 Review
Review

Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Centro de Investigación Biomédica en Red (CIBEREHD), 028029 Madrid, Spain
ORCID: https://orcid.org/0000-0002-0842-0451
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Centro de Investigación Biomédica en Red (CIBEREHD), 028029 Madrid, Spain
4Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
ORCID: https://orcid.org/0000-0003-3422-2990
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Centro de Investigación Biomédica en Red (CIBEREHD), 028029 Madrid, Spain
Email: lcondedelarosa@gmail.com
ORCID: https://orcid.org/0000-0001-9392-7320
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB), CSIC, 08036 Barcelona, Spain
2Liver Unit, Hospital Clínic i Provincial de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Centro de Investigación Biomédica en Red (CIBEREHD), 028029 Madrid, Spain
4Keck School of Medicine, University of Southern California, Los Angeles, CA 90033, USA
Email: carmen.garcia@iibb.csic.es
ORCID: https://orcid.org/0000-0002-2652-6102
Explor Dig Dis. 2024;3:382–413 DOI: https://doi.org/10.37349/edd.2024.00057
Received: April 06, 2024 Accepted: July 01, 2024 Published: September 10, 2024
Academic Editor: Marco Falasca, Curtin University, Australia
The article belongs to the special issue Mitochondria and Lipid Signalling in Liver Diseases
Lipids are intricate biomolecules responsible for the building up of biological membranes. Besides this structural function, they also display crucial roles in signaling, acting as second messengers that activate specific pathways. Mitochondria are fundamental for cells as they participate in several pivotal functions, such as ATP synthesis, cell survival, metabolic pathways, and calcium homeostasis. Thus, the lipid composition of mitochondrial membranes can affect specific proteins and impact vital functions of mitochondria, such as oxidative phosphorylation and dynamics. The liver possesses a critical function in lipid homeostasis, involving the generation, oxidation, and trafficking of free fatty acids (FFA), triglycerides (TG), cholesterol, and bile acids (BAs). Mitochondria play a key role in lipid storage regulation in hepatocytes, which can control liver function. Their diverse tasks are affected by the lipid composition of mitochondrial membranes, characterized by low cholesterol content and enrichment of specific lipids such as cardiolipin. As mitochondria determine the bioenergetic status of cells and are key regulators of cell viability, alterations of mitochondrial lipid composition can contribute to the induction and progression of chronic diseases, including alcohol-related liver disease (ARLD) and metabolic dysfunction-associated steatotic liver disease (MASLD), two of the most common forms of liver diseases characterized by steatosis, necroinflammation, and fibrosis, which can progress to hepatocellular carcinoma (HCC). Thus, the disruption of lipid metabolism and membrane composition of mitochondria are characteristic features of cancer cells, and altered mitochondrial lipid composition may be a critical player in the progression of chronic liver diseases toward HCC. This review will address the mechanisms whereby alterations of mitochondrial lipid composition lead to the onset and progression of chronic liver diseases. Thus, a better characterization of the alterations of lipid composition in mitochondria may be a crucial step to design strategies and novel therapeutic opportunities for the treatment of MASLD and ARLD.
Mitochondria are intracellular double membrane-bound organelles that supply energy for the intracellular metabolism in eukaryotic cells. Mitochondria are crucial for the assembly of iron-sulfur clusters, calcium homeostasis, and metabolism of carbohydrates, lipids, and proteins. Mitochondria are also pivotal in cellular survival by regulating strategic pathways involved in the intrinsic or death receptor-mediated cell death. Their structure and membrane composition are unique (Figure 1). The existence of double encircling membranes, the outer (OMM) and the inner (IMM) membrane, is the manifestation of the endosymbiotic foundation of mitochondria about two million years ago [1, 2]. The presence of the OMM and IMM defines two different spatial regions: the intermembrane space (IMS) and the mitochondrial matrix, where mitochondrial DNA (mtDNA) and ribosomes are present and participate in the transcription and translation of proteins encoded by mitochondria [3, 4]. The IMS, localized between OMM and IMM, controls important functions, such as protein sorting, redox balance with glutathione (GSH) reduction and oxidation, cytochrome c (Cyt C) release, and apoptotic cascade activation [4, 5]. Distinctive characteristics and particular functions distinguish mitochondrial membranes from other membrane bilayers. Even at the specific contact sites between the OMM and IMM, there exists a different protein and lipid composition [6, 7].
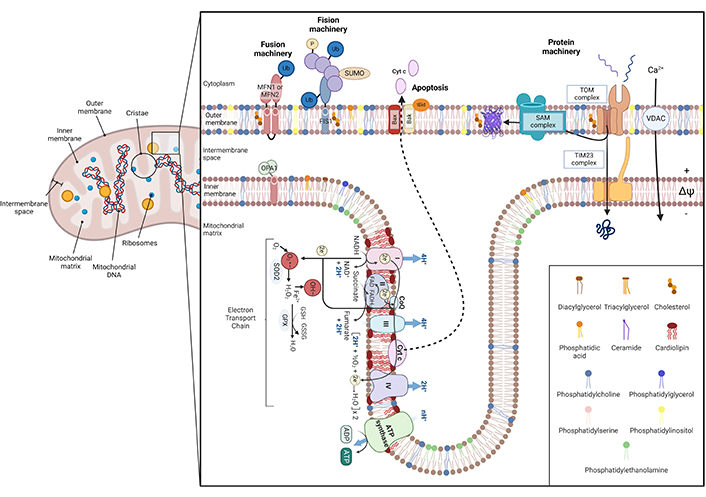
Schematic illustration of mitochondrial architecture and function. Mitochondria are conformed by two bilayers separated by the intermembrane space: the outer mitochondrial membrane (OMM) and the inner mitochondrial membrane (IMM) [1–3]. The IMM surrounds the mitochondrial matrix, where mitochondrial DNA (mtDNA) and ribosomes are located [4], and it includes the inner boundary membrane (IBM) and mitochondrial cristae. Each bilayer has a specific lipid composition and IMM is specially enriched in proteins [7]. Concretely, mitochondrial cristae contain the respiratory chain complexes and the F1F0-ATP synthase, harboring oxidative phosphorylation to obtain energy. The overflow of electrons between each complex generates reactive oxygen species (ROS) that are reduced by mitochondrial antioxidants, such as glutathione (GSH) [8]. ROS can impair mitochondrial constituents, such as mtDNA, phospholipids, and proteins, leading to cytochrome c (Cyt c) detachment from IMM and releasing it into the cytosol via Bax/Bak oligomerization [4, 5, 9]. Furthermore, ROS production can be enhanced by an altered calcium (Ca2+) homeostasis. Mitochondrial dynamics depend on fusion/fission processes. Fusion relies on mitofusin 1/2 (MFN1/2) of the OMM, and optic atrophy protein 1 (OPA1), placed at IMM. In contrast, fission is regulated by dynamin-related protein 1 (Drp-1), which interacts with fission protein 1 (FIS1) [10, 11]. Mitochondria also have a protein machinery to translocate proteins into the matrix. This machinery is composed of the translocase of the outer membrane (TOM) channel the sorting and assembly machinery (SAM) in the OMM and the translocase of the inner membrane (TIM23) in the IBM [12–17]. Ub: ubiquitin; SUMO: small ubiquitin-like modifier; tBID: proapoptotic truncated BID; VDAC: voltage-dependent anion channel; SOD2: superoxide dismutase 2; GPX: glutathione peroxidase; GSSG: glutathione disulfide; O2•−: superoxide; OH–: hydroxyl radical
Note. Adapted from “Electron Transport Chain”, “Mitochondrial Membrane (Phospholipid Bilayers)”, and “Protein Import into the Mitochondria”, by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates
Mitochondrial membranes exhibit predominantly phospholipids, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), and phosphatidylserine (PS), and low levels of sphingolipids and sterols [18–21]. A unique mitochondrial characteristic consists in the presence of cardiolipin (CL), a negatively charged phospholipid located in the IMM, which plays an essential role in the maintenance of membrane integrity and cristae morphology [22]. Its particular conical shape allows for hexagonal structure formation that generates strongly curved areas within the IMM. Moreover, CL is involved in the generation and preservation of protein-protein and protein-membrane interactions [23, 24], conservation of the mitochondrial respiratory chain complexes [25, 26], organization of supercomplexes [27], and assembly of F1F0-ATP synthase dimers. Therefore, compromised CL biosynthesis is linked to damaged cristae morphology and consequently modifies mitochondria shape and dynamics [28–30], respiration, and capability to cope with energy demands through oxidative phosphorylation (OXPHOS) [31, 32]. CL can also flip to the OMM, serving as a signaling molecule to activate mitophagy and apoptotic signaling pathways [33, 34] under stress conditions.
