 Review
Review

Affiliation:
Department of Gastroenterology and Hepatology, Zhongshan Hospital, Fudan University, Shanghai Institute of Liver Diseases, Shanghai 200032, China
ORCID: https://orcid.org/0009-00021-4300-0743
Affiliation:
Department of Gastroenterology and Hepatology, Zhongshan Hospital, Fudan University, Shanghai Institute of Liver Diseases, Shanghai 200032, China
Email: guo.jinsheng@zs-hospital.sh.cn
ORCID: https://orcid.org/0000-0002-9980-8725
Explor Dig Dis. 2024;3:226–240 DOI: https://doi.org/10.37349/edd.2024.00049
Received: January 11, 2024 Accepted: April 22, 2024 Published: June 18, 2024
Academic Editor: Jean Francois D. Cadranel, GHPSO, France
The article belongs to the special issue Viral Hepatitis
Hepatitis C viral infections present a significant global health challenge, carrying substantial economic implications. These infections manifest in various clinical forms, including acute and chronic hepatitis, liver cirrhosis, hepatic failure, and hepatocellular carcinoma (HCC). Liver cirrhosis and HCC emerge as the primary contributors to mortality in hepatitis virus-induced liver diseases. To alleviate the public health impact of this disease, it is imperative to enhance the diagnosis and treatment rates among hepatitis C virus-infected individuals. The advent of direct-acting antivirals (DAAs), especially pan-genotypic regimens such as a combination of sofosbuvir and velpatasvir, has shown remarkable progress in achieving hepatitis C cure. However, potential obstacles, such as drug adverse effects and resistance-associated substitutions (RASs), warrant attention. Managing chronic hepatitis C (CHC) requires tailored treatment plans, vigilant monitoring, and judicious re-treatment strategies.
Over 71 million individuals worldwide are affected by chronic hepatitis C virus (HCV) infection [1]. HCV successfully evades the host’s immune response in 50% to 90% of acute infections, leading to the development of chronic infections in majority of cases. The natural history of hepatitis C exhibits significant heterogeneity, influenced by a complex interplay of viral, host, and environmental factors.
In chronic HCV infection, approximately 2% to 24% of individuals progress to cirrhosis over a span of 20 years. Among these, an annual incidence of approximately 3% is observed for decompensated cirrhosis, characterized by complications due to hepatic dysfunction and portal hypertension such as ascites, hepatic encephalopathy, variceal hemorrhage, and hepatorenal syndrome [2]. Additionally, hepatocellular carcinoma (HCC) occurs at an annual rate of 1% to 4% [3]. HCV infection is associated with increased overall and liver-related mortality rates. Notably, complications of HCV-related cirrhosis remain a primary indication for liver transplantation [4, 5]. However, with the introduction of highly efficient direct-acting antivirals (DAAs), a reduction in the frequency of these complications is anticipated by 2030 [6].
HCV is a single-stranded positive-sense RNA virus belonging to the Flaviviridae family. The HCV genome comprises approximately 9,600 nucleotides and consists of an open reading frame (ORF) that encodes a large viral polyprotein of approximately 3,000 amino acids. HCV polyprotein is processed into nucleocapsid (C, core protein), envelope 1 (E1) and envelope (E2), viroporin protein (p7), and six nonstructural proteins including NS2 (cysteine protease), NS3 (serine protease and RNA helicase, providing enzymatic functions for replication and evasion of innate immune responses), NS4A (NS3 protease cofactor), NS4B, NS5A (RNA-binding site essential for replication and assembly, interacting with the HCV replication complex), and NS5B (RNA-dependent RNA polymerase, RdRp) [7–11].
HCV exhibits inherent high mutation rates, resulting in significant genetic heterogeneity throughout its entire genome. Based on this substantial genetic variation, the viral sequences are categorized into eight major genotypes (denoted by numbers, sharing 60% to 70% sequence similarity) and over 86 subtypes (designated with lowercase letters, with sequence similarity ranging from 77% to 80%) [12–14]. Furthermore, within the host, HCV exists in a quasi-species form, characterized by closely related yet heterogeneous HCV RNA sequences that arise due to mutations during the viral replication process. The generation of quasi-species may represent a mechanism by which the virus escapes host immune responses and establishes persistent infection. An increased number of quasi-species is also associated with rapid progression to cirrhosis and the occurrence of HCC [15–18].
