MCAT) to an acyl carrier protein (ACP), resulting in malonyl-ACP. Thus, a major function of mitochondrial ACP (mtACP) is the biosynthesis of lipoic acid. The initial condensation reaction, malonyl-ACP condensation with acetyl-ACP or acyl-ACP, produces 3-ketoacyl-ACP. The 3-ketoacyl-ACP is reduced to 3-hydroxyacyl-ACP. Hydroxyacyl-thioester dehydratase type 2 (HTD2) then dehydrates 3-hydroxyacyl-ACP to trans-2,3-enoyl-ACP. Finally, mitochondrial trans-2-enoyl-CoA reductase (MECR) reduces trans-2,3-enoyl-ACP to acyl-ACP. Octanoyl-ACP is shuttled into the lipoic acid synthesis pathway [55, 56] in which lipoyltransferase 2 (LIPT2) transfers ocyanoyl moiety. Iron-sulfur enzyme lipoic acid synthetase (LIAS) catalyzes the final step in the de novo biosynthesis of lipoic acid. Lipoic acid is an essential cofactor for mitochondrial metabolism by lipoylation of enzymes such as PDH, α-KG dehydrogenase, and oxoacid dehydrogenase. LIPT1 targets protein lipoylation. Therefore, the knockdown of mtFASII components results in reduced cellular lipoic acid content and protein lipoylation levels [57]. Application of exogenous lipoate does not alleviate the effects of mtFASII knockdowns on protein lipoylation, indicating that a mitochondrial origin of fatty acids may be required for lipoylation to occur [58]. In addition, ETC activity is highly regulated by mtFAS function independent of protein lipoylation [59].
The mitochondrion is involved in the synthesis of amino acids, such as glutamine, glutamate, alanine, proline, and aspartate. The glutamine metabolites are distributed into macromolecules throughout the cell, including TCA cycle intermediates (important in bioenergetics), amino acids, nucleotides, glutathione, and lipids. In mitochondria, glutaminase (GLS) converts glutamine into glutamate and ammonia. Reciprocally, glutamine synthetase (GS) condenses glutamate and ammonia to make glutamine. Glutamate is converted into α-KG by transaminase or glutamate dehydrogenase (GDH) [60, 61]. When mitochondrial α-KG is low due to citrate being exported to cytosol for lipogenesis, activation of glutaminolysis converts glutamine to glutamate to replenish mitochondrial α-KG [62]. Glutamine crosses the plasma and mitochondrial membrane via glutamine transporters (SLC1A5) [63]. GS has been reported to have activity in cytosol and mitochondria in the liver [64].
The branched chain amino acids (BCAAs) leucine, isoleucine, and valine generate acetyl-CoA and succinyl-CoA as an alternative source of cellular energy especially in myocytes and adipocytes during exercise and fasting, respectively [65, 66]. Proline and ornithine metabolism are centered in mitochondria. The mechanisms underlying the compartmentalization of proteinogenic amino acids, such as proline and glutamate, are sparse [67].
Compared to other subcellular membranes, mitochondrial membranes, particularly IMM, are cholesterol-poor. Thus, cholesterol metabolism (oxysterol synthesis in hepatocytes, and steroidogenesis in the adrenal gland) is controlled through the delivery of cholesterol to the IMM [68]. Mitochondria receive cholesterol from late endosomes and lysosomes or from the ER membrane [69]. The transport of cholesterol from the OMM to the IMM occurs via non-vesicular transport by the steroidogenic acute regulatory protein 1 (StARD1, also known as StAR) [70]. The ER is the main organelle responsible for cholesterol synthesis [71]. Cholesterol is imported to mitochondrial membranes by multiprotein complexes that incorporate Tom40 acting at ER-mitochondria contact sites. In the absence of Tom40, the mitochondria-associated membrane (MAM) complex fails to assemble and mitochondrial cholesterol transport is ablated [72]. Cholesterol import to the mitochondria from the late endosomes and lysosomes is mediated by the interaction of metastatic lymph node-64 (MLN-64), Niemann-Pick disease type C1 (NPC1), and NPC2 [73]. Subsequently, IMM localized cytochrome P450 family 27 subfamily A member 1 (CYP27A1) converts cholesterol to oxysterols such as (25R)26-hydroxycholesterol and 3β-hydroxy-5-cholesten-(25R)26-oic acid. Oxysterols accumulate in mitochondria and are transferred to the cytoplasm to enter into bile acid synthesis (acidic pathway). Transcription of CYP27A1 is suppressed by bile acids but activated by hepatocyte nuclear factor 4α (HNF4α) [74]. Reactive adduct modification of CYP27A1 is identified [75], but this needs further characterization of enzyme activity. The responsible transporter of oxysterol is not known. In addition, autooxidation of cholesterol by free radicals and ROS generates many oxysterols [76, 77]. Oxysterol inhibits cholesterol synthesis by directly silencing the expression of two enzymes, CYP51A1 and squalene synthase, via the nuclear hormone receptor for oxysterols, the liver X receptor α (LXRα) [77, 78]. The role of oxysterol in cholesterol-associated steatohepatitis and cancer was recently reviewed [79].