Another feature of mitochondria is the high content of proteins, the majority of which are implicated in OXPHOS and organized in respiratory chain complexes, including the F1F0-ATP synthase and the presence of solute carriers in IMM [35–37]. Most mitochondrial proteins are generated as precursors on cytosolic ribosomes and then transferred and organized into distinct organelle compartments by protein translocases [12–16]. Moreover, mitochondria continuously experience fusion and fission, which implicate transitory disruption of the classical membrane bilayer and mixture of phospholipids. This event promotes the interchange of mitochondrial content, including mtDNA, which is critical for the generation of ATP in the OXPHOS [10, 11]. Besides, the IMM splits into inner borders and cristae to define specific invaginations where the respiratory chain machinery is located, which can undergo extensive transformation to release Cyt C during apoptotic signaling [9]. The mitochondrial electron transport chain (ETC) is one of the principal sources of reactive oxygen species (ROS) [8], principally due to complex I (CI, NADH coenzyme Q reductase) and complex III (CI, ubiquinol Cyt C reductase) activities [38, 39]. During electron trafficking to molecular oxygen (O2) for ATP production in the ETC, electron leakage at these two complexes is accepted by O2, generating superoxide anion (O2•−) as a side product of OXPHOS, that subsequently dismutates to hydrogen peroxide (H2O2) [40, 41].
The endoplasmic reticulum (ER) is responsible for lipid biosynthesis in cells, arising as the main source of essential components of membrane bilayers such as phospholipids and cholesterol, which are distributed to different intracellular compartments, including mitochondria. Unlike other lipid species, CL is synthesized within mitochondria where it plays essential roles in mitochondrial function (see below), Thus, the communication between mitochondria and the ER via mitochondria—ER membrane contact sites is crucial to mitochondrial lipid biosynthesis and calcium exchange [42].
In this review, we will provide evidence for the contribution that alterations in mitochondrial membrane lipid composition have on the development and progression of metabolic liver diseases, including metabolic dysfunction-associated steatotic liver disease (MASLD) and alcohol-related liver disease (ARLD).
Lipid composition influences the biophysical properties of membrane bilayers. In turn, membrane fluidity controls diverse membrane-associated processes, including membrane—protein contacts and function, signal transduction, and membrane fusion and fission events that ultimately regulate organelle biogenesis and growth, dynamics, and vesicular trafficking [43, 44]. In addition, lipids can act as signaling intermediates that trigger specific signaling pathways, as best illustrated by the hydrolysis of sphingomyelin by sphingomyelinases.
Focusing on the molecular organization of membrane lipids, they can generally be classified as phospholipids, sphingolipids, and sterols [45]. Phospholipids, the most abundant lipids in bilayers, are formed by a glycerol backbone with one (or two, in CL) phosphate group(s) together with fatty acid (FA) side chains [46]. The main phospholipids of mitochondrial membranes are PC and PE, accounting for 40–45% and 25–30% respectively of the total lipid composition, with phosphatidylinositol (PI) and CL representing 10–15%, while PS is the less abundant phospholipid (3–5%) [18–21]. The amount of these lipids differs between the IMM and OMM. For example, the IMM is enriched in PE and CL [21, 24].
In contrast, sphingolipids are characterized by a sphingoid backbone formed by the condensation between palmitoyl-CoA with serine [47]. Sphingosine is the singular sphingoid precursor, consisting of the palmitoyl-CoA-serine backbone. Fatty acyl chains of varying length acylate the sphingosine backbone to form sphingolipids of which ceramides are the most intensively studied. Ceramides are a heterogeneous family of sphingolipids that exhibit a large degree of FA of different lengths determined by 6 ceramide synthases (CerS1–CerS6), which exhibit different affinity towards short or long-chain FA. Ceramides, like most sphingolipids, are synthesized in the ER and then undergo exchange and transport to different membrane bilayers in the cell, including mitochondria through the ER-mitochondria contact sites [48, 49].
Not only does the presence of diverse fatty acyl chains with different lengths and saturation regulates the flexibility and stiffness of the bilayer, but also the existence of cholesterol is key in membrane fluidity regulation [50]. For instance, although the amount of cholesterol in mitochondrial membranes is low compared to its abundance in plasma membranes, the OMM is relatively enriched in this sterol compared to IMM.
Fatty acyl chains in the backbone of lipids can be oxidized by lipid peroxidation, which results in membrane bilayer damage thereby affecting cell integrity and organelle performance [51–54]. Oxidative damage to the mitochondrial lipidome induces mitochondrial dysfunction [55]. As mentioned above, CL is crucial for OXPHOS [23, 27, 56, 57], mitochondrial dynamics [58], ATP production, and apoptosis [59, 60]. Due to this key role in mitochondrial physiology, decreased CL levels caused either by low biosynthesis of CL, as in the rare genetic disorder of Barth syndrome, or CL loss by lipid peroxidation due to its susceptibility to ROS attach, compromise mitochondrial function with low OXPHOS activity and ATP generation [61].
Mitochondrial dynamics are controlled by lipid composition via diacylglyceride (DAG), PE, PS, and cholesterol, promoting negative membrane bends essential for mitochondrial fusion [62]. In addition, the import of proteins is pivotal for mitochondrial synthesis and activity. Many mitochondrial proteins are produced as precursors on cytosolic ribosomes and then are transferred to mitochondria. Phospholipids possess important functions in protein trafficking through and into mitochondrial membranes. Concretely, CL, PE, and PC distinctly influence protein translocases of both OMM and IMM, such as the translocase of the OMM (TOM) complex activity, which depends on OMM phospholipids [17], and the dynamic translocase of the inner membrane 23 (TIM23) and S-adenosylmethionine carrier (SAM) complexes. The association of phospholipids-proteins in specific domains displays a central task in the biogenesis and function of mitochondria [63, 64]. Modifications in the content of lipids within the organelle induce expanded, fragmented, and dysfunctional mitochondria, which is associated with diverse diseases [65].
The liver is a central hub for lipid metabolism and the disruption of hepatic lipid homeostasis is a key step in metabolic liver disease development. As liver mitochondria oxidize FA to obtain energy by ETC, the loss of mitochondrial function due to changes in mitochondrial membrane lipid composition can contribute to the accumulation of lipids, resulting in steatosis. This first phase of MASLD can progress to advanced stages of the disease, characterized by mitochondrial dysfunction, generation of oxidative stress, hepatocellular damage, inflammation, and fibrosis. In Table 1, we have summarized recent results on how changes in different lipids (from mitochondrial membranes or as a result of mitochondria-lipid interaction), influence the development of liver diseases.