The distribution of HCV genotypes varies geographically across the whole world. In China, HCV genotypes 1b accounts for approximately 56.8% of the patients, and 2a are relatively common [19]. HCV genotypes may also play a role in disease progression and the complications of chronic HCV infection. Genotype 3 of HCV is associated with a more rapid liver fibrosis progression, an increased risk of cirrhosis, and HCC [20].
Hepatitis C is an infectious liver disease primarily transmitted through exposure to HCV infected body fluids. The routes of HCV transmission include percutaneous and non-percutaneous transmission. The transmission route of percutaneous transmission (e.g., bloodborne and needlestick infection) is particularly associated with intravenous drug use (IVDU), where the frequency of infection among people who inject drugs (PWID) ranges from 57% to 90% [21, 22]. Other common modes of the transmission include blood transfusions and chronic hemodialysis. Occupational transmission may occur when healthcare workers are exposed to infected patients, and occasionally vice versa. HCV can also be transmitted through non-percutaneous route such as sexual contact and during childbirth. In addition, up to one-third of HCV infection cases have an unknown source of transmission. These infections may stem from undisclosed or undetected percutaneous exposure, such as through non-commercial tattooing and body piercing practices where equipment is improperly sterilized, reused, or shared.
The pathogenesis of hepatitis C involves both the viral associated mechanisms (direct viral infection of hepatocytes and hepatotoxicity caused by viral proteins) and the immune-mediated host mechanisms. HCV infection elicits immune responses from the host, including initial innate responses and subsequent adaptive immunity. Simultaneously, HCV has the capacity to disrupt both innate and subsequent adaptive immune responses at multiple levels, which may be crucial for establishing chronic infection. This includes impairing NK cell-mediated cytotoxicity and cytokine production, inhibiting the transcription and activation of interferon response genes, and manipulating T-cell immune responses in the liver, leading to both direct clearance of infected cells and suppression of viral replication through the secretion of antiviral cytokines [23]. Both viral products and immune responses may play roles in the development of hepatocellular damage [24, 25].
HCV-specific antibodies (HCV-Ab) are not protective and appear to be independent of the stage of infection or immune response. Furthermore, administration of high-titer HCV-enriched or HCV-specific IgG has limited impact on viral levels or duration in the human body [26].
The presence of fibrosis as a scar forming response to repeated liver cell damage is a significant risk factor for hepatitis C progression. Liver biopsies may reveal faster fibrosis progression in individuals with higher activity grades and elevated serum transaminase levels. The risk factors associated with the rapid progression of HCV include [27–33]: host factors such as advanced fibrosis stage, higher inflammatory grades, older age at infection, male gender, under immunosuppression, organ transplantation, as well as alcohol consumption, non-alcoholic fatty liver disease, obesity, insulin resistance, smoking, and increased liver iron load and viral factors such as genotype 3, co-infection with HBV or HIV.
Symptoms are not observed in almost all cases. Even among symptomatic patients, most clinical symptoms are nonspecific. The commonly reported symptoms include fatigue, nausea, abdominal pain, loss of appetite, mild fever, itching, muscle pain, and jaundice, the latter being the most specific liver-related symptom, occurring in 50% to 84% of clinically apparent acute HCV infections [34]. In contrast to other hepatitis virus infections, acute liver failure (ALF) due to HCV is rare and has only been reported in isolated cases [35]. Clinical presentation tends to be more evident in individuals with heavy alcohol consumption or co-infection with HBV or HIV, and the clinical course can be more severe.
In patients with chronic HCV infection, serum alanine aminotransferase (ALT) levels are typically elevated. ALT levels may fluctuate, and up to half of the patients may have normal ALT levels at specific times. Prolonged normal ALT levels, which are more common in females, are associated with lower serum HCV RNA levels and milder inflammation and fibrosis in liver biopsy specimens [36].
Most patients with chronic hepatitis C (CHC) are asymptomatic until the late stage of liver cirrhosis. Patients diagnosed with chronic infection may complain of nonspecific symptoms such as fatigue, vague abdominal discomfort, or depression, with consistently lower health-related quality of life (HRQOL) scores in all aspects when compared to HCV-negative individuals [37]. Improvement in HRQOL scores is observed in patients who achieve sustained virological response (SVR) with antiviral therapy [38–41]. Less common symptoms include arthralgia, sensory disturbances, myalgia, Sjögren’s syndrome, nausea, loss of appetite, and difficulty concentrating. The severity of these symptoms may, but not exclusively, correlate with the severity of underlying liver disease. Late-stage chronic HCV infection can progress to decompensated cirrhosis, leading to complications of cirrhosis and HCC.
