 Review
Review

Affiliation:
Department of Biochemistry, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry 605006, India
Affiliation:
Department of Biochemistry, Jawaharlal Institute of Postgraduate Medical Education and Research (JIPMER), Puducherry 605006, India
Email: balasubramaniyan.v@jipmer.edu.in
ORCID: https://orcid.org/0000-0003-1708-4864
Explor Dig Dis. 2024;3:163–174 DOI: https://doi.org/10.37349/edd.2024.00045
Received: August 23, 2023 Accepted: February 20, 2024 Published: May 11, 2024
Academic Editor: Chiara Raggi, University of Florence, Italy
The article belongs to the special issue Fibrosis and Hepatobiliary Cancer
The most common type of liver cancer is hepatocellular carcinoma (HCC) causes a lower survival rate even after systemic treatment. Previous studies have shown evidence that various molecular and epigenetic mechanisms are involved in the transition of HCC from normal liver cells. Epigenetics plays an important role in maintaining genomic stability in normal liver cells. Apart from the mutation of genes, epigenetic factors are involved in HCC progression. miRNA tends to be a major epigenetic factor involved in regulating major cell cycle pathways. miRNA regulates the HCC progression by inhibiting the major apoptotic pathways and favors angiogenesis and tumor microenvironment. Apart from regulating major pathways, miRNA appears to regulate tight junction integrity. Tight junction proteins appear to be strong barrier proteins involved in cell adhesion and integrity. Disturbance in cell adhesion and integrity leads to the major dysregulation of cell cycle resulting in cancer progression. Moreover, dysregulation of tight junction integrity was observed in the pathogenesis of HCC. The regulation of tight junction proteins via miRNA were studied in various diseases. Here, we discussed the regulation of tight junction proteins via miRNA in HCC progression. This review may focus on the significance of miRNA in cellular function and its role in regulating tight junction proteins that impact in HCC progression which opens a new approach to develop a new diagnostic marker for an early detection of HCC and novel therapeutic approach against HCC.
Liver cancer appears to be the sixth record prevalent type of cancer and the world’s fourth biggest cause of cancer-related mortality. About 90% of cases of liver cancer are hepatocellular carcinoma (HCC). Major risk factors for causing HCC include hepatitis B and C virus, cirrhosis patients after the clearance of hepatitis viral infections, non-alcoholic fatty acid liver disease (NAFLD) and non-alcoholic steatohepatitis (NASH) [1]. Exposure to Aflatoxin B1 (AFB1) and chronic alcohol abuse are the other major risk factors that cause liver cirrhosis and HCC [2]. Available treatment options such as surgical approach and radiological and systemic therapy account for poor response to HCC patients. Despite improved advancement in the treatment options, overall survival rates of HCC patients 5-year overall survival remain low [3]. The low survival rate of HCC patients is mostly attributable to late detection. Hence, the early detection of HCC and robust treatment options are currently required to improve the overall survival of HCC patients. A greater understanding of the molecular pathways underlying the beginning and progression of HCC is critical for making early diagnoses and developing novel treatment strategies. This review intends to describe the molecular mechanism behind the regulation of tight junction (TJ) of miRNA, which opens the void of new approaches in treatment options and early diagnostic markers in HCC.
The existing treatment appears to be challenging against HCC due to the development of resistance. Hence studying the heterogenicity of HCC hinge on genetics, epigenetics, transcriptomics, proteomics and metabolomics could reveal the mechanism behind it. HCC consist of cancerous cells surrounded with tumor microenvironment, resulting in the progression of carcinogenesis [4]. The tight association between cancerous cells and the tumor microenvironment opens the door to epigenetic mechanisms that can alter chromatin to make DNA more accessible, thus altering gene expression [5]. As a consequence, a synergistic combination of genetics and epigenetics could serve as the cause of HCC heterogenicity [6].