Mitochondrial methylenetetrahydrofolate dehydrogenase 2 (MTHFD2) is among the major contributors of NADPH [80, 81]. The cytosolic isoform favors NADP+ production [82]. The MTHFD isoforms are involved in adaptive response to survive oxidative stress. Upon ETC inhibition, the mitochondrial MTHFD2 is activated for NADPH/NADP+ balance [83]. NADPH is required for the reduction of glutathione, an antioxidant. The co-factor tetrahydrofolate (THF) is the carrier for de novo nucleotide (purine) synthesis. Activated THF molecules are generated through an oxidative/reductive cycle that catabolizes serine to generate glycine in the mitochondria and synthesizes serine in the cytosol [84, 85]. Thus, the pathway involves de novo nucleotide synthesis, amino acids, and redox homeostasis. Mitochondrial MTHFD2 converts 5,10-methylene-THF to 10-formyl-THF which is then hydrolyzed to formate, the main carbon source for cytosolic 1C metabolism. MTHFD2 expression is regulated by the mammalian target of rapamycin complex 1 (mTORC1) and is critical for growth and proliferation [86].
Gluconeogenesis is a predominantly cytosolic process occurring when liver glycogen stores start to deplete during fasting for more than 8 h. However, the initiating step by PC occurs inside the mitochondria. PC converts pyruvate to OAA which is in turn converted to malate by MDH. Malate is exported from the mitochondria for the remaining steps of gluconeogenesis. The mechanism driving the export of malate for gluconeogenesis is unknown. It is unclear if the pathway is activated in conditions such as nutrient deprivation or hypoxia. However, in fatty liver, impaired ketogenesis, inefficient respiratory chain, and increased acetyl-CoA oxidation promote hyperglycemia [9, 10]. In the cytosol, malate is converted to OAA which is converted by phosphoenolpyruvate (PEP) carboxykinase (PCK) to PEP for gluconeogenesis (Figure 5). The mitochondrial isoform of this enzyme, PCK2, is not involved in gluconeogenesis. A comprehensive review of gluconeogenesis was recently updated [87–90].
Dietary fat is distributed mainly to adipose tissue and muscle. In the liver, de novo lipogenesis is a major contributor to hepatic TAG accumulation. Dietary carbohydrates and FFA redistributed from adipose tissue lipolysis are the primary sources of de novo lipogenesis in the liver, contributing to 1–3% of the total fat balance in humans consuming a typical diet. Although lipogenesis occurs in the cytoplasm (ER), mitochondria are the primary source of acetyl-CoA, the building block for lipogenesis in the liver. Acetyl-CoA is not transported across mitochondrial membranes. However, mitochondrial citrate is transported to the cytoplasm and converted by cytosolic ACLY to acetyl-CoA for fatty acid synthesis in the cytoplasm.
The liver is a key organ in cholesterol homeostasis. The increased lipogenesis in the liver generates increased cholesterol synthesis which occurs in the cytoplasm [91]. Mitochondrial-derived acetyl-CoA is essential for the synthesis of cholesterol and bile acids in the liver. Bile acid synthesis is a major catabolic pathway of hepatic cholesterol. Under physiological conditions, the bile acid synthesis occurs via two main pathways. In humans, the neutral (or classical) pathway accounts for more than 90% of bile acid production, whereas the acidic (or alternative) pathway accounts for a smaller portion. The cholesterol 7α-hydroxylase (CYP7A1), localized in the ER of hepatocytes, is the rate-limiting enzyme. It hydroxylates the carbon-hydrogen bond of cholesterol at the 7-alpha position. CYP7A1 gene expression is negatively regulated by bile acids returning to the liver via enterohepatic circulation. In the alternative pathway, StARD1, a mitochondrial cholesterol transporter, and CYP27A1 limit mitochondrial oxysterol synthesis. CYP7B1 in cytoplasm is the rate-limiting enzyme in oxysterols to bile acid synthesis. Oxysterols can be excreted after sulfation or glucuronidation [92]. Oxysterol, on the other hand, activates LXRs, and synthetic LXR agonists increase the transcription of sterol regulatory element binding protein 1c (SREBP1c) and its lipogenic genes [93]. Update on hormonal and circadian regulation of bile acid homeostasis have been recently updated [94].
Mitochondrial metabolism adapts to the signals of metabolic and stress sensors at post-translational or transcriptional levels in response to conditions such as changes in the ratio of NADH/NAD+ or adenosine monophosphate (AMP)/ATP, Ca2+ influx from ER to mitochondria, redox balance, and changes in the level of amino acids, succinyl-CoA, acetyl-CoA, and methyl donors. The broader and conceptual review of metabolic sensors has been described [95]. This review focuses on signal activations that regulate mitochondrial metabolism and that are closely associated with mitochondria.