Patients with chronic HCV infection may be accompanied by extrahepatic manifestations, including type II and type III cryoglobulinemia (found in 19% to 50% of cases), characterized by polyclonal IgG plus monoclonal IgM and polyclonal IgG plus polyclonal IgM, respectively. However, clinical manifestations of cryoglobulinemia are reported in only 5% to 10% of patients and are more common in those with cirrhosis [42]. The symptoms and signs of CHC patients include fatigue, joint pain, arthritis, purpura, Raynaud’s phenomenon, vasculitis, peripheral neuropathy, and renal disease. Patients may be test positive for rheumatoid factors and exhibit cryoglobulins, as well as low serum complements levels. Hepatitis C patients can develop glomerular diseases, often presenting as cryoglobulinemic glomerulonephritis, membranoproliferative glomerulonephritis, and membranous nephropathy [43–46]. Other extrahepatic manifestations include autoimmune thyroiditis, B-cell non-Hodgkin lymphoma, late-onset porphyria cutanea tarda, lichen planus, and Sjögren’s syndrome [47]. The spectrum of extrahepatic manifestations may negatively impact the overall survival of HCV-infected individuals.
The primary goal of treating CHC is to achieve a cure, eliminate or alleviate HCV-related liver damage and extrahepatic manifestations, reverse liver fibrosis, prevent or delay fibrosis progression to cirrhosis, prevent the happening of decompensated cirrhosis, liver failure, or HCC, improve long-term survival rates of patients, enhance the quality of life, and prevent the further transmission of HCV.
Until 2011, the standard treatment for HCV infection was a combination of pegylated interferon (Peg-IFN) and ribavirin (RBV). However, interferon-based therapy had a relatively low overall cure rate (54% to 56%) and significant toxicity, leading to discontinuation of treatment in 14% of patients [48]. Additionally, many patients were not eligible for or could not tolerate interferon-based therapy, therefore limiting their treatment options. Subsequently, interferon-free regimens consisting of new-generation DAAs were developed and are now a preferred treatment for HCV infection. Interferon-based treatment is no longer recommended for the management of HCV. DAAs include agents targeting various steps in the HCV life cycle (Figure 1), such as NS3/NS4A protease inhibitors (-previrs: glecaprevir, grazoprevir), NS5A inhibitors (-asvirs: daclatasvir, ledipasvir, ombitasvir, pibrentasvir, elbasvir, velpatasvir), and NS5B polymerase inhibitors (nucleotide and non-nucleotide NS5B inhibitors: sofosbuvir, dasabuvir). The combinations of DAAs target different and complementary stages of the HCV life cycle, resulting in a paradigm shift in the treatment of HCV infection, with SVR rates exceeding 95% and minimal toxicity. The selection of interferon-free DAA regimen depends on factors including HCV genotype/subtype, the presence or absence of cirrhosis, and prior treatment history. Some regimens cover all genotypes (pan-genotypic), and appropriate regimens and treatment duration are applicable to special populations including HCV and HIV co-infection [49], decompensated liver disease, HCV recurrence post-liver transplantation [50], and patients with advanced kidney disease [51].