TJs in epithelia and endothelia govern the transport of ions, water, and chemicals along the paracellular route. TJs possess a dynamic structure that helps cells to adapt to their surroundings [7, 8]. Intercellular protein-protein interaction connects plasma membrane of adjacent cells to form TJs. TJ consists of two structure models, including protein and protein-lipid hybrid models. According to the protein model, the junctional diffusion barrier is constructed by transmembrane proteins on both sides interacting in a homotypic or heterotypic way. Membrane hemi fusions produced by inverted lipid micelles and balanced by transmembrane proteins are proposed in the hybrid model [9]. The instability of TJ protein expression or TJ integrity affects cellular functions leading to various diseases, including skin, intestinal and lung diseases, and even cancer [10]. TJs are made up of transmembrane proteins such as claudins (CLDNs), TJ-associated marvel proteins (TAMPs) like occludin (OCLN), junctional adhesion molecules (JAMs), and cytosolic proteins that link transmembrane components to the cytoskeleton through the junctional plaque [11].
Other TJ proteins have been found external of TJs, including the basolateral membrane, the cytoplasm, and the nucleus. Apparently, non-junctional TJ proteins appear to play a key role in cellular functions such as propagation, cell adhesion, migration, and incursion. Non-junctional TJ proteins are not dispersed randomly across the membrane. Instead, it interacts with defined targets within the membrane or their phosphorylation. Non-junctional CLDNs exhibit to be stabilised in distinct domains within the basolateral membrane and subsidize cell adhesion via extracellular matrix interactions [12]. Likewise, CLDNs can influence the expression and action of matrix metalloproteases (MMPs), which are involved in matrix remodelling. These functions lead to the contribution to epithelial to mesenchymal transition (EMT), a process in which polarised epithelial cells lose contact with nearby cells, allowing them to migrate [13]. EMT plays a vital role in organogenesis (EMT type 1), homeostasis, inflammation, and fibrosis (e.g., wound healing, fibrogenesis; EMT type 2), whereas EMT type 3 promotes cancer, incursion, and metastasis [14].
The previous and our studies identified the role of TJ proteins in HCC [15, 16]. Altered expression in CLDN family subgroups perturbed during HCC. Overexpression of CLDN1, 4, 5, 7, and 10 were observed in HCC. Low CLDN5 levels and high CLDN7 levels were discovered to be independent predictive variables [17]. Also, in HCC, CLDN10 overexpression was associated with poor patient outcomes and tumour recurrence [18]. CLDN14 downregulation in HCCs, on the other hand, coincides with progressive tumour stage and poor overall survival, whereas CLDN3 expression is lowered in HCC [19, 20]. CLDN1 has been demonstrated to induce EMT in normal liver and HCC cells, causing them to become invasive. The c-Abl-RasRaf-1-ERK pathway is involved in this process, as are the transcription factors Slug and Zeb1 [21]. A CLDN1-specific antibody suppresses the hepatitis C virus (HCV)-induced rise in ERK1/2 phosphorylation in human liver tissue, confirming the functional involvement of these signalling pathways [22]. Bouchagier et al. [17] reported that an upregulation of OCLN in HCC tumours relative to non-neoplastic liver tissues was associated with a better prognosis. Downregulation of zonula occludens-1 (ZO-1) were observed in HCC patient with hepatectomy with poor prognosis [23].
Non-coding RNAs, chromatin remodeling, and DNA methylation are three major genetic regulatory mechanisms that govern chromatin remodeling and gene transcription regulation, and the efficient interaction between these epigenetic processes dictates cell phenotype [24]. miRNAs or miRs are 21–30 nucleotide-long endogenous interfering RNAs stimulate target gene deprivation or limit target gene translation by complementary base pairing, resulting in post-transcriptional control of target genes [25]. Cancer, metabolic disorders, inflammatory diseases, and cardiovascular disease largely depend on miRNA control [26]. Respectively miRNA inhibits target genes by attaching to mRNAs’ untranslated region (UTR). These complexes are implicated in RNA-mediated interference, and in-vertebrates, mRNA transcripts are generally not sliced by a miRNA-associated RNA-induced silencing complex (RISC) but relatively experience translational suppression and destruction by deadenylation [27].