Changes in mitochondrial lipid composition in different disease models of MASLD, ARLD, and HCC
| Lipids | Mitochondrial changes | Disease outcomes | Disease model | |
|---|---|---|---|---|
| Fatty acids | ↑ mitochondrial fatty acid β-oxidation at early stages | ARLD [66]MASLD [67] | Mice | |
| ↓ mitochondrial fatty acid β-oxidation at advanced stages | ARLD [68–70]MASH [67, 68, 71] | Mice | ||
| ↓ nicotinamide adenine dinucleotide (NAD+/NADH) levels in mitochondria | ARLD [69]MASLD [68] | |||
| ↑ carnitine palmitoyltransferase-1 in mitochondrial membrane | MASLD [67] | Mice | ||
| ↑ lipid peroxidation in mitochondria | ARLD [72]MASLD [71]MASH [71] | RatHepG2 | ||
| ↓ ETC coupling (CI, CIV) | MASLD [71]MASH [67, 71] | Mice | ||
| ↓ mitophagy mediated by NLRP3 activation and AMPK inhibition | MASLD [73, 74] | MiceCells | ||
| ↑ mitochondrial attachment to lipid droplets because of diacylglycerol-O-acyltransferase-2 increased activity | MASLD [67] | |||
| ↑ lipid peroxidation in mitochondria | ARLD [72]MASLD [71]MASH [71] | RatHepG2 | ||
| ↓ mesh due to altered mitochondrial membrane composition | ARLD [75, 76]MASLD [68] | Rat | ||
| Glyceride | Diacylglycerides | ↑ pyroptosis via NLRP3 activation | MASH [77] | MiceHuman |
| Triglycerides | ↑ mitochondrial oxidative flux | MASLD [68] | ||
| ↓ membrane fluidity if the cholesterol/triglycerides ratio is altered | ARLD [76] | Rat | ||
| ↑ tumor anabolism | HCC [78] | |||
| Phospholipid | Cardiolipin | ↑ NLRP3 and apoptosis by CL peroxidation and redistribution from IMM to OMM | ARLD [69, 75, 76]MASLD [68] | Rat |
| ↓ ETC complex activity (CI, CIII, CIV, and ADP/ATP carrier) | ARLD [69]MASLD [67, 71, 79] | Rat | ||
| ↑ mPTP opening and cytochrome c release by Bcl-2 family proteins interaction (Bax) | ARLD [80]MASLD [67, 71]MASH [80] | Rat | ||
| Phosphatidylcholine | ↓ mitochondrial ROS production by CYP2E1 inhibition | ARLD [72] | ||
| ↑apoptosis due to changes in mitochondrial phosphatidylcholine redox state and through JNK activation | ARLD [76]MASLD [68, 79]MASH [68, 77] | MiceRatHuman | ||
| Phosphatidylethanolamine | ↓ membrane fluidity | ARLD [76] | ||
| Sphingolipid | Ceramide | ↑mitochondrial ROS generation and apoptosis by TNFα/Fas signaling | ARLD [70, 75, 81]MASLD [68, 73]MASH [81] | PMH |
| ↓ ETC (CIII) | ARLD [70]MASH [67] | Mice | ||
| ↓ mitochondrial fatty acid β-oxidation | MASLD [68]MASH [67] | Mice | ||
| ↓ mitophagy through NLRP3 activation | MASLD [73] | |||
| ↓ mitochondrial membrane permeabilization | HCC [82] | Cell line | ||
| ↑ mitochondrial depolarization | MASLD [82] | |||
| Ganglioside | ↑ ETC (CIII) | MASH [67] | ||
| Sterol | Cholesterol | ↑ mitochondrial ROS production | ARLD [75, 83]MASH [83] | CellsHuman |
| ↓ ETC (CI) | MASLD [84]ARLD [69]HCC [83] | |||
| ↑survival by a defective assembly of the apoptosome | HCC [80, 83] | Rat | ||
| ↓ mitochondrial membrane permeabilization | ARLD [75, 76, 83]MASLD [84]MASH [67, 83, 85]HCC [80, 83] | HepG2MiceRatsMonkeysHuman | ||
| ↓ mitochondrial protein transport (SLC25A11) by TNFα and Fas-induced apoptosis | ARLD [69, 75, 80, 83, 86]MASH [80, 83, 85, 87] | PMHMiceHuman | ||
| ↑ mitochondrial fusion (megamitochondria) | ARLD [69] | Mice | ||
| ↑ mPTP by JNK-dependent proinflammatory pathway | ARLD [75]MASH [68] | PMH | ||
| ↑ alternative (acidic) bile synthesis pathway | MASLD [84]MASH [88, 89]HCC [88] | PRHMice | ||
| Lipid droplets | ↓ motility and fusion rates of peridroplets mitochondria | MASLD [67] | ||
| ↑ megamitochondria through fusion-fission rates alteration | ARLD [66] | Mice | ||
| ↑ function of cytosolic mitochondria | MASLD [90]HCC [90] | |||
MASLD: metabolic dysfunction-associated steatotic liver disease; ARLD: alcohol-related liver disease; HCC: hepatocellular carcinoma; MASH: metabolic-associated steatohepatitis; ETC: electron transport chain; NLRP3: NLR family pyrin domain containing 3; IMM: inner mitochondrial membrane; OMM: outer mitochondrial membrane; mPTP: mitochondrial permeability transition pore; ROS: reactive oxygen species; JNK: c-Jun N-terminal kinase; TNFα: tumor necrosis factor-alpha
MASLD is a multifaceted spectrum of liver alterations linked to genetic, epigenetic, and environmental risk factors that affect lipid homeostasis, altering the generation, oxidation, and release of free fatty acids (FFA), cholesterol, triglycerides (TG), and bile acids (BAs). First considered as a consequence of “two-hits” in a simplistic initial hypothesis, the pathogenesis of this complex liver disease was later recognized in the “multiple hits” hypothesis in which the accumulation of lipids in hepatocytes in the onset of MASLD sensitizes the liver to the action of several hits [91–93]. Therefore, steatosis can progress to advanced forms, such as metabolic-associated steatohepatitis (MASH), where lipid deposition coexists with inflammation, liver injury, and fibrosis. Then, it can progress to cirrhosis and culminate in hepatocellular carcinoma (HCC) [94, 95]. MASLD is recognized as the most prevalent chronic liver disease worldwide (25% occurrence of normal population) and more than 50% of type 2 diabetic and overweight patients suffer it [96]. Hence, the current definition of MASLD includes steatotic, overweight, type 2 diabetes, or metabolic dysregulated patients [97, 98].
During steatosis, impaired lipid and glucose metabolism arises principally from high-fat diet (HFD) consumption, resulting in diverse lipid accumulation (FFAs, DAG, TG, ceramides, and cholesterol). The complex process associated with mitochondrial proteome alteration and mitochondrial dysfunction is considered a major player in the transition from MASLD to MASH [99]. Mitochondria play a central role in the regulation of lipid metabolism and hence disruption of mitochondrial function contributes to hepatic steatosis [100].
Mitochondrial lipid variation has been described in patients at different stages of the MASLD continuum, with increased CL and ubiquinone in early MASLD but increased acylcarnitine in MASH [101]. The impact of these changes on mitochondrial function during MASLD is not well established. Patients with MASH display reduced activities of the respiratory chain complexes, which correlated with serum tumor necrotic factor-alpha (TNFα), and insulin resistance (IR) [101]. There are findings either indicating impaired respiration or increased mitochondrial mass but lower maximal respiration in obese subjects with MASH, which contrasts with reports of increased hepatic mitochondrial function in the same category of patients [102]. In line with the impaired mitochondrial performance, increased hepatic oxidative stress and oxidative DNA damage have been reported in parallel with reduced antioxidant defense [74, 103].