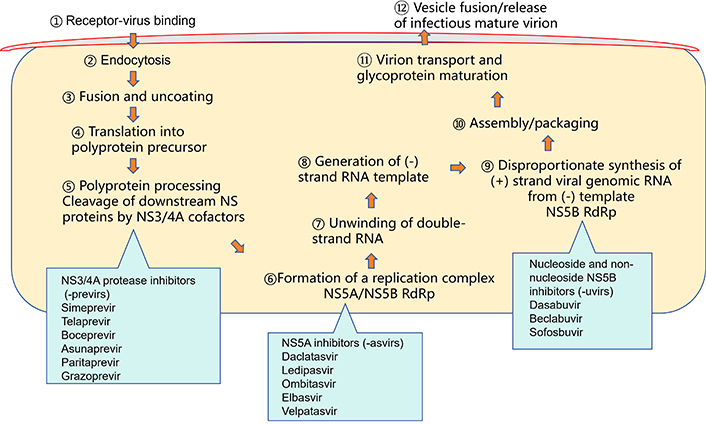
Schematic representation of the hepatitis C virus life cycle and the targets for antiviral therapies. RdRp: RNA-dependent RNA polymerase; NS3: serine protease and RNA helicase; NS4A: NS3 protease cofactor; NS5A: RNA binding site; NS5B: RNA-dependent RNA polymerase
Antiviral therapy can prevent clinical endpoints of CHC. Patients with advanced liver fibrosis who achieve SVR with DAAs experience a significant reduction in liver-related mortality and the development of decompensated cirrhosis [52]. The use of highly effective and well-tolerated interferon-free DAA regimens, coupled with the clear benefits of clearing HCV in terms of reducing all-cause and liver-related mortality rates and improving quality of life, suggests that antiviral therapy should be considered for all individuals with CHC. Public health programs aimed at eliminating the public health hazards of hepatitis C, such as the “Action Plan for Eliminating the Public Health Hazards of Hepatitis C (2021–2030)” proposed by Chinese National Health Commission, the Ministry of Science and Technology, and the Ministry of Industry and Information Technology, emphasize the importance of expanding testing and improving treatment access, particularly in high-risk populations, to achieve the goal of hepatitis C elimination [53–55].
Early treatment yields greater benefits, with a more significant increase in the benefits of achieving SVR in individuals with early liver fibrosis (Metavir stage less than F2/F3) [56]. Treatment reduces all-cause and liver-related mortality rates, prevents ongoing transmission, improves quality of life, and improves or prevents extrahepatic complications, including diabetes, cardiovascular disease, kidney disease, and B-cell non-Hodgkin lymphoma [57, 58].
Key points for the treatment of hepatitis C are summarized in Table 1.
| Key points | Description |
|---|---|
| Universal treatment | All hepatitis C virus (HCV) RNA-positive individuals, regardless of the presence of liver cirrhosis, concurrent chronic kidney disease, or extrahepatic manifestations, should receive antiviral therapy. |
| Treating the high-risk populations | Patients with advanced liver fibrosis or cirrhosis, significant extrahepatic manifestations, HCV recurrence after liver transplantation, and those at high risk of accelerating liver disease progression should be prioritized for immediate treatment. |
| Pre-treatment assessment | Before initiating antiviral treatment, assess the severity of liver disease, kidney function, HCV RNA quantification, hepatitis B surface antigen (HBsAg) and hepatitis B virus (HBV) core antibody status, comorbidities, and concomitant medication use. HCV genotyping may be necessary in some cases. |
| Monitoring during treatment | Monitor treatment efficacy and safety throughout the course of therapy. Patients receiving direct-acting antivirals (DAAs) should undergo clinical adverse event assessments at each visit. Alanine aminotransferase (ALT) levels should be monitored at baseline, at weeks 4, 12, and 24 of treatment, or as clinically indicated. |
| Treatment endpoint | The treatment endpoint is sustained virologic response after 12 weeks of ending antiviral therapy (SVR12). This is achieved when sensitive detection methods (with a lower limit of detection ≤ 15 IU/mL) show that HCV RNA is undetectable. |
| Retreatment of previous failures | For patients with prior antiviral treatment failures, it is crucial to ascertain the previous treatment regimen, the clinical type of treatment failure (non-response, relapse, or breakthrough), and the presence of cirrhosis. The selection of a DAA combination regimen should avoid targeting the same viral points as previous treatments, based on drug availability and DAA target differences. |
The primary goal of antiviral therapy for HCV is to achieve SVR, defined as the continuous absence of detectable HCV RNA for at least 12 weeks after completing treatment. SVR is the hallmark of HCV infection cure, which has been demonstrated to persist in 99% of patients over a follow-up period of ≥ 5 years [58]. SVR signifies the absence of detectable HCV RNA, whereas HCV antibodies may still be present. There will be a significant improvement in liver function and histology in patients achieving SVR. The Food and Drug Administration (FDA) of America has approved quantitative or qualitative nucleic acid testing (NAT) with a lower limit of detection of ≤ 25 IU/mL. It’s important to note that SVR is not equivalent to virologic responses during or immediately after the treatment; rather, it refers to the sustained virologic response observed at least 12 weeks after the completion of therapy. Patients can become re-infected if they persist in risk behaviors. Therefore, caution should be taken for this situation when evaluate SVR and the patients should be educated during treatment.