The regulation of mRNA expression is greatly influenced by miRNA, but their specific activities remain unclear. Fascinatingly, the expression of nearly 30% of all human genes were regulated by miRNAs, many of which are connected with tumors or located in unstable genome sections [28]. This indicates the role of miRNA in human carcinogenesis consisting of two types of miRNAs, including oncogenic miRNAs (oncomiRs) and tumor suppressor miRNA (Figure 1). OncomiRs promote carcinogenesis by suppressing tumor suppressor expression, whereas tumor suppressor miRNAs decrease oncogene production in normal cells but are absent in cancer cells [29]. Of note, miR-15 and miR-16, were discovered to be changed in cancer and are linked to BCL2, a protein that inhibits apoptosis, is frequently deleted from chromosomes [30].
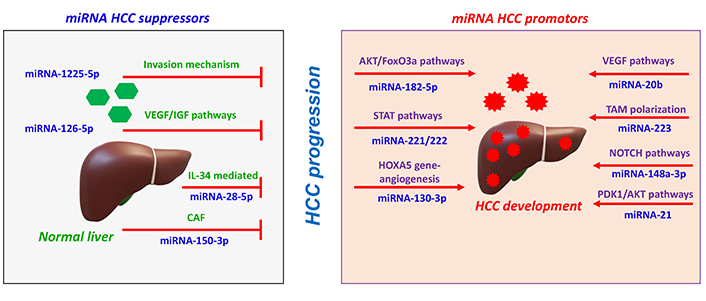
Regulations of miRNAs in HCC cells. i) miRNA suppresses tumor progression by inhibiting incursion mechanism, major pathways such as vascular endothelial growth factor/insulin growth factor (VEGF/IGF) pathways, interleukin-34 (IL-34) mediated pathways. ii) miRNA promotes HCC progression by promoting major pathways protein kinase B/forkhead box O3a (AKT/FoxO3a) pathways, signal transducer and activator of transcription (STAT) pathways, homeobox A5 (HOXA5) gene expression, VEGF pathways, neurogenic locus notch homolog protein (NOTCH) pathways, and phosphoinositide-dependent protein kinase-1 (PDK1)/AKT pathways. CAF: cancer-associated fibroblast; TAM: tumor-associated macrophage
miRNAs can switch on or switch off in HCC, regulating major cellular functions, including propagation, migration, incursion, and apoptosis [31]. For instance, in surgically treated HCC patients, miR-182-5p targets the 3’ UTR of FoxO3a, resulting in the activation of AKT/FoxO3a pathways leading to the propagation and migration of HCC cells [32]. Similarly, miR-1225-5p reduces the MMP-9 expression, inhibiting propagation, migration and incursion of HCC [33]. Other miRNAs regulate the hepatocyte apoptosis pathways. miR-221/222 targets the apoptosis-related factor p53, which is the STAT3 and nuclear factor (NF-κB), resulting in the development of HCC [34].
miRNA tends to regulate the angiogenesis process intricate in the activation of related pathways. For instance, the expression of endothelial markers such as CD31 and CD34 were regulated by miR-130b-3p. HOXA5 gene expression were targeted by miR-130b-3p, thereby endorsing HCC angiogenesis resulting in increased tumor growth and worse overall survival [35]. Similarly, miR-20b controls the expression of hypoxia-inducible factor-1α (HIF-1α) and VEGF to encourage HCC survival during stress conditions [36]. Apart from the activation of angiogenesis process, few miRNAs involving in the inhibition process. miR-126 inhibits the expression of serum alanine aminotransferase and alpha-fetoprotein (AFP), and VEGF leading to the inhibition of HCC angiogenesis [37].