MASH patients also accumulate microtubule-associated protein 1A/1B-light chain 3 (LC3-II) and sequestosome-1 (p62), a marker of Mallory-Denk bodies, which are associated with disease complications [104]. As mitochondrial quality control is regulated by mitophagy, intervention in this process modulates the implication of mitochondria in MASLD progression. In this regard, knocking down the cell survival regulator macrophage stimulating 1 (MST1), promoted PTEN-induced kinase 1 (PINK1)/Parkin-related mitophagy and reduced HFD-associated liver damage [105]. Besides, optic atrophy type 1 protein (OPA1) inhibition prevented mitophagy intermediates overload and mitigated methionine-choline-deficient (MCD) diet-promoted liver damage [106].
Mitochondrial morphology changes during MASLD in experimental models, with the appearance of fragmented morphology in parallel with Ca2+ increment, reduced OXPHOS activity, and increased ROS generation. Mitochondrial ROS generation has detrimental effects on the development of MASLD [107]. For instance, elevated ROS levels induce the production of cytokines and c-Jun N-terminal kinase (JNK) activation, which plays a feed-forward loop in mitochondrial dysfunction. This scenario leads to impaired β-oxidation, promoting FA accumulation and ATP depletion in hepatocytes while worsening insulin signaling. Damaged mitochondria accumulation within hepatocytes drives necrotic cell death and the leakage of DNA-enhanced mitochondria-induced danger-associated molecular patterns (DAMPs), such as mtDNA, N-formyl peptides, and ATP, which promote the inflammasomes NLR family pyrin domain containing 3 (NLRP3) and absent in melanoma 2 (AIM2) through toll-like receptors (TLRs) recognition [108–110]. It is also important to mention that the translocation of CL to the OMM acts as a docking site to recruit and bind inflammasome components like NLRP3. Furthermore, these signals promote TLR9 activation on Kupffer cells (KCs) and hepatic stellate cells (HSCs), and formyl peptide receptor 1 (FPR1) stimulation, which induces interferon regulatory factor (IRF) and nuclear factor kappa β (NFκβ) action [111]. In turn, the generation of inflammatory cytokines and activation of fibrogenic players establish a chronic inflammatory environment that participates in the progression of MASLD towards MASH and fibrosis (Figure 2). DAMPs have been detected in the plasma of MASH patients, linking mitochondrial dysfunction and inflammation during MASH [112]. Mitochondrial injury also promotes the depletion of nicotinamide adenine dinucleotide (NAD+/NADH) levels, which regulate an adaptive reaction to increased FFA hepatic levels [113] together with an augmented synthesis of mitochondrial free cholesterol and the JNK-dependent proinflammatory routes [114, 115]. The last promotes leakage of mitochondrial components via the mitochondrial permeability transition pore (mPTP) induction and hepatocyte death [116].
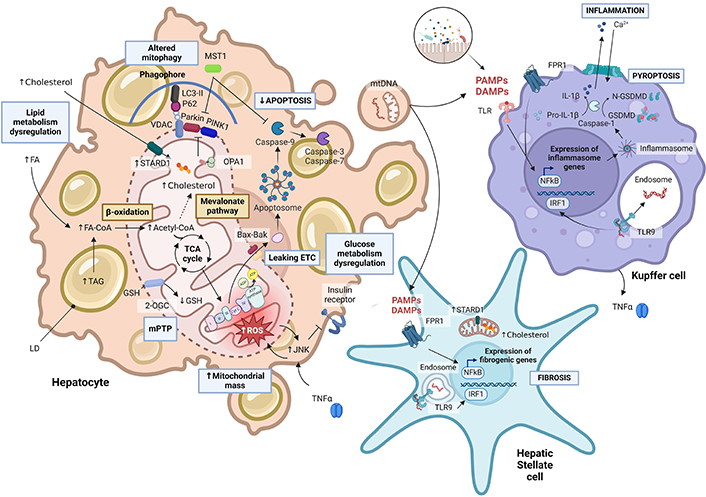
Mitochondrial dysfunction in metabolic-associated steatohepatitis-hepatocellular carcinoma (MASH-HCC) progression. In the cytoplasm, fatty acids bound to CoA (FA-CoA) are transported through the outer (OMM) and inner mitochondrial membranes (IMM), for β-oxidation. The resulting acetyl-CoA is metabolized in the tricarboxylic acid cycle (TCA) or used in the mevalonate pathway to synthesize cholesterol, which can also come from diet and be transported to the mitochondria via steroidogenic acute regulatory protein (STARD1) transporter [87, 88]. STARD1 is not only overexpressed in hepatic cells but also stellate hepatic cells. Changes in the fluidity of the mitochondrial membrane due to cholesterol deposition lead to reduced activity of mitochondrial proteins such as the 2-oxoglutarate carrier (2-OGC). Alterations in OXPHOS, reactive oxygen species (ROS) production [91], the low levels of mitochondrial glutathione (mGSH), and tumor necrosis factor-alpha (TNFα) signaling [101, 117] stimulate c-Jun N-terminals kinase (JNK) action, leading to altered glucose metabolism, apoptosis via mitochondrial permeability transition pore (mPTP) formation [87] and the apoptosome formation by cytochrome c release. In the transition between MASH and HCC, mitophagy is compromised: as the mitochondrial membrane potential shrinks, PTEN-induced kinase 1 (PINK1) recruits Parkin, which ubiquitinates the voltage-dependent anion channel (VDAC) [108–110]. The overexpression of macrophage stimulating 1 (MST1) enhances repressed Parkin, leading to mitochondrial fission activation and mitophagy inhibition [104, 105]. A high-fat diet can lead to intestinal microbiota dysbiosis. By entering the portal circulation, pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) bind to toll-like receptors (TLR) from Kupffer cells and activate nuclear factor kappa B (NFκβ) [108–110, 118]. This initiates the gene transcription of inflammasome components, such as NOD-, LRR-, and pyrin domain-containing protein 3 (NLRP3). Active caspase 1 promotes pro-interleukin-1β (pro-IL-1β) and gasdermin D (GSMD) cleavage into their mature forms. The amino terminal fragments of N-terminal of gasdermin D (GSDMD) form pores at the cell membrane by oligomerization for pyropoptosis. In turn, IL-1β release promotes attraction and activation of further immune cells to the liver as well as stimulates HSC [77, 119, 120]. PAMPs and DAMPs can bind also to formyl peptide receptor 1 (FPR1), stimulating fibrogenic gene transcription. Foam hepatocytes release vesicles containing mitochondrial DNA (mtDNA) and intact mitochondria. Once DAMPs are internalized, TLR9 recognizes mtDNA at the endosomes of Kupffer cells and hepatic stellate cells, inducing proinflammatory cytokines secretion [112, 121, 122], such as TNFα [101] and IL-1β, which can further enhance hepatic damage. LD: lipid droplets; TAG: triacylglycerides; LC3-II: microtubule-associated protein 1A/1B-light chain 3; IRF1: interferon regulatory factor 1; OPA1: optic atrophy type 1 protein; Cyt C: cytochrome c; ROS: reactive oxygen species; ETC: electron transport chain
Note. Adapted from “Suppression of Inflammasome by IRF4 and IRF8 is critical for T cell Priming”, by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates
Concerning the transition from steatosis to MASH, pioneering observations pointed at free cholesterol increment. Its putative trafficking to mitochondria has emerged as an important player as shown in a cohort of patients with established MASH, which exhibited increased free cholesterol content and high expression of the steroidogenic acute regulatory protein (STARD1) compared to patients with simple steatosis [87]. These findings have been identified in the progression of MASLD towards advanced stages like HCC [122–124]. Thus, the cholesterol deposition at IMM by STARD1 has important negative consequences for the mitochondrial status, including the depletion of a crucial antioxidant defense like GSH, which reflects the defect in the activity of the 2-oxoglutarate carrier (2-OGC; SLC25A11) due to its sensitivity to cholesterol-promoted modifications in membrane fluidity [125, 126], and the impairment in the assembly of respiratory supercomplexes and thus OXPHOS [117, 127]. Hence, STARD1 activation reflecting increased mitochondrial cholesterol (mChol) levels [128], account for the mitochondrial GSH (mGSH) depletion found in MASH patients [129, 130], who also presented mitochondria ultrastructural abnormalities [131] and mitochondrial dysfunction [132]. In addition, the accumulation of cholesterol in KCs and HSCs accounts for stimulated inflammation and fibrosis, characteristic of the advanced stages of MASLD [87, 121, 122, 133]. Overall, the evidences in patients and experimental models indicate that the type rather than the amount of fat is a key player in MASLD progression, with the increase in cholesterol and in particular in mitochondrial membrane emerging as a putative new target for intervention to prevent progression of MASLD.