Both c-Jun N-terminal kinase (JNK) and p38 stress-activated kinases are activated by mitogen-activated protein (MAP) kinase (MAPK) cascade [MAPK kinase kinase (MAP3K)→MAPK kinase (MAP2K)→MAPK] in response to various chemical, environmental, and biological stress including metabolic stress. Hepatic JNK is activated by meal but activated phosphorylated-JNK (P-JNK) decreases shortly after meal (after enteric emptying). However, p38 is phospho-activated in fasting [96, 97]. Both kinases are predominantly localized in the cytoplasm. The activated P-JNK, but not phospho-p38 (P-p38), translocates and binds with OMM protein SH3 domain-binding protein 5 (SAB/SH3BP5) releasing tyrosine-protein phosphatase non-receptor type 6 (PTPN6) from SAB to the intermembrane space. PTPN6 inactivates phospho-Src (P-Src, active form) which is required to maintain mitochondrial respiration. Inactivation of Src by PTPN6 on Src-binding protein docking protein 4 (DOK4) occurs on the IMM leading to decreased ETC activity and increased production of ROS. ROS then activates MAPK cascade—MAP3K such as apoptosis signal-regulating kinase 1 (ASK1) which activates MAP2K such as MAP2K 4 (MKK4) which then activates JNK. Thus, the feed-forward activation pathway is described as the JNK-SAB-ROS activation loop [98–102]. Therefore, JNK and SAB interaction sustains the P-JNK activation followed by decreased ATP synthesis (decreased oligomycin-inhibitable respiration) and may result in an incremental increase of NADH leading to allosteric inhibition of enzymes in the TCA cycle such as PDHC, CS, IDHC, OGDHC [6]. Initial post-prandial JNK activation is suggested through cell membrane-associated Src and mixed lineage kinases (MLKs) activation [103]. Continue JNK activation (sustain P-JNK) through P-JNK-SAB mediated modulation of mitochondrial metabolism is suggested because the hepatocyte-specific deletion of SAB before the hepatic accumulation of TAG or in established nonalcoholic steatohepatitis (NASH) abolishes P-JNK and decreases hepatic lipogenic genes expression, hepatic steatosis, inflammation, fibrosis, and improved metabolic intolerance and energy expenditure [104]. Moreover, SAB peptide or JNK inhibitor prevents palmitic acid-induced P-JNK interaction with SAB and inhibits mitochondrial bioenergetics, implying the P-JNK effect on mitochondrial metabolism [104]. However, further exploration is required to uncover the direct and indirect mechanism of modulation of mitochondrial metabolism when P-JNK interacts with SAB, especially during nutrition excess.
AMP-activated protein kinase (AMPK) primarily resides in the cytoplasm. Mitochondrial localization of AMPK has been reported and supported by the presence of two AMPK substrates at the OMM, mitochondrial fission factor (MFF) and acetyl-CoA carboxylase 2 (ACC2) [105, 106]. AMPK is the key sensor of coordinated upregulation of the enzymes of β-oxidation, ketogenesis, gluconeogenesis, and peripheral lipolysis in fasting and starvation [107].
AMPK is inhibited by overnutrition, excess amino acids (high protein diets) especially leucine, inflammation, and hypersecretion of certain anabolic hormones, such as insulin, during obesity. Under energy (ATP)-shortage conditions lipid and sterol synthesis is inhibited by activated AMPK through inhibitory phosphorylation of ACCs, which catalyzes the first step in de novo lipid synthesis, and inhibitory phosphorylation of HMG-CoA reductase (HMGCR), which catalyzes the rate-limiting step in cholesterol synthesis. AMPK inhibition of ACCs, especially ACC2 on mitochondria decreases malonyl-CoA inhibition of long-chain fatty acyl-CoA transporter mitochondrial CPT-I and thus enhances mitochondrial β-oxidation [108, 109]. AMPK is a dominant negative regulator of mTORC1. AMPK inhibition of mTORC1 in the liver activates ketogenesis in fasting. mTORC1 inhibition removes the nuclear receptor co-repressor 1 (NCoR1), and recruits transcriptional coactivators and histone acetyltransferases to the peroxisome proliferator-activated receptor α (PPARα)-regulated genes for oxidation and ketogenesis [110, 111]. On the other hand, mTORC1 activates SREBP and lipogenesis [112]. In the liver, PPARα strongly upregulates fibroblast growth factor 21 (FGF21) which activates hepatic lipolysis and ketogenesis in hepatocytes. In addition, under low ATP conditions, cyclic AMP (cAMP)-activated AMPK phosphorylates MFF and unc-51 like autophagy activating kinase 1 (ULK1) to facilitate the segregation of damaged mitochondria and their removal by mitophagy [113]. Moreover, AMPK activation of insulin induced gene 1 (INSIG) inhibits the cleavage of SREBP1c and reduces lipogenic gene expression and lipid accumulation [114]. Re-activation of AMPK suppresses hepatic steatosis but its downregulation does not promote fatty liver development [115]. Overall, AMPK activity is a keynote in the regulation of fatty acid transport to mitochondria and gene expression of mitochondria β-oxidation and ketogenesis.
Two-thirds of mitochondrial proteins contain lysine acetylation sites. Mitochondrial acetylation is thought to occur through the chemical reaction between lysine residues and acetyl-CoA. However, a definitive mitochondrial acetyltransferase has not been identified [116]. Overall, the acetylation of mitochondrial enzymes causes inhibition of enzyme activity, and thus oxidative metabolism is inhibited [117]. NAD+ dependent mitochondrial deacetylase SIRT3 is the principal enzyme for deacetylation, and SIRT4 and 5 contribute to the reversal of succinylation [49, 118, 119]. SIRT3 deacetylation activates PDHC subunits in pyruvate metabolism. Acetylation and succinylation of HMGCS2 inhibit the enzyme activity and ketogenesis in mitochondria. During fasting, SIRT3 removes acetyl groups to activate enzymes involved in fatty acid oxidation, such as long-chain acyl-CoA dehydrogenase (LCAD) [120], which acts to activate ketogenesis in the liver. Therefore, mitochondrial SIRTs promote fatty acid oxidation, glucose oxidation, and ketone body production [119].