miRNA supports the regulation of tumor microenvironment, which favors the development of HCC. Tumor microenvironments comprise heterogeneous mesenchymal cells, including macrophages, CAFs, endothelial cells, and neutrophils that favor HCC development [38]. M2 macrophage are the TAMs that supports HCC development with poor diagnosis [39]. Tumor-derived exosomes may influence TAM polarization via carrying miRNAs [40]. miR-223 carried by micro vesicles derived from macrophage mediates monocyte differentiation [41]. miR-148a-3p initiates the monocyte differentiation to macrophage through the NOTCH signaling pathway in the presence of granulocyte-macrophage colony-stimulating factor [42]. Moreover, the lowering of miR-28-5p levels, which inhibited HCC metastasis via IL-34-mediated TAMs infiltration, suggests that the feedback loop of miR-28-5p-IL-34-macrophage governs HCC metastasis [43]. Similarly, miR-150-3p from CAFs-derived exosomes transferred to HCC inhibits HCC migration and incursion [44]. In hematopoietic stem cells, miR-21 targets phosphatase and tensin homolog (PTEN) to initiate the PDK1/AKT signaling pathway, and activate CAFs. This promotes the release of angiogenic cytokines such as VEGF, bFGF, and transforming growth factor beta (TGF-β) in HCC progression [45, 46].
The mammalian target of rapamycin (mTOR) is a well-preserved serine-threonine kinase which plays a vital role in signal transduction of cell growth and metabolism [47]. Activating mTOR pathways leads to various diseases, including cardiovascular disease, diabetes and cancer [48]. In HCC, 50% of cases are due to the stimulation of the mTOR pathway, leading to the downregulation of IGF or epidermal growth factor (EGF) [49]. Various studies reported that miRNA activates the mTOR pathway in HCC cells (Table 1).
Role of miRNAs and their target proteins in HCC
| S.No | miRNAs | Target proteins | Target pathway | Cellular function in HCC | HCC severity | Reference(s) |
|---|---|---|---|---|---|---|
| 1 | miR-758-3p | ↓ MDM2↑ p53, AKT, and PRAS40 | mTOR pathway | Inhibits cell propagation, migration and incursion | Reduced HCC progression | [50] |
| 2 | miR-142 | ↓ TGF-β | mTOR pathway | Cell metabolism, propagation, EMT and neo-angiogenesis | Promotes HCC progression and angiogenesis process | [51] |
| 3 | miR-199b-5p | ↑ N-cadherin↓ TGF-β1 | mTOR pathway | Inhibitory possessions on EMT | Reduced HCC progression | [52] |
| 4 | miR-187, miR-497, miR-99a, and miR-592 | ↑ IGF-1R | mTOR pathway | Cell propagation, migration and incursion | Promotes HCC progression | [53–55] |
| 5 | miR-10a, miR-106b, miR-99a, miR-148a, miR-125b, miR-30e, miR-199a, miR-199a3p, miR-24, miR-122, and miR-215 | ↓ β-catenin and c-myc↑ APC | Wnt pathway | Inhibits cell propagation | Reduced HCC progression | [58] |
| 6 | miR-18a | ↓ KLF4, a negative regulator of β-catenin expression | Wnt pathway | Propagation and migration of human HCC | Promotes HCC progression | [59] |
| 7 | miR-195 | ↑ AEG-1 | Wnt pathway | Cell propagation, metastasis, angiogenesis and chemoresistance | Promotes HCC progression, angiogenesis and drug resistance | [60] |
| 8 | miR-320a | ↓ β-catenin, c-myc, cyclin D1, and DKK-1 | Wnt pathway | Inhibition of HCC cell propagation and incursion through c-myc suppression | Inhibits HCC progression | [61] |
| 9 | miR-98-5p | ↓ IGF2BP1 | Wnt pathway | Cell propagation | Promotes HCC progression | [62] |
| 10 | miR-214-3p | ↑ PIM-1 | JAK-STAT pathway | Cell propagation | Promotes HCC progression | [65] |
| 11 | miR-30e | ↓ JAK1 | JAK-STAT pathway | Inhibition of cell migration, propagation proliferation and incursion of HCC cells | Inhibits HCC progression | [66] |
| 12 | miR-340 | ↓ JAK-1 | JAK-STAT pathway | Inhibits the cell migration, propagation of HCC | Inhibits HCC progression | [67] |
| 13 | miR-140-5p and miR-200 | ↑ Pin-1 | JAK-STAT pathway | HCC cell growth and migration | Promotes HCC progression | [69] |