HCC is the main type of liver cancer and the second principal cause of cancer-related demise in the world due to the delay in diagnosis and modest therapeutic strategies, becoming a global health concern [134, 135]. HCC is the final stage of chronic liver diseases caused by several etiologies, including viral hepatitis, ARLD, or MASH [136, 137]. The pathogenesis of HCC is complex and multifactorial and, besides genetic factors and DNA damage, a plethora of players, including lipotoxicity, mitochondrial dysfunction, oxidative stress, inflammation, ER stress, and the disruption in Ca2+ homeostasis induce the perfect milieu for tumor development [136] (Figure 3).
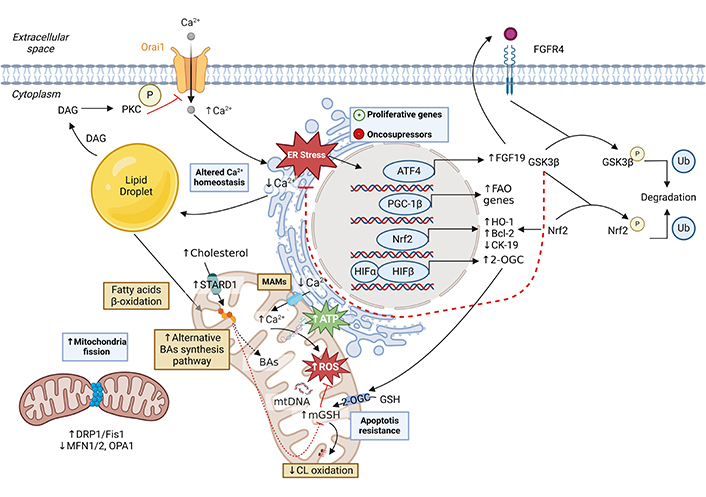
Mitochondrial changes in hepatocellular carcinoma (HCC) development. Lipid accumulation in the liver results in an alteration of Ca2+ homeostasis [138]. The loss of the cation in the endoplasmic reticulum (ER) and consequent increase in the cytoplasm and mitochondrial matrix of the hepatocyte leads to the creation of reactive oxygen species (ROS) that causes mutations in nuclear and mitochondrial DNA (mtDNA), thus activating proliferative genes and inhibiting oncosuppressor genes [138]. Among them, nuclear factor erythroid 2-related factor 2 (Nrf2) activation defends from oxidative stress, reduces cytokeratin 19 (CK-19) expression, and promotes the ER stress response mediated by the fibroblast growth factor 19 (FGF19) pathway [139, 140]. Also, the overexpression of proliferator-activated receptor-gamma coactivator-1 beta (PGC-1β) induces fatty acid oxidation gene expression and promotes tumor growth and anabolic metabolism. The main lipids in HCC are cholesterol and cardiolipin (CL) [78]. Increased cholesterol in the inner mitochondrial membrane (IMM) due to overexpression of the STARD1 transporter, alters membrane fluidity, endowing tumor cells with reduced permeability and thus increased resistance to chemotherapy [84, 141–143]. Furthermore, the acidic pathway of bile acid (BA) synthesis is overactive. Although membrane stiffness affects the activity of the glutathione transporter 2-oxoglutarate (2-OGC), stabilization of hypoxia-inducible factor 1 (HIF-1) promotes its overexpression, thus maintaining glutathione (GSH) levels in the mitochondrial matrix [125, 126]. The high antioxidant capacity of tumor cells prevents oxidation of CL, keeping the electron transport chain stable and preventing the release of cytochrome c [28, 80]. In turn, cholesterol accumulation increases the synthesis of lipotoxic BAs resulting from the acid pathway of BA synthesis in mitochondria [88]. In HCC, mitochondrial dynamics are altered by overexpression of dynamin-related protein 1 (DRP1) and mitochondrial fission 1 protein (Fis1) and decreased expression of mitofusin 1/2 (MFN1/2), leading to increased mitochondrial fission [144–146]. DAG: diacylglyceride; PKC: protein kinase C; MAMs: mitochondria-associated membranes; ATF4: activating transcription factor 4; HO-1: heme oxygenase 1; GSK-3β: glycogen synthase kinase-3 beta; FGFR4: fibroblast growth factor receptor 4; OPA1: optic atrophy type 1 protein; Ub: ubiquitin. Created with BioRender.com
Metabolic variations are abundant in cancer pathogenesis. HCC cells subsist in a potently abundant fat milieu [78, 147]. Peroxisome proliferator-activated receptor gamma co-activator 1β (PGC-1β) controls hepatic oxidative metabolism, mitochondria biogenesis, and antioxidant defense mechanisms. It also plays a pivotal function in cancer progression, supporting metabolic modifications by lipogenic enzyme activation, driving tumor growth and anabolism, and activating gene expression in FA and TG production [78]. Elevated PGC-1β levels activate the expression of ROS scavengers, such as superoxide dismutase (SOD), catalase, glutathione peroxidase (GPX), glutathione reductase (GR), thioredoxin (Trx) and peroxiredoxins (Prx), reducing ROS overload and subsequent apoptosis. In contrast, PGC-1β knockdown protects mice from cancer development [78].
Linked to the progression of MASLD, unphysiological overload of mChol has been found in HCC and has been associated with tumor growth and malignancy [141, 142, 148]. As previously described, mChol promotes a reduction of the mitochondria membrane fluidity [84, 149], increasing the mitochondrial membrane order of HCC cancer cells [143, 150–152]. Paradoxically, this event does not decrease mGSH in cancer cells because of the adaptive overexpression of 2-oxoglutarate transporter (2-OGC or SLC25A11) through hypoxia-inducible factor-1 (HIF-1) stabilization [125, 126, 152]. Consequently, ATP production is supported via OXPHOS and glycolysis [152, 153], which is an intriguing vis-à-vis the reported negative effect of mChol in the assembly of mitochondrial respiratory supercomplexes to support OXPHOS [84]. Therefore, this scenario favors tumor growth by the synergism between protection against mitochondrial OMM permeabilization and defense against oxidative stress [117, 154]. In line with these effects of mChol in cancer cell survival, recent findings indicated that mChol overload promotes sorafenib resistance of HCC cells [155], validating the critical function of mChol in HCC progression.
As alluded to above, CL is vital for mitochondrial ATP production in the respiratory chain and for preserving IMM organization [28, 80]. Oxidative modifications of CL, due to ROS attack to double bonds of its FA constituents, not only influence CI, CIII, and complex IV (CIV) function [156–158] but also regulate programmed cell death by controlling Cyt C leakage and by attaching to the B-cell lymphoma 2 (Bcl-2) family protein Bid to promote Bax and Bak oligomerization and subsequent OMM permeabilization [34, 71, 79, 159, 160]. Thus, the outcome of mChol acting as a proapoptotic versus an antiapoptotic factor in HCC cells depends on the oxidized status of CL, which in turn depends on the mGSH levels. In stages pre-HCC like early MASLD, mChol accumulation causes mGSH depletion and CL peroxidation, which overall contribute to hepatocellular cell death and mitochondrial dysfunction. However, in established HCC, CL is intact due to increased expression of SLC25A11, contributing to apoptosis resistance and tumor growth promotion.