Cytoplasmic cAMP produced by soluble adenylyl cyclase (sAC) in response to starvation activates protein kinase A (PKA) on mitochondria resulting in mitochondrial elongation because PKA phosphorylation of dynamin-related protein 1 (DRP1) at S637 inhibits mitochondria fission [121]. The mechanism is mediated by PKA binding to mitochondrial A-kinase anchoring protein 1 (AKAP1) localized at the outer leaflet of the IMM. PKA is also identified in the mitochondrial matrix. However, cytoplasmic cAMP does not have access to matrix PKA. Intriguingly, the CO2/HCO3–-mito-sAC-cAMP-mito-PKA signaling cascade was identified as wholly contained in mitochondria and the cascade is completely compartmentalized [122]. CO2/HCO3–-inducible mito-sAC increases intramitochondrial cAMP leading to activation of mito-PKA which phospho-activates some of the subunits of cytochrome c (CYCS) oxidase subunit IV (COXIV) and increases bioenergetics. Therefore, mito-PKA synchronizes mitochondrial tricarboxylic metabolism and bioenergetics, and hepatic expression of PKA-inhibiting peptide in the liver accelerates TAG accumulation [121–126]. Further studies will be intriguing to uncover the mito-PKA targets of mitochondrial metabolism.
Liver mitochondrial metabolism switches between the TCA cycle, β-oxidation, and ketogenesis in compliance with mitochondrial coenzymes and metabolites, signals from metabolic sensors, substrates availability, and hepatic and whole-body metabolic needs which vary diurnally or with stress such as exercise, infection, temperature, and hepatotoxins.
Mitochondrial metabolic pathways are tightly regulated by intermediate metabolites, substrates, and coenzymes such as acetyl-CoA, pyruvate, fatty acyl-CoA, amino acid, NAD+/NADH, and ADP/ATP. Mitochondria coordinately synchronize with non-mitochondrial metabolic pathways through activated JNK, AMPK, and PKA. Mitochondrial utilization of metabolites varies depending on the type of diet consumed (e.g., HFD), and the time between meals (e.g., fasting or starvation). Mitochondria metabolizes one molecule of pyruvate with preexisting one molecule of OAA to produce one molecule of citrate which enters the TCA cycle and produces 3 NADH and 1 FADH2 which generate 22 ATP in coupling with the ETC and ATP synthesis. However, the entry molecule to the TCA cycle could be malate from cytoplasm via malate-OAA or citrate-malate shuttle, succinate from cytoplasm via malate-succinate shuttle, α-KG from the conversion of cytoplasmic glutamate. During high-fat and high-carbohydrate diet consumption, excess intermediate metabolites such as acetyl-CoA are produced in mitochondria because of upregulated β-oxidation and glycolysis. Mitochondrial acetyl-CoA is converted to citrate by the first reaction of TCA cycle and then exported into the cytoplasm via citrate transport protein and stored in the cytoplasm as TAG via lipogenesis [127]. This process results in a leak in the TCA cycle and could cause insufficient conversion of α-KG to OAA. Mitochondrial OAA is replenished by carboxylating pyruvate into OAA by PC, which is activated by high mitochondrial acetyl-CoA during hepatic lipogenesis [11, 128]. In addition, cytosolic malate could be transported into mitochondria and converted to OAA [129]. High expression and activity of PC leads to gluconeogenesis and has been associated with metabolic reprogramming and increased tumor progression [130].
A depletion of OAA relative to acetyl-CoA is the key step for the production of ketone bodies, acetoacetyl-CoA, and β-HB, within the liver when replenishing of OAA is not available such as low/no carbohydrate HFD [131]. Excess amounts of acetyl-CoA converted from fatty acyl-CoA inhibit pyruvate metabolism via inhibition of PDH but increase ketone bodies [132]. Mitochondria contain ACOTs, similar to cytoplasmic and peroxisomal ACOTs (12 and 8), which convert excess acetyl-CoA to acetate, a SCFA. Acetate crosses the mitochondrial membrane to cytoplasm where it is converted to acetyl-CoA by ACSS2 for fatty acid synthesis and cholesterol synthesis [133]. During nutrient excess: A high carbohydrate diet stimulates adipose tissue lipogenesis via insulin and SREBP-targeted adipogenic gene expression [134]. Postprandial dietary fat from the gut deposits in adipose tissue and muscle. High fructose diet consumption facilitates hepatic de novo lipogenesis. During fasting and starvation: When liver glycogen storage is depleted in fasting or starvation, FFA delivered to the liver from adipose and muscle by lipolysis is the source of metabolic substrates for gluconeogenesis, β-oxidation, ketone, and amino acid synthesis [90].