| 14 | miR-21 | ↓ PTEN | PDK1/AKT pathway | Promotes cell migration and incursion of HCC by targeting KLF5 | Promotes HCC progression | [70] |
| 15 | miR-221 | ↑ CDKN1B/p27 and CDKN1C/p57 | Apoptosis pathway | Promotes cell propagation | Promotes HCC progression | [71] |
MDM2: mouse double minute 2; PRAS40: proline-rich Akt substrate 40 kDa; APC: adenomatous polyposis coli; KLF4: Kruppel-like factor 4; AEG-1: astrocyte elevated gene-1; DKK-1: Dickkopf-1; IGF2BP1: insulin-like growth factor 2 mRNA-binding protein 1; PIM-1: proviral integration site for Moloney murine leukemia virus 1; JAK: Janus kinase; Pin-1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1; CDKN1B: cyclin-dependent kinase inhibitor 1B; ↑: resembles upregulation; ↓: resembles downregulation
Restoration of miR-758-3p in HCC inhibits cell propagation, migration and incursion. This is due to the downregulation of the expression of MDM2, mTOR and upregulates p53, AKT, and PRAS40 expression [50]. miR-142 identified as a direct target of TGF-β, which regulates cell metabolism, propagation, EMT and neo-angiogenesis. TGF-β appears to be one of the majors signaling cascades of the mTOR pathway [51]. miR-199b-5p was found to be the target of N-cadherin, which inhibits cell migration, incursion and metastasis in xenograft tumors [51]. The overexpression of miR-199b-5p leads to the inhibition of TGF-β1-induced AKT phosphorylation [51]. Furthermore, inhibiting phosphoinositide 3-kinase (PI3K)/AKT signalling inhibits TGF-β1-induced N-cadherin overexpression in HCC cells [51]. The inhibitory possessions on EMT and the TGF-β1 signaling pathway suggest the adoption of miR-199b-5p as a possible treatment method for HCC [52]. miR-187, miR-497, miR-99a, and miR-592 resemble to target IGF-1R, which is responsible for cell propagation, migration and incursion through the PI3K/AKT/mTOR pathway in HCC [53–55]. In addition, miR-187 was found to be downregulated in HCC promotes cell propagation and incursion [55]. Similarly, miR-592 was found to be under expressed in HCC tissues and cell lines were associated with lymph node metastases [54].
The Wnt pathway regulates the degradation and phosphorylation of β-catenin and is involved in major cell cycle processes during development and tissue homeostasis [56]. In this context, several miRNAs have been shown to regulate the Wnt pathway (Table 1). In HepG2 cell lines, several RNA interferences mediate the knockdown of β-catenin, resulting in decreased propagation in vitro [57].
Ashmawy et al. [58], found that 11 miRNAs (miR-10a, miR-106b, miR-99a, miR-148a, miR-125b, miR-30e, miR-199a, miR-199a3p, miR-24, miR-122, and miR-215) were suppressed in HCC patients. However, miR-10a, miR-30e, miR-215, miR-125b, and miR-148a were reported to be linked with the Wnt/β-catenin pathway such as β-catenin, APC, and c-myc [58]. The expression of miR-18a was upregulated in tissues, leading to the propagation and migration of human HCC. miR-18a inhibits KLF4, which is a negative regulator of β-catenin expression [59]. Furthermore, miR-195 was under expressed in both HCC cell lines and HCC tissues. miR-195 targets AEG-1 expression, which triggers the PI3K/AKT, NF-κB, and Wnt/β-catenin signalling pathways that supports propagation, metastasis, angiogenesis and chemoresistance [60]. miR-320a was found to be under expressed in HCC tissues. miR-320a targets the expression levels of β-catenin, c-myc, cyclin D1, and DKK-1. The overexpression of miR320a leads to the supression of HCC cell propagation and incursion through c-myc suppression [61]. The under expression of miR-98-5p was found in HCC tissues are linked to tumor growth, lymph node metastasis, and clinical stage. The under expression of miR-98-5p is related with the upregulation of IGF2BP1, which supports cell propagation in HCC [62].