The transport and metabolism of cholesterol in IMM acts as an alternative pathway for the generation of BAs generation in the so-called mitochondrial acidic pathway [161–163]. BAs are synthesized in hepatocytes predominantly by the classic pathway, which is controlled by 7α-hydroxylase (CYP7A1), and to a lesser extent by the mitochondrial alternative pathway, which is determined by the availability of cholesterol in the IMM for its metabolism by sterol 27-hydroxylase (CYP27A1). As the transport of cholesterol to IMM is determined by STARD1, the overexpression of this carrier in MASLD dictates a unique role for STARD1 in stimulating BA synthesis in mitochondria. BAs are not only essential for fat digestion but they also regulate gene expression and promote inflammation. Thus, the expression of STARD1 favors the switch in the synthesis of BAs from the classic to the alternative pathway, and the generation of mitochondrial-derived chenodeoxycholic acid and its taurine forms have been described as a crucial step in the MASLD-driven HCC development [88].
ARLD encompasses a spectrum of hepatic alterations as the result of the ingestion of elevated doses of alcohol that comprises from steatosis, the first stage, to alcoholic hepatitis, cirrhosis, and HCC. Alcohol oxidative metabolism induces several events that are involved in ARLD progression, including the disruption in the balance between fat synthesis and degradation that underlies the onset of steatosis and the alterations in mitochondrial function and morphology.
Hepatic ethanol metabolism initiates with the enzyme alcohol dehydrogenase (ADH) transforming alcohol to acetaldehyde, which is then metabolized to acetate by the acetaldehyde dehydrogenase (ALDH). In addition, alcohol is also metabolized via the microsomal system cytochrome P450, CYP2E1, to acetaldehyde. Although the ADH has a higher affinity for alcohol than CYP2E1, the chronic consumption of alcohol induces the expression of CYP2E1 and becomes the preferential metabolic pathway for the metabolism of alcohol. In turn, acetaldehyde and the derived malonaldehyde can form protein adduct due to their reactivity, which can be enclosed by KCs, endothelial, and HSCs. This activates an inflammatory reaction that contributes to ARLD progression [164]. The molecular mechanisms that promote the deleterious effects of alcohol metabolism to cause ARLD are intricate and multifactorial and include disruption of lipid and methionine metabolism, alterations in mitochondrial dynamics, respiration, membrane structure, mtDNA oxidation, and ROS production [69], whose final impact in ARLD onset is determined by genetic and environmental factors.
Lipid organization influences membrane conformation and modifies the function of proteins embedded in the bilayer. Two major modifications have been documented in the lipid configuration of hepatic mitochondria from rodents fed with ethanol: elevated cholesterol content and depleted CL level. Changes in lipid composition directly affect the physical properties of the bilayer, as exemplified by the relative ratio of PC to PE and particularly by the cholesterol/phospholipid molar ratio, which is a pivotal factor of membrane fluidity. The length and saturation of the fatty acyl chains of the PC/PE molecular species is another factor that regulates physical membrane properties, but especially the increase in cholesterol in a particular bilayer restricts the rotation of acyl chains and determines the fluidity of the bilayer, which in turn can affect the activity of mitochondrial membrane proteins [75]. In this regard, alcohol consumption stimulates cholesterol content in hepatocytes and STARD1 expression, resulting in the trafficking and accumulation of cholesterol in mitochondrial membranes. Interestingly, the increase in the expression of STARD1 occurs in a zonal-dependent fashion with the predominant expression in the perivenous zone of the liver, coinciding with the predominant expression of CYP2E1 and the site of injury as a consequence of alcohol consumption [165]. The zonal-dependent increase in mChol by STARD1 causes mGSH depletion through impairment of the SLC25A11 function and the onset of oxidative stress, thus contributing to the pericentral damage caused by alcohol intake [125, 166]. As mentioned above, although mChol is the precursor for the synthesis of BAs in mitochondria and cholestasis is an accompanying complication of patients with alcoholic hepatitis, it remains to be established whether or not the increase in mChol by alcohol feeding in the pericentral zone contributes to the cholestatic manifestations in ARLD.
Mitochondria are dynamic organelles that relocate in the cytoskeleton and control their morphology and function by a fine-tuned and highly regulated fusion/fission process, which is pivotal for the preservation of mitochondrial tasks and the management of metabolism and cellular signaling [167]. The equilibrium between fission and fusion impacts mitochondrial morphology and adapts their function to diverse stresses. It is also associated with cellular division, apoptosis, and autophagy. Mitochondrial dynamics are regulated by mitochondria-shaping proteins (MSP), of which mitofusin 1 and 2 (MFN1/2) and OPA1 are involved in mitochondrial fusion, while the cytosolic dynamin-related protein 1 (Drp-1) has a crucial role in mitochondrial fission [168–170].
ARLD patients exhibit alterations in the morphology of hepatic mitochondria, with the pioneering description of the presence of megamitochondria (large and elongated mitochondria) as a result of an elevated mitochondrial fusion activity, which triggers mitochondrial elongation and is related to the induction of OXPHOS activity and mtDNA relocation in the mild stage of the ARLD spectrum [171]. In addition, patients with alcoholic hepatitis displayed an elevated expression of Drp-1 in severe stages of the disease, an event that correlates with the data described in human precision-cut hepatic slices incubated with different ethanol concentrations [169]. Thus, chronic alcohol ingestion induces mitochondrial hyperfragmentation through Drp-1 overexpression, promoting more serious hepatic damage [169], pointing at Drp-1 inhibition as a potential therapy to regenerate the balance between mitochondrial dynamics. In line with this possibility, Drp-1 genetic knockdown protected against alcohol-induced hepatotoxicity in VL-17A cells and abolished the growth impairment induced by ethanol exposure. Besides, mice with Drp-1 liver-specific deletion fed the acute-on-chronic alcohol model exhibited less liver injury and the appearance of megamitochondria, with similar findings observed in MASLD following Drp-1 inhibition, which caused decreased steatosis and oxidative stress [170, 172]. Thus, based on this existing evidence it is clear that the presence of megamitochondria is a positive adaptive reaction to alcohol intake and their presence in liver biopsies is a clinical and histological factor linked to a better prognosis in AH patients [173–176]. However, the elimination of megamitochondria via mitophagy is not an efficient process due to the size of this type of mitochondria, raising the question of whether the presence of megamitochondria is a transient process during the early adaptation to alcohol intake or not, which in case of persistence may contribute to mitochondrial maladaptation and disruption in the innate immune reaction that increases liver injury in the late phase of chronic ARLD [177–179]. The influence that the increase in cholesterol accumulation in mitochondria and subsequent change in membrane fluidity has on mitochondrial dynamics in ARLD remains to be fully established.
In comparison with nuclear DNA, mtDNA is highly susceptible to free radical attack since is not protected by histones, and it is located near the IMM, the main cellular ROS source. Mitochondria display several mtDNA copies, and while some degree of mtDNA oxidation can occur, this may not necessarily compromise mitochondrial function, providing that intact copies are still available [180]. Cells mainly remove damaged mtDNA and replicate intact copies to preserve the global mtDNA pool integrity. Thus, a remarkable transient decline of murine hepatic mtDNA was reported within hours after acute ethanol exposure, which was further prominent in older animals. This is consistent with the appearance of mtDNA fragments in the serum of patients with ARLD [181].