Mitochondria membrane lipids play a vital role in mitochondrial biogenesis, bioenergetics, and metabolism, in addition to protein import and assembly, membrane architecture, and mitochondrial dynamics [135]. The liver mitochondria mainly consist of 34–55% phosphatidylcholine, 19–36% phosphatidylethanolamine (PE), 12–23% cardiolipin, and small fractions of phosphatidylinositol, phosphatidylserine, lysophospholipids, sphingomyelin, and phosphatidic acid [136, 137]. The OMM is rich in phosphatidylcholine and PE [138]. PE is highly enriched in inner membranes [139]. Cardiolipin, an important phospholipid in the IMM, has four fatty acids rather than two and makes the membrane especially impermeable to ions. Cardiolipin is synthesized via condensation of phosphatidylglycerol (PG) and cytidine diphosphate (CDP)-diacylglycerol catalyzed by cardiolipin synthase 1 (CRLS1) in the matrix side of the IMM [137]. The majority of mitochondrial lipids are synthesized in the ER and transported to the mitochondria, but the synthesis of cardiolipin and PE in the IMM highlights the importance of mitochondrial lipid metabolism [140, 141].
The lipid compositions of mitochondrial membrane change during liver metabolic stress such as in HFD-fed mice, diabetes mice, and progression of NASH in humans. Cardiolipin is important in the efficient coupling of the respiratory chain and ATP synthase [142]. Deletion of CRLS1 (decreased cardiolipin) or lysophosphatidylglycerol acyltransferase 1 (LPGAT1), an acyltransferase that catalyzes the remodeling of PG, exacerbates the hepatic steatosis in diet-induced NASH model [143, 144]. Cardiolipin has a methylene bridge between two double bonds of fatty acids. Thus, cardiolipin is highly sensitive to ROS-induced oxidative damage [145]. In fact, cardiolipin level decreases in established hepatic steatosis [143], but, increases in streptozotocin (STZ)-induced diabetes models, suggesting the compensatory adaptation at preserving mitochondrial bioenergetic function [146–148]. In addition, cardiolipin changes facilitate the initiation and completion of apoptosis. Flipping of cardiolipin from the IMM to the OMM is a hallmark of upcoming CYCS leakage and apoptotic cell death. Flipping facilitates the contact between cytoplasmic caspase 8 and BH3 interacting domain death agonist (Bid) to form a truncated Bid (tBid). Besides, loss of cardiolipin from the IMM confers complete disorganization of mitochondrial respiratory complexes. Cardiolipin also regulates mitochondrial fission through positive interaction with the DRP1 the major effector of mitochondrial division. Other lipids best described in apoptosis have been reviewed recently [138, 149].
Compared to other subcellular membranes, the IMM is cholesterol-poor [68]. Mitochondria require cholesterol for biogenesis and membrane fluidity and for the biosynthesis of steroids, oxysterols, and bile acids. Cholesterol is dynamically transported to mitochondria primarily by non-vesicular mechanisms. Sterol transfer proteins (STPs) bind cholesterol in their hydrophobic pockets to transfer across the aqueous cytosol [150, 151]. Cholesterol is situated in the membrane in between the phospholipids. The rigid rings of the cholesterol molecule interact with the hydrophobic tails of the phospholipids. This stabilizes them and makes them more rigid as well. This also prevents the membrane from being permeable to water and water-soluble small molecules [152, 153]. Thus, the fluidity of the mitochondrial membrane is determined by its cholesterol content. The higher the cholesterol content in membranes, the lower its fluidity, and vice versa [154]. At high temperatures, cholesterol stabilizes the membrane by raising the melting point to keep the same degree of fluidity. At low temperatures, cholesterol prevents clustering by intercalating between the phospholipid bilayer [155]. Mitochondrial cholesterol levels are elevated at low temperatures [156]. Cholesterol increases bilayer thickness, presumably due to the ordering of the acyl chains of phospholipids [70, 157].
The liver adapts to whole-body metabolic needs through the hormonal and neurogenic regulation of liver mitochondrial metabolism. Insulin increases hepatic glycolysis and lipogenesis but suppresses gluconeogenesis. Glucagon counteracts insulin action [90]. Insulin directly stimulates mitochondrial glucose oxidation without modulating glucose uptake or glycolysis, through activating mitochondrial PDH, the rate-limiting enzyme of pyruvate metabolism in mitochondria [158]. Insulin stimulates lipogenesis by activating glucose import, regulating the levels of G3P and lipoprotein lipase (LPL) [134, 159]. Insulin markedly reduces the activity of AMPK both by increasing glucose uptake and oxidation and by protein kinase B (Akt)-mediated phosphorylation of AMPK (Ser485/491) leading to protein phosphatase 2A (PP2A) mediated dephosphorylation at Thr172 and deactivation of AMPK [160]. Metabolic dysfunction-associated steatohepatitis (MASH) is associated with insulin resistance. Insulin resistance is suggested to promote the progression from simple fatty liver to MASH because insulin stimulates lipogenic enzymes via SREBP1c [161–163], but it needs further evidence to explore the contribution of insulin resistance mechanism in MASH progression.