The JAK-STAT pathway plays a vital role in stem cell maintenance, hematopoiesis and immune response. The JAK-STAT pathways are activated in many cancers and neoplastic cells through mutations in STATs or lower expression of negative regulators [63].
PIM-1 is a serine/threonine kinase protein involved in various human cancers such as breast cancer, leukemia, lymphoma, osteosarcoma, and colorectal cancer. STAT proteins activate the PIM-1 expression, resulting in cell propagation and cell survival [64]. In this context, miR-214-3p targets the PIM-1 expression and promotes the HCC progression [65]. miR-30e restored in HCC, leading to downregulation of JAK1 expression resulting in the inhibition of cell migration, propagation and incursion of HCC cells [66]. miR-340 targets the JAK-1 expression, which inhibits the cell migration, propagation of HCC [67]. Pin-1 expression appears to be high in 70% of human HCCs and is associated with greater tumor size and poor prognosis [68]. miR-140-5p and miR-200 target the expression of Pin-1. The downregulation of miR-140-5p and miR-200 leads to the overexpression of Pin-1, resulting in HCC cell growth and migration (Table 1) [69–71].
miRNA tends to target the TJ proteins in order to disturb the cellular junction resulting in HCC progression (Table 2). For instance, miR-99b endorses the migration and metastasis of HCC cells by aiming CLDN11 mRNA, inhibiting its expression [72]. Gao et al. [73], reported that IGF2BP3/IMP3 binds to overlapping IGF2BP3 RNA cross-linkage and miR-191-5p targeting sites, promoting the creation of a miR-191-5p induced RNA induced silencing complex. This leads to the suppression of ZO-1 expression resulting in HCC incursion. The inhibition of IGF2BP3 or miR-191-5p increases ZO-1 expression and decreased cellular invasiveness [73]. Similarly, miR-122 induces the increased expression of E-cadherin, ZO-1 and α-catenin expression favouring the increased cell adhesion, and cytoskeleton depolymerization in HCC [74].
miRNA targeting TJ protein expression in HCC
| S.No | miRNA | Target TJ protein | Role in HCC | HCC severity | Reference |
|---|---|---|---|---|---|
| 1 | miR-99b | ↓ CLDN11 | Migration and metastasis of HCC | Promotes HCC progression | [72] |
| 2 | miR-191-5p | ↑ ZO-1 | Decreased cellular invasiveness | Inhibits HCC progression | [73] |
| 3 | miR-122 | ↑ E-cadherin, ZO-1 and α-catenin | Increased cell adhesion, and cytoskeleton depolymerization in HCC | Inhibits HCC progression | [74] |
| 4 | miR-103 | ↓VE-Cad, p120-catenin (p120) and ZO-1 | Enhances the transendothelial incursion of tumor cells | Promotes HCC progression | [75] |
| 5 | miR- 638, miR-663a, miR-3648, and miR-4258 | ↓ VE-Cad and ZO-1 | Increased rates of hepatic and pulmonary metastasis and endothelial junction integrity loss favours the metastatic cancer cells | Promotes HCC progression and metastasis | [76] |
| 6 | miR-23a-3p | ↓ TJP-1, OCLN, and CLDN5 | HCC cell migration, incursion and vascular permeability | Promotes HCC progression | [77] |
VE-Cad: vascular endothelial cadherin; TJP-1: TJ protein 1; ↑: resembles upregulation; ↓: resembles downregulation
A higher serum miR-103 level implies that the HCC has progressed metastatic stage. miR-103-expressing hepatoma cells promote tumour cell trans endothelial incursion, resulting in increased vascular permeability in the tumour, greater levels of exosomal miR-103 and tumour cells in blood circulation, and increased rates of hepatic and pulmonary metastasis. The molecular mechanism reveals that miR-103 secreted by hepatoma cells delivered into endothelial cells via exosomes and diminished the endothelial junction integrity by supressing the VE-Cad, p120 and ZO-1 expression resulting in tumor cell migration by limiting p120 expression in hepatoma cells [75]. Similarly, exosomal miRNAs such as miR- 638, miR-663a, miR-3648, and miR-4258 inhibits the VE-Cad and ZO-1 resulting in endothelial junction integrity loss favours the metastatic cancer cells in HCC [76].