Alcohol intake and its oxidative metabolism induce oxidative stress, reflecting the unbalance between ROS generation and its scavenging by compromised antioxidant defense strategies. This disrupts mitochondrial organization and function, and subsequently drives to ROS formation in a vicious cycle of injury, which is more evident with aging. The “mitochondrial theory of aging” proposes that alterations in mtDNA overload compromise cellular energy metabolism, affecting cellular life span [182]. Besides O2•− and H2O2 generated by changes in cholesterol homeostasis in mitochondria by ethanol intake, these species can also generate hydroxyl radical (OH–) by the Fenton reaction, which can attack mtDNA, leading to the release of purine and pyrimidine bases from mtDNA and strand breakdown [183]. Moreover, OH– directly modifies purine and pyrimidine bases, generating 8-oxoguanine (8-oxoG), which is regularly used as an indicator of free radical damage to DNA. It has been demonstrated that 8-oxoG and mtDNA strand break accumulation were remarkably increased in the livers of long-term ethanol-fed rats [184]. Since mtDNA encodes for 13 constituents of the respiratory chain machinery, ethanol-driven mtDNA harm via ROS production can injure the mitochondrial respiratory complexes [185]. Moreover, alcohol impairs mitochondrial ribosomes leading to a reduction in mitochondrial protein synthesis, and ROS can oxidize intramitochondrial proteins [186], contributing to alcohol-induced liver injury.
Damaged mitochondria can activate the cascade of apoptosis and induce an innate and sterile inflammatory reaction by releasing apoptotic factors and/or mtDNA. In turn, mtDNA triggers cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS), which elevates secondary messenger cyclic 2’3’-cGAMP generation that initiates stimulator of interferon genes (STING), leading to IRF3 and IRF7 activation [187, 188]. This cGAS-IRF3 route is activated in experimental ARLD models and is positively associated with disorder severity in ARLD patients [189].
Alcohol-induced structural changes in mitochondria reflect alterations in mitochondrial OXPHOS. Although primary findings in alcohol-fed rat hepatic mitochondria have revealed depleted respiration via the downregulation of CIII and V synthesis [186, 190], recent findings described an opposing effect in mice, in which alcohol intake elevated state III respiration in hepatic mitochondria [66]. Remarkably, this increment in state III was more dramatic in the intragastric ethanol-fed model, which is associated with increased hepatic damage and advanced stage of ARLD, compared to the milder effect observed in oral ethanol consumption models. The impact of alcohol ingestion on mitochondrial performance is linked to the regeneration of NAD+ from NADH oxidation to stimulate oxidative alcohol metabolism. The transfer of electrons from NADH to the respiratory machinery to oxidize it to NAD+ in murine mitochondria stimulates the generation of ROS from the increased consumption of O2 in the ETC. Interestingly, the elevated respiration in mice vs rats correlates with the species-dependent susceptibility to ethanol-induced liver damage. That fact matches the mitochondrial regeneration of NAD+ from NADH, indicating that augmented respiration links ethanol consumption with elevated metabolism and ROS production. The importance of putative variations of mitochondrial respiration to ARLD patients is not well defined. Indirect evidence indicates a reduced mitochondrial function in ARLD patients, who exhibited a decreased peak exhalation of 13CO2 from 2-keto[1-13C]isocaproic acid whereas aminopyrine breath assay and galactose removal capability were not modified [191]. Besides, in patients with high alcohol consumption, ARLD severity was linked to mtDNA fragment expression in hepatic tissue [192, 193]. Hence, these data suggest that ethanol ingestion is related to mitochondrial respiration disturbances, correlating with the severity of ARLD progression.
As alluded to above, a consequence of NAD+ regeneration from NADH oxidation, which is required for endured ethanol metabolism, is the increase in the leakage of electrons transported directly to O2 to produce O2•− [72]. In addition to this event, the consumption of ethanol further increased ROS generation via its metabolism through CYP2E1, as besides ER, it is also found in mitochondria and ethanol consumption induces its expression [194, 195]. In mitochondria, the principal defense against O2•− production is SOD2, which generates H2O2, a powerful oxidant that forms reactive radicals via the Fenton reaction. Mitochondrial H2O2 detoxification can occur via the GSH redox cycle and the Prx-III systems. Reduced GSH level is critical for this GSH redox cycle which requires the participation of GPX-4, while the mitochondrial oxidized form of Prx-III, generated after the reduction of H2O2, is regenerated by Trx2 [196]. Mitochondria do not generate GSH de novo, therefore, they depend on cytosolic GSH to provide mGSH to the mitochondrial matrix. mGSH acts as a cofactor for GPX4 to detoxify H2O2 and other FAs-derived peroxides [190]. As mentioned above, the transport of cytosolic GSH through SLC25A11 is susceptible to changes in cholesterol-dependent modifications in membrane fluidity [190, 196, 197]. Although the relative importance between mGSH/GPX and the Prx-III/Trx2 systems in detoxifying H2O2 remains to be established, both antioxidant systems are interconnected, and mGSH depletion determines the efficiency of the Prx-III/Trx2 pair. Thus, alcohol-induced mChol trafficking may result in the perturbation of membrane physical properties, which can alter efficient antioxidant defense mechanisms, leading to alterations in the mechanisms of mitochondrial respiration.
The ER regulates the transport and maturation of membrane and secretory proteins, post-translational protein processing, and Ca2+ homeostasis. ER stress leads to lipid overload, inflammation, and cell death [198], and has been also described to play a key role in hepatic steatosis via sterol regulatory element-binding proteins (SREBPs) activation (Figure 4). Disturbances in protein folding or modifications in lipid homeostasis lead to the initiation of the unfolded protein response (UPR) that induces the expression of the transcription of chaperones (GRP78/BiP) and the ER-associated degradation (ERAD) mechanisms [199–201] to reestablish homeostasis. Alcohol ingestion promotes ER stress via diverse mechanisms, such as ceramide production via the acid sphingomyelinase (ASMase) activation, the production of acetaldehyde as a result of protein adducts formation in the ER, and also via oxidative stress [202]. A crucial mechanism of ethanol-promoted ER stress is the disruption of methionine metabolism, followed by a rise in homocysteine levels. In this regard, it has been demonstrated that feeding mice with betaine reduced ethanol-induced ER stress, liver steatosis, and hepatic damage [203]. In addition, ER stress can control mitochondrial function. Thus, Ca2+ homeostasis disturbances in the ER affect mitochondria due to the transport of Ca2+, which induces mitochondrial mPTP and cellular damage [70]. ER and mitochondria display physical interaction via mitochondrial-associated membranes (MAMs) defining the boundary of ER and mitochondria, which operate as a channel for the transfer of ions and lipids. In addition, ER stress can regulate cholesterol overload in mitochondrial membranes via STARD1 upregulation [86], a pivotal participant in metabolic hepatic disorders such as MASH and ARLD as mentioned above [87, 88]. Moreover, another connection between methionine metabolism and mitochondrial crosstalk has been revealed in ARLD. Methionine adenosyltransferase 1A (MAT1A) is normally located in the cytosol and nucleus, although new data described MAT1A also present in mitochondria, where it protects mitochondrial proteome and regulates mitochondrial performance [204]. Hepatic tissue from ARLD patients and mice fed alcohol displayed a remarkable reduction in MAT1A localization in mitochondria, facilitated by the isomerase peptidyl-prolyl cis/trans isomerase (PIN1) and the casein kinase (CK2). Prevention of PIN1-MAT1A interaction increases MAT1A levels in mitochondria and protects against alcohol-promoted mitochondrial malfunction and fat storage. However, whether the advantageous outcomes of targeting mitochondrial MAT1A results from SAM local generation to induce stronger methylation and elevated mitochondrial proteins remains to be established vis-à-vis the direct transport of cytosolic SAM to mitochondrial fraction by a transport system that is insensitive to changes in mitochondrial membrane fluidity [205].