The thyroid hormone [triiodothyronine (T3)] is the major endocrine regulator in a hypermetabolic state. T3 also has profound impacts on mitochondrial function and turnover by regulating processes such as mitogenesis, proton leak, oxidative phosphorylation (OXPHOS), ROS generation [164, 165], autophagy, and β-oxidation [166, 167]. Thyroid hormones stimulate mitochondrial replication and energy production by switching metabolism from the glycolytic pathway to more efficient OXPHOS [168] and by regulating NADH shuttles in the liver and cardiac mitochondria [169]. Thyroid hormones regulate serum levels of cholesterol by stimulating cholesterol biosynthesis through inducing HMGCR, cholesterol export [primarily as low-density lipoprotein (LDL) and very LDL (VLDL)], reverse transport from peripheral tissues, hepatic reuptake via LDL receptors (LDLRs), and cholesterol conversion into bile acids in the liver [170, 171]. The past and updated reviews are recommended for the direct and indirect effects of thyroid hormone on mitochondria [172–177].
The sympathetic nervous system stimulates hepatic gluconeogenesis, whereas the parasympathetic system does not. Catecholamines, particularly norepinephrine, are the primary activators of fasting-induced peripheral (adipose) lipolysis, while cortisol, glucagon, growth hormone (GH), and adrenocorticotropic hormone (ACTH) also activate lipolysis [178]. Dietary compounds, such as caffeine and calcium, also stimulate lipolysis. Leptin inhibits lipogenesis increases lipolysis, and thus reduces fat pad size [179]. In the liver, leptin stimulates fatty acid oxidation and glucose uptake. Thus, leptin-mediated increases in energy expenditure and basal metabolic rate contribute to the improvement in steatosis [180].
Among different forms of liver metabolic stress dietary, alcohol, and genetic causes are briefly discussed here.
Mitochondrial metabolites, such as acetyl-CoA, succinyl-CoA, acetate, NAD+/NADH, and S-adenosyl-L-methionine (SAM) which are capable of directly affecting the posttranslational and epigenome regulation, switch according to nutrients and whole-body metabolic response through hormone and neurogenic signals. Consumption of very low amounts of carbohydrates with an adequate protein content diet (ketogenic diets, fasting) will force the liver to produce ketone bodies. Excessive production of acetyl-CoA by β-oxidation and/or depletion of oxalacetate are both capable of triggering liver ketogenesis. However, the rate of ketogenesis is not linearly dependent on hepatic acetyl-CoA concentration [181, 182]. The amount of acetyl-CoA available can modulate acetylation-mediated post-translational modifications such as histone, transcription factors, and kinases, resulting in gene activation for de novo lipogenesis and consequential transcription of pro-growth genes [183]. Most of the dietary fat (chylomicron) is deposited in adipose tissue and FFAs are redistributed to the liver and further metabolized in mitochondria to acetyl-CoA which are building blocks for hepatic de novo lipogenesis. Intriguingly, dietary sugars, fructose versus glucose have opposing effects on HFD-induced metabolic changes. Fructose supplementation increases fatty acid synthesis mediated via upregulation of SREBP1c and carbohydrate response element binding protein (ChREBP) target genes, while glucose supplementation upregulates ChREBP target genes [184, 185]. Dietary sugar (glucose, fructose) supplementation in the chow diet causes hyperacetylation of mitochondrial proteins including acyl-CoA dehydrogenase, long chain (ACADL) and hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex subunit A/B (HADHA/B), thereby inhibiting β-oxidation [186]. Mitochondrial protein acetylation is increased in chronic HFD feeding (13 weeks) but not in acute HFD feeding (1 week). Chronic HFD feeding reduces ACADL enzyme activity by increased acetylation which is enhanced in SIRT3 knockout. However, the direct role of mitochondrial protein acetylation in de novo lipogenesis is difficult to reconcile in SIRT3 knockout or HFD-fed mice because of the absence or loss of SIRT3 leading to aberrant upregulation of stearoyl-CoA desaturase 1 (SCD1) [187]. Notably, fructose intake triggers de novo lipogenesis independent of ACLY-mediated citrate to acetyl-CoA metabolism. Dietary fructose is converted by the gut microbiota to acetate which is taken up by the liver and irreversibly catalyzed to acetyl-CoA by the hepatic cytosolic enzyme ACSS2 [188]. Fructose in hepatocytes is converted to fructose-1-phosphate which is converted by aldolase B to DHAP and then G3P, a backbone of TAG synthesis. GA3P derived from fructose metabolism eventually gives rise to citrate as a result of oxidative stress-induced inhibition of aconitase [189]. Thus, dietary fructose-derived citrate provides substrates for fatty acid synthesis [190–193].
Cholesterol content in the diet increases mitochondrial oxysterol production which is highly dependent on CYP27A1 expression activated by HNF4α [74]. However, dietary intake and endogenous enzymatic synthesis of oxysterols account slightly for the increased oxysterol levels, because the nonenzymatic cholesterol oxidation pathway is responsible for the changes in oxysterol metabolism [194]. Furthermore, HFD induces cytoplasmic retention of HNF4α in hepatocytes [195], and HNF4α prevention of hepatic TAG accumulation could be due to other actions of HNF4α such as promoting hepatic TAG lipolysis, fatty acid oxidation, and VLDL secretion [196].