Infiltration of M2 macrophage promotes EMT, vascular permeability and angiogenesis. M2-macrophage derived exosomes contains higher miR-23a-3p. MiR-23a-3p targets the expression of PTEN and TJP-1. Upon transfer of miR-23a-3p into HCC cells, which inhibits the expression of E-cadherin and upregulates the N-cadherin and vimentin expression resulting in HCC cell migration and incursion. M2 derived miR-23a-3p downregulates TJP-1, OCLN, and CLDN5 expression levels resulting in vascular permeability [77].
In conclusion, TJ proteins possess as a significant barrier integrity in association with miRNA resulting in cell propagation, migration, incursion in HCC. However, miRNA has the tendency to regulate the cell cycle progression involving in major signalling pathways such as mTOR, JAK-STAT, Wnt, PDK/AKT pathways, etc. Notably, TJ proteins serve as an imperative factor in HCC progression where the dysregulation of TJs were prominently observed in human HCC patients. Nevertheless, reports suggest that miRNA influences the expression of TJs proteins such as CLDN, ZO-1, E-cadherin, VE-Cad, N-cadherin, vimentin at early or late stages of HCC progression which could help to develop a novel biomarker to screen early HCC prognosis. Certainly, future studies are to construct the detailed pathways in regulating TJs protein via miRNA and subsequent development of drugs to target TJs and miRNA.
AKT/FoxO3a: protein kinase B/forkhead box O3a
CAF: cancer-associated fibroblast
CLDNs: claudins
EMT: epithelial to mesenchymal transition
HCC: hepatocellular carcinoma
HOXA5: homeobox A5
IGF2BP1: insulin-like growth factor 2 mRNA-binding protein 1
IL-34: interleukin-34
JAK: Janus kinase
KLF4: Kruppel-like factor 4
mTOR: mammalian target of rapamycin
NOTCH: neurogenic locus notch homolog protein
OCLN: occludin
PDK1: phosphoinositide-dependent protein kinase-1
PI3K: phosphoinositide 3-kinase
PIM-1: proviral integration site for Moloney murine leukemia virus 1
Pin-1: peptidyl-prolyl cis-trans isomerase NIMA-interacting 1
PTEN: phosphatase and tensin homolog
STAT: signal transducer and activator of transcription
TAM: tumor-associated macrophage
TGF-β: transforming growth factor beta
TJP-1: tight junction protein 1
TJ: tight junction
VE-Cad: vascular endothelial cadherin
VEGF/IGF: vascular endothelial growth factor/insulin growth factor
ZO-1: zonula occludens-1
SBS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. BV: Conceptualization, Investigation, Validation, Writing—original draft, Writing—review & editing, Supervision. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the Indian Council of Medical Research (ICMR) extramural research grant [5/13/83/2020/NCD/III]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Patrizia Pontisso, Maurizio Parola
Anna Fichera, Mirella Fraquelli
Lorenzo Mainardi ... Chiara Raggi