Achieving cure from HCV infection yields numerous health benefits for patients, including improved liver inflammation (reduced transaminase levels), decreased fibrosis progression rate, resolution of liver cirrhosis, a > 70% reduction in the risk of HCC development, a 90% reduction in liver-related mortality risk, and diminished liver transplantation requirements [61]. Achieving SVR also reduces symptoms and mortality rates associated with severe extrahepatic manifestations [62], such as cryoglobulinemic vasculitis, non-Hodgkin lymphoma, and other lymphoproliferative disorders, with over 75% of cases achieving complete or partial remission upon successful HCV treatment [63]. The reduction in disease severity leads to a decrease in overall mortality rates. Patients who achieve SVR experience an improvement in their quality of life across physical, emotional, and social well-being dimensions. Conversely, those who do not achieve SVR see their HRQOL continuously deteriorate, along with sustained liver damage, fibrosis progression, and the risk of transmitting HCV infection, necessitating consideration for re-treatment. Patients should be monitored for signs and symptoms of decompensated cirrhosis. In cases of treatment failure, patients may carry one or more drug-resistant variants, and resistance-associated substitutions (RASs) testing should be considered if retreatment is contemplated. The retreatment should consider 3 classes DAA regimen in combination including NS3, NS5A, and NS5B inhibitors.
DAAs can interact with other medications co-administered for comorbid conditions, potentially leading to DDIs that may result in severe adverse reactions. DDIs may occur at various stages of drug metabolism, involving drug transporters and cytochromes. Foods can also influence the absorption of DAAs. Commonly encountered drugs that interact with DAAs include antiarrhythmic drugs such as amiodarone, antiepileptic and barbiturate drugs (e.g., carbamazepine, oxcarbazepine, phenobarbital, and phenytoin), antipsychotic medications, antituberculosis drugs (e.g., rifampin and rifapentine), anticoagulant medications, and antidiabetic agents [64]. Some important DDIs with commonly used HCV DAAs (i.e., sofosbuvir, sofosbuvir/velpatasvir, sofosbuvir/velpatasvir/voxilaprevir) are listed in Table 2. Prior to commencing treatment, healthcare providers should carefully review drug package inserts and utilize online tools (e.g., https://www.hep-druginteractions.org/) to ascertain potential interactions.
Key drug-drug interactions between hepatitis C virus (HCV) direct-acting antiviral agents and clinically commonly used drugs [59, 60]
| Class/MOA | Drugs | SOF | SOF/VEL | SOF/VEL/VOX |
|---|---|---|---|---|
| Antiretroviral drugs | ||||
| NRTIs | Tenofovir disoproxil fumarate (TDF) | |||
| Tenofovir alafenamide (TAF) | ||||
| NNRTIs | Efavirenz | |||
| Etravirine | ||||
| Nevirapine | ||||
| Protease inhibitors | Atazanavir/ritonavir | |||
| Atazanavir/cobicistat | ||||
| Darunavir/ritonavir | ||||
| Lopinavir/ritonavir | ||||
| Entry/Integrase inhibitors | Elvitegravir/cobicistat/emtricitabine/TDF | |||
| Elvitegravir/cobicistat/emtricitabine/TAF | ||||
| Lipid-lowering drugs | ||||
| HMG-CoA reductase inhibitors | Atorvastatin | |||
| Bezafibrate | ||||
| Fenofibrate | ||||
| Fluvastatin | ||||
| Gemfibrozil | ||||
| Lovastatin | ||||
| Pitavastatin | ||||
| Pravastatin | ||||
| Rosuvastatin | ||||
| Simvastatin | ||||
| Selective cholesterol adsorption inhibitor | Ezetimibe | |||
| Antihyperglycemic drugs | ||||
| Glucagon-like peptide-1 (GLP-1) receptor agonists | Dulaglutide | |||
| Liraglutide | ||||
| Sodium-glucose cotransporter-2 (SGLT2) inhibitors | Empagliflozin | |||
| Central nervous system drugs | ||||
| Antipsychotics | Paliperidone | |||
| Cardiovascular drugs | ||||
| Antiarrhythmics | Amiodarone | |||
| Digoxin | ||||
| Hypertension | ||||
| β-adrenergic antagonist | Carvedilol | |||