Alcohol (ethanol) is primarily oxidized in the liver by alcohol dehydrogenase with NAD+ and by CYP2E1 to acetaldehyde and NADH in the cytosol. Alcohol dehydrogenase-derived NADH is transported into the mitochondria by substrate shuttles. Acetaldehyde is oxidized to acetate by NAD+ dependent acetaldehyde dehydrogenase generating NADH in mitochondria. Long-term alcohol consumption causes mitochondrial enlargement (mega-mitochondria) and structural changes and impairs mitochondrial functional respiration. Mitochondrial morphology is regulated by mitochondrial fission proteins such as DRP1, and MFF and fusion proteins such as mitofusin-1 (MFN1), and MFN2. Intriguingly, alcohol decreases DRP1 expression by impairing transcription factor EB activity. Mega-mitochondria formation is accelerated in alcohol-fed liver-specific DRP1 knockout mice because of inefficient removal of damaged mitochondria by mitochondrial fission followed by mitophagy. Importantly, the results implied that mega-mitochondria is an adaptive response to increased mitochondrial metabolism in alcohol consumption, but the defect in the removal of damaged mitochondria due to decreased expression of DRP1 in alcohol consumption contributes to alcohol-induced liver injury [197]. In chronic alcohol consumption, inefficient mitochondrial electron transfer reactions and high NADH lead to increased production of mitochondrial ROS when mitochondrial glutathione is reduced, or mitochondrial DNA and translation are defective. Then, impaired mitochondrial metabolism and increased lipid oxidation lead to hepatic steatosis [2, 198–201]. Studies showed that alcohol causes mitochondrial membrane depolarization in nearly all hepatocytes in the liver as early as 1 h and peaks at 6–12 h [202, 203]. Furthermore, alcohol induces StARD1 expression in perivenous hepatocytes and increases mitochondrial cholesterol accumulation and loss of mitochondrial ultrastructure architecture [204]. Acetate, the alcohol metabolite, is converted by ACSS1 in mitochondria and ACSS2 in the cytosol to acetyl-CoA, directly supplementing substrates for fatty acid synthesis. In addition, chronic alcohol consumption shifts the substrate of oxidation from carbohydrates to lipids and increases acetyl-CoA availability because of increased adipose tissue lipolysis [205]. Acetyl-CoA and acetylation reprogram circadian metabolism and hepatic metabolic enzymes via SREBP-driven transcription [206]. Multiple pathways of SREBP activation by alcohol, including induction of SREBP1c maturation by acetaldehyde, downregulation of AMPK and SIRT1, and signal transducer and activator of transcription 3 (STAT3) suppression of SREBP1c expression have been described [207, 208].
Mitochondrial FAO disorders (FAODs) are a heterogeneous group of defects in fatty acid transport and mitochondrial β-oxidation [48]. FAODs are inherited as autosomal recessive disorders [209] approximately one in every 5,000 to 10,000 live births. The most common FAOD is medium-chain acyl-CoA dehydrogenase deficiency (MCADD) causing accumulation of MCFAs in the blood which can lead to a metabolic crisis with hypoglycemia and lethargy. Without rapid treatment, this can result in liver damage, brain damage, and death [210, 211].
The role of mitochondria in metabolism has been expanding ever since the discovery of the TCA cycle. The integration of several metabolic pathways, the presence of shuttles, and signal transduction from cytoplasmic sensors constantly determine the flow of mitochondrial metabolites which are continuously changing with influx and efflux of intermediary metabolites. Among metabolites, mitochondrial acetyl-CoA level plays a key role in lipogenesis and cholesterol biosynthesis in the liver via providing substrate for lipogenesis and modulating enzyme activity, epigenetic, and transcriptional activity. The direct action of stress kinases on mitochondrial metabolic flow needs further exploration. The critical role of mitochondria in lipogenesis and cholesterol biosynthesis is conceptualized in Figure 6. This review provides an overview of the regulation of metabolic flow in the progression of steatotic liver disease.
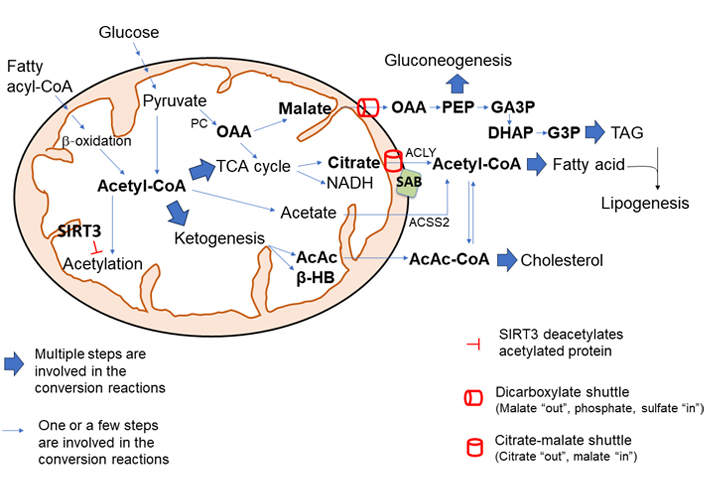
The central role of mitochondria in lipogenesis and cholesterol synthesis. The level of mitochondrial acetyl-CoA plays a pivotal role in mitochondrial metabolic shift. High mitochondrial acetyl-CoA upregulates the synthesis of citrate which is exported out of mitochondria into the cytosol via citrate-malate shuttle (SLC25A1) and conversion by ACLY to acetyl-CoA, which is the building block for fatty acid synthesis and cholesterol synthesis. Excess mitochondrial malate, generated by activation of PC by acetyl-CoA, is exported out of mitochondria via dicarboxylate shuttle (SLC25A10) and provides intermediate metabolites for gluconeogenesis and TAG synthesis. Ketogenic activity is largely activated by high levels of acetyl-CoA in mitochondria. AcAc, a ketone body, is condensed to acetoacetyl-CoA which provides substrates for cholesterol and fatty acid synthesis. Overall, the acetylation of mitochondrial enzyme inhibits its activity, and deacetylation by intramitochondrial deacetylase SIRT3 increases mitochondrial metabolism and bioenergetics. Moreover, JNK interaction with OMM protein SAB could make two major changes in mitochondrial metabolism: JNK interaction with SAB inhibits mitochondrial respiration and increases NADH/NAD+ and ROS production; the interaction increases the SAB and CS (and possibly SLC25A1) association. Therefore, hyper nutrition diet along with increased mitochondrial acetyl-CoA, JNK activation, and interaction with SAB could enhance the citric acid synthesis and could export citric acid out of the mitochondria into cytosol for de novo fatty acid synthesis and cholesterol biosynthesis
AcAc: acetoacetate
ACC2: acetyl-CoA carboxylase 2
ACLY: ATP-citrate lyase
ACO: aconitase
ACOT: acyl-CoA thioesterases
ACP: acyl carrier protein
ACSS1: acyl-CoA synthases short-chain family member 1
AMP: adenosine monophosphate
AMPK: adenosine monophosphate-activated protein kinase
cAMP: cyclic adenosine monophosphate
cGPD: cytosolic glycerol-3-phosphate dehydrogenase
CPT-I: carnitine palmitoyl transferase-I
CS: citrate synthase
CYP27A1: cytochrome P450 family 27 subfamily A member 1
DHAP: dihydroxyacetone phosphate
DRP1: dynamin-related protein 1
ER: endoplasmic reticulum
ETC: electron transport chain
FAD: flavin adenine dinucleotide
FADH2: flavin adenine dinucleotide (reduced)
FAO: fatty acid β-oxidation
FAODs: fatty acid β-oxidation disorders
FFAs: free fatty acids
G3P: glycerol-3-phosphate
GA3P: glyceraldehyde-3-phosphate
GOTs: glutamate-oxaloacetate transaminases
HFD: high-fat diet
HMG: hydroxymethylglutaryl
HMGCS2: hydroxymethylglutaryl-CoA synthase 2
HNF4α: hepatocyte nuclear factor 4α
IDH: isocitrate dehydrogenase
IDHC: isocitrate dehydrogenase complex
IMM: inner mitochondrial membrane
JNK: c-Jun N-terminal kinase
LCFAs: long-chain fatty acids
LDL: low-density lipoprotein
LXRα: liver X receptor α
MAP2K: mitogen-activated protein kinase kinase
MAPK: mitogen-activated protein kinase
MASH: metabolic dysfunction-associated steatohepatitis
MCFAs: medium-chain fatty acids
MDH: malate dehydrogenase
MFF: mitochondrial fission factor
mGPD: mitochondrial glycerol-3-phosphate dehydrogenase
mtFASII: mitochondrial fatty-acid synthesis type II
MTHFD2: methylenetetrahydrofolate dehydrogenase 2
mTORC1: mammalian target of rapamycin complex 1
NASH: nonalcoholic steatohepatitis
OAA: oxaloacetate
OGDH: oxoglutarate dehydrogenase
OGDHC: oxoglutarate dehydrogenase complex
OMM: outer mitochondrial membrane
PC: pyruvate carboxylase
PDH: pyruvate dehydrogenase
PDHC: pyruvate dehydrogenase complex
PE: phosphatidylethanolamine
PEP: phosphoenolpyruvate
P-JNK: phosphorylated-c-Jun N-terminal kinase
PKA: protein kinase A
PTPN6: tyrosine-protein phosphatase non-receptor type 6
ROS: reactive oxygen species
SAB/SH3BP5: SH3 domain-binding protein 5
sAC: soluble adenylyl cyclase
SCFAs: short-chain fatty acids
SDH: succinate dehydrogenase
SIRTs: sirtuins
SLC25A1: solute carrier family 25 member A1
SREBP1c: sterol regulatory element binding protein 1c
StARD1/StAR: steroidogenic acute regulatory protein 1
TAG: triglyceride
TCA: tricarboxylic acid
THF: tetrahydrofolate
α-KG: α-ketoglutarate
β-HB: β-hydroxybutyrate
Sanda W: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Supervision. TAT: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Validation. NK: Writing—review & editing, Validation. NW, AA, ZTW, SHW, EHP, CK, JS, SA, PPD, HWL, Sean W, SK, Samuel W, KKS, and GK: Resources, Visualization. FWMA: Resources, Visualization, Writing—review & editing, Validation. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Daniel L. Pouliquen
Vicent Ribas
Laura Fàbrega ... Carmen Garcia-Ruiz