| Calcium channel blockers | Diltiazem | |||
| Angiotensin II receptor blockers | Losartan | |||
| Enalapril | ||||
| Immunosuppressants | ||||
| Cyclosporine | ||||
| Sirolimus | ||||
| Tacrolimus | ||||
| Everolimus | ||||
| Antiplatelets and anticoagulants | ||||
| Antiplatelets | Clopidogrel | |||
| Dabigatran | ||||
| Ticagrelor | ||||
| Rivaroxaban | ||||
| Apixaban | ||||
| Edoxaban | ||||
| Anticoagulants | Warfarin | |||
| Anticonvulsants | ||||
| Carbamazepine | ||||
| Clonazepam | ||||
| Eslicarbazepine | ||||
| Lorazepam | ||||
| Sodium channel blockers | Oxcarbazepine | |||
| Phenytoin | ||||
| Primidone | ||||
| GABA potentiation | Phenobarbital | |||
| Gastrointestinal drugs | ||||
| Gastrointestinal agents | Aluminium hydroxide | |||
| Antacids | ||||
| Anti-inflammatory agents | ||||
| Aminosalicytates | Sulfasalazine | |||
| Histamine H2 receptor antagonist | Cimetidine | |||
| Famotidine | ||||
| Ranitide | ||||
| Proton pump inhibitors | Esomeprazole | |||
| Lansoprazole | ||||
| Pantoprazole | ||||
| Rebeprazole | ||||
HMG-CoA: 3-Hydroxy-3-methylglutaryl coenzyme; MOA: mechanism of action; NNRTIs: non-nucleoside reverse transcriptase inhibitors; NRTIs: nucleoside reverse transcriptase inhibitors; SOF: sofosbuvir, NS5B-polymerase inhibitor; VEL: velpatasvir, 2nd generation of NS5A inhibitor; VOX: voxilaprevir, 2nd generation of protease inhibitor. Green: no clinically significant interaction expected; orange: potential interaction which may require a dosage adjustment, altered timing of administration or additional monitoring; red: these drugs should not be co-administered
Note. Adapted with permission from “EASL recommendations on treatment of hepatitis C: Final update of the series” by European Association for the Study of the Liver. J Hepatol. 2020;73:1170–218 (https://www.journal-of-hepatology.eu/article/S0168-8278(20)30548-1/fulltext). © 2020 European Association for the Study of the Liver.
Sofosbuvir/velpatasvir for example, which is a pangenotypic anti-HCV oral pellet conformulated with fixed-dose combinations of sofosbuvir 400 mg/velpatasvir 100 mg for adults, is indicated for chronic HCV genotype 1–6 infection without cirrhosis or with compensated cirrhosis and in combination with RBV for those with decompensated cirrhosis. The most common adverse reactions (≥ 10%, all grades) with sofosbuvir/velpatasvir in adults and pediatric patients 6 years of age and older were headache and fatigue; and when used with RBV in adults with decompensated cirrhosis were fatigue, anemia, nausea, headache, insomnia, and diarrhea [65–67].
Amiodarone is not recommended for use with sofosbuvir/velpatasvir due to the risk of symptomatic bradycardia, particularly in patients also taking beta blockers or with underlying cardiac comorbidities and/or with advanced liver disease. Risk of reduced therapeutic effect due to concomitant use of sofosbuvir/velpatasvir with P-glycoprotein (P-gp) inducers and/or moderate to strong inducers of CYP2B6, CYP2C8 or CYP3A4: Rifampin, St. John’s wort, and carbamazepine are not recommended for use with sofosbuvir/velpatasvir as they may significantly decrease sofosbuvir and/or velpatasvir plasma concentrations. Coadministration of sofosbuvir/velpatasvir is not recommended with topotecan due to increased concentrations of topotecan. Coadministration of sofosbuvir/velpatasvir is not recommended with proton-pump inhibitors, phenobarbital, phenytoin, rifabutin, rifapentine, efavirenz, and tipranavir/ritonavir due to decreased concentrations of sofosbuvir and/or velpatasvir.
The following questions should be always considered when assessing potential DDIs:
1) Does the concomitant drug undergo hepatic metabolism?
2) Are these drugs substrates or inducers of P-gp/breast cancer resistance protein (BCRP), organic anion-transporting polypeptide (OATP), or cytochrome P450-3A4 (CYP3A4), or other transporters?
3) Does the patient receive drugs with a narrow therapeutic range?
4) Is monitoring of drug levels feasible?
5) Can alternative drugs be considered, and how should they be administered?
The most common pathway influencing drug metabolism often involves the induction or inhibition of CYP450 enzymes, resulting in altered drug exposure levels. Protease inhibitors, particularly those acting as strong CYP450 inhibitors like ritonavir, tend to interact with other drugs [68]. Protease inhibitors are metabolized through hepatic pathways and represent a common source of DDIs. Conversely, most NS5B and NS5A inhibitors have no or minimal effects on CYP450 enzymes, leading to fewer clinically significant pharmacokinetic interactions with other medications. Sofosbuvir, for instance, is less prone to DDIs because its metabolism does not depend on cytochromes. Therefore, the FDA of America issued a warning on August 28, 2019, cautioning against the use of DAA regimens containing protease inhibitors in patients with moderate to severe hepatic impairment due to the potential for hepatic decompensation as a result of elevated drug levels and limited safety data. Close monitoring through regular visits (telephone or in-person) is recommended for adverse effects and potential drug interactions during treatment. Patients taking antidiabetic medications should be informed about the possibility of hypoglycemic symptoms. Patients on warfarin should be aware of potential changes in coagulation status, and necessitate monitoring of international normalized ratio (INR). If ALT elevations reach ≥ 10 times the baseline during DAA treatment, the therapy should be promptly discontinued, especially if accompanied by hepatic symptoms or signs or in conjunction with elevated bilirubin, alkaline phosphatase, or INR. ALT elevations of < 10 times the baseline in asymptomatic patients should trigger close monitoring with repeat measurements every 2 weeks. If elevations persist, discontinuation of DAA therapy should be considered.
HBV reactivation is a recognized complication of immunosuppressive therapy. HBV reactivation can occur not only in patients with active and occult HBV infection (hepatitis B surface antigen positive, with or without detectable HBV DNA) but also in those with prior infections (hepatitis B surface antigen-negative, HBV DNA-negative, but positive for antibodies against hepatitis B core antigen).
A systematic review and meta-analysis of HBV reactivation in HCV-HBV co-infected individuals treated with interferon-based and DAA-based interferon-free regimens demonstrated similar HBV reactivation rates in interferon-based regimens (14.5%) and DAA-based regimens (12.2%) [69]. However, patients receiving the two treatment modalities exhibited different clinical presentations, with HBV reactivation occurring earlier and more severely during DAA treatment. DAAs specifically target HCV proteins involved in the replication cycle without known direct immunomodulatory properties or anti-HBV activity. Thus, the potential mechanisms of HBV reactivation in this context remain unclear.
Patients with a history of HBV infection (current or past) should be monitored for HBV during DAA therapy. Any patient with a sudden elevation in serum transaminase levels during active DAA treatment should raise suspicion of HBV reactivation. For hepatitis B surface antigen-positive patients who have not received suppressive HBV therapy at baseline and did not achieve the treatment target of HBV DNA suppression, two measures should be considered: 1) Initiating prophylactic HBV antiviral therapy for patients with low or undetectable HBV DNA levels. If this approach is chosen, prophylactic therapy should continue for 12 weeks after completing DAA treatment. 2) Immediate monthly monitoring of HBV DNA levels after DAA treatment. If the patient’s prior HBV DNA was undetectable or unquantifiable, antiviral treatment should be initiated if HBV DNA levels exceed 10 times the baseline or >1,000 IU/mL.
DAA resistance, including baseline resistance or on-treatment selective resistance, can impact the success of a particular DAA treatment regimen or the choice of retreatment options after DAA failure [70, 71]. Two factors should be considered for the formation of resistance barriers: genetic barriers which involve the need for multiple amino acid substitutions to confer resistance, and the fitness of RASs, which refers to the ability of RASs to maintain replication and be selected within the quasi-species.
Resistance can be categorized into four types: 1) Response-relapse, where serum HCV RNA becomes negative during antiviral therapy but turns positive shortly after discontinuation; 2) Response-breakthrough, where HCV RNA becomes negative during antiviral therapy but turns positive before discontinuation; 3) Partial response, characterized by a reduction in HCV RNA levels by > 1 log10 but without reaching negativity during antiviral therapy; and 4) Non-response, where HCV RNA levels remain relatively unchanged during antiviral therapy.
Most treatment failures involve relapse rather than virologic breakthrough. Viral resistance mutations often occur when drug levels fall below therapeutic concentrations, creating selective pressure for resistant variants to become dominant and selectively grow under antiviral drug treatment. Baseline RASs, some of which can be detected before treatment, especially in regimens containing NS5A inhibitors, appear to play a more critical role than on-treatment viral mutations [72]. Under the selective pressure of DAAs, previously dominant high-replicating wild-type strains are eradicated, while low-replicating mutant strains with RASs present at low levels before treatment continue to survive during treatment at lower replication levels. After discontinuation, these mutant strains with RASs can eventually lead to relapse [73]. Baseline resistance, particularly NS5A RASs, forms the foundation of DAA resistance and should be emphasized and optimized in clinical practice regarding the use of NS5A inhibitors (Table 3) [74, 75]. The usual time for sequencing of RASs is in patients about to be retreated having failed an initial DAA regimen.
Resistance associated substitutions and genotypes/subtypes in direct-acting antivirals (DAAs) agents non-responding hepatitis C virus (HCV) infected patients
| DAA | Amino acid substitutions detected in DAA failing patients | HCV genotype/subtype associated with SVR failure |
|---|---|---|
| SOF | 159F | 1a, 1b, 2a/b/c, 3a |
| 237G | 1a, 3a, 4a/d | |
| 282R/G/T | 1a, 1b, 2a/b/c, 3a, 4a/d | |
| 316F/H/N | 1a, 1b | |
| 320F | 1a | |
| 321A/I | 1a, 1b, 3a, 4a/d | |
| VEL | 24R | 1a |
| 28T/V | 1a | |
| 30E/K/H/V/K/L/R | 1a, 3a | |
| 31M/V/P | 1a, 1b, 2a/b/c, 3a, 4a/d | |
| 32L | 1a | |
| 58D | 1a | |
| 92K | 3a | |
| 93H/N/R/S/T/W | 1a, 1b, 2a/b/c, 3a, 4a/d | |
| VOX | 36L | 1a |
| 43S | 1b | |
| 80K/R | 1a, 4a/d | |
| 156T/V | 1a,1b |
SOF: sofosbuvir, NS5B-polymerase inhibitor; VEL: velpatasvir, 2nd generation of NS5A inhibitor; VOX: voxilaprevir, 2nd generation of protease inhibitor; SVR: sustained virological response
The treatment goals for chronic HCV infection are aligned with the aim to eliminate the public health impact of viral hepatitis by 2030. These goals include reducing the incidence of new infections by 90% and decreasing the mortality rate by 65%. To achieve these targets, it is essential to diagnose over 90% of infected individuals and treat more than 80% of those diagnosed [6, 55]. The treatment landscape for chronic HCV infection has evolved significantly with the introduction of highly effective and well-tolerated DAAs. The use of pan-genotypic DAA regimens is a primary recommended approach to meet these treatment goals. Individualized treatment plans, careful monitoring, and appropriate retreatment strategies are important considerations in the management of CHC. When embarking on the treatment of hepatitis C, healthcare providers should closely monitor virologic responses, consider potential drug interactions, and select an appropriate treatment regimen based on the individual patient’s profile. This ensures treatment success and patient safety.
ALT: alanine aminotransferase
CHC: chronic hepatitis C
DAAs: direct-acting antivirals
DDIs: drug-drug interactions
HBV: hepatitis B virus
HCC: hepatocellular carcinoma
HCV: hepatitis C virus
HIV: human immunodeficiency virus
HRQOL: health-related quality of life
NS3: serine protease and RNA helicase
NS4: NS3 protease cofactor
NS5A: RNA binding site
NS5B: RNA-dependent RNA polymerase
RASs: resistance-associated substitutions
RBV: ribavirin
SVR: sustained virological response
HX: Writing—review & editing. JG: Writing—review & editing, Investigation, Supervision.
Prof. Jinsheng Guo who is a Guest Editor of Exploration of Digestive Diseases, had no involvement in the decision-making or the review process of this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was funded by the National Fund of Nature Science of P.R. China [91129705, 81070340] and Shanghai Pujiang Talent Program [09PJ1402600]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Laura C. McCoullough ... Peter A. Revill
Chunzheng Li ... Xianguang Yang
Juntian Yao ... Youhua Xie