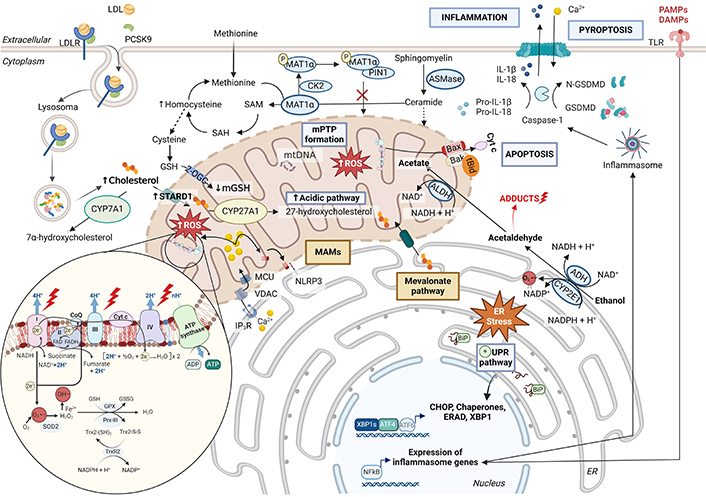
Alterations in mitochondrial function during alcohol-related liver disease (ARLD) development. Alcohol is metabolized by alcohol dehydrogenase (ADH), leading to acetaldehyde and subsequently to acetate by acetaldehyde dehydrogenase (ALDH). CYP2E1 can also metabolize alcohol in the endoplasmic reticulum. This process, together with the adducts formed by acetaldehyde and its by-products, causes high endoplasmic reticulum stress [69, 164]. Also contributing to this phenomenon is the overexpression of acid sphingomyelinase (ASMase) and the consequent increase in ceramides, oxidative stress, and alterations in methionine metabolism [75, 86]. ER stress promotes the activation of the three branches of the unfolded protein response (UPR) and the overexpression of the cholesterol transporter steroidogenic acute regulatory protein (STARD1) [199, 200, 202]. Thus, in alcoholic steatohepatitis, there is an increased accumulation of cholesterol in the inner mitochondrial membrane (IMM). This cholesterol can come from both the diet and the mevalonate pathway. Its deposition in the IMM together with the decrease in cardiolipin causes increased mitochondrial membrane stiffness, reducing the activity of the 2-oxoglutarate carrier (2-OGC) transporter and, consequently, mitochondrial glutathione (mGSH) levels [75, 206]. In turn, cholesterol induces increased bile acid synthesis through the alternative route, further affecting mitochondrial antioxidant levels. The reduction of mGSH levels together with the high production of reactive species due to the alteration of the mitochondrial electron transport chain leads to an alteration of the glutathione peroxidase (GPX)-peroxiredoxin-3 (Prx-III)-thioredoxin (Trx-2) system [190, 196, 197]. In ARLD, the action of peptidyl-prolyl cis/trans isomerase NIMA-interacting 1 (PIN1) and casein kinase 2 (CK2) are reduced and, consequently, methionine adenosyltransferase 1A (MAT1α) is able to metabolize methionine to S-adenosylmethionine (SAM), which is transported to the mitochondria [204–206]. Calcium (Ca2+) homeostasis is altered in this environment and, consequently, mitochondrial permeability transition pore (mPTP) is stimulated. This whole scenario triggers inflammasome activation, IL-1β and IL-18 releasing to recruit immune cells, and leads to a translocation of cardiolipin from the IMM to the outer mitochondrial membrane (OMM) in the mitochondria-associated membranes (MAMs) [120, 207–209]. LDL: low-density lipoprotein; LDLR: LDL receptor; PCSK9: proprotein convertase subtilisin/kexin type 9; SOD2: superoxid dismutase 2; O2•−: superoxide; OH–: hydroxyl radical; CYP7A1: cholesterol 7α-hydroxylase; CYP27A1: cytochrome P450 family 27 subfamily A member 1; VDAC: voltage-dependent anion channel; MCU: mitochondrial calcium uniporter; IP3R: inositol 1,4,5-trisphosphate receptor; NLRP3: NLR family pyrin domain containing 3; SAH: S-adenosylhomocysteine; IL: interleukin; GSDMD: N-terminal of gasdermin D; TLR: toll-like receptor; PAMPs: pathogen-associated molecular pattern molecules; DAMPs: damage-associated molecular patterns; XBP1: X-box binding protein 1; ERAD: endoplasmic-reticulum-associated protein degradation; ATF4: activating transcription factor 4; ATF6: activating transcription factor 4; CHOP: C/EBP homologous protein; Cyt c: cytochrome c
Note. Adapted from “Electron Transport Chain”, “PCSK9 Inhibitors”, and “Suppression of Inflammasome by IRF4 and IRF8 is Critical for T cell Priming”, by BioRender.com (2024). Retrieved from https://app.biorender.com/biorender-templates
Inflammasome is known to be activated in ARLD [207] and it consists of a set of intracellular multiprotein oligomers [NLRP3, caspase-1, interleukin (IL)-1β] found in the cytosol. Inflammasome senses the pathogen-associated molecular patterns (PAMPs) and DAMPs, RNA viruses, pore-forming toxins, cholesterol crystals and uric acid [120, 208], and triggers the generation and release of pro-inflammatory cytokines, as IL-1β and IL-18 [119, 209]. Diverse processes can activate NLRP3 inflammasome at MAMs, such as ROS generation by dysfunctional mitochondria [210], impaired mitophagy [118], mtDNA oxidation [120], and disturbances of the intestinal barrier that trigger the translocation of DAMPs and PAMPs. CL, located in MAMs, flips from the IMM to the OMM triggering NLRP3 initiation [211]. Furthermore, in MAMs, the trafficking of lipids and Ca2+ between ER and mitochondria takes place via voltage-dependent anion channel (VDAC) [212], which enables the NLRP3 inflammasome assembly [73]. Then, the association between ethanol metabolism and NLRP3 initiation is orchestrated via mitochondrial disruption, since mitochondria participate in the oxidative metabolism of ethanol, the initiation of ROS production, and oxidative stress. Thus, the crosstalk between mitochondria and inflammasome promotes ARLD progression and arises as a possible target for therapeutic intervention.
Lipids are intricate biomolecules that determine the structural and physical properties of membranes, which in turn can regulate multiple signaling pathways. The multifaceted mitochondrial double membrane participates in ATP generation, oxidative stress, and cell death regulation. The lipid composition of the mitochondrial membrane, particularly the IMM, is distinctive in performing its numerous functions. Oxidative stress and apoptosis are well-recognized outcomes from the alterations of CL levels and redox status. Hence, genetic disorders disrupting enzymes involved in the synthesis and maturation of CL result in the disruption of mitochondrial organization with defective ATP generation and early cell death. Furthermore, modified fat metabolism and membrane content are critical characteristics of cancer cells. Alterations in mitochondrial membrane lipid configuration and fluidity are hallmarks of solid tumors, and CL with highly saturated acyl chains has been described in cell death-resistant cancer cell lines. In this regard, the mChol pool regulates metabolism and redox biology and contributes to the development of hepatic disorders, such as MASLD, MASH, ARLD, or HCC. Upregulation of STARD1 expression is responsible for the cholesterol transport and accumulation in mitochondria and its expression is induced by ER stress, thus establishing a link between ER stress and mitochondrial dysfunction in metabolic liver diseases, MASLD and ARLD. In the liver, cholesterol accumulation alters mitochondria membrane fluidity and prevents the trafficking of GSH from the cytosol to the mitochondrial matrix promoting oxidative damage through excess ROS formation. mChol also raises BAs synthesis in the mitochondrial alternative pathway, which influences liver tumorigenesis. Thus, although changes in lipid composition can have a significant impact in membrane physical properties, which in turn affect mitochondrial function, the cumulative evidence points to a crucial role for cholesterol accumulation in orchestrating these deleterious effects in mitochondria via impaired antioxidant defense and subsequent ROS generation, affecting essential unique lipid components like CL. In addition to promoting ROS generation, increased cholesterol in mitochondria may fuel the synthesis of oxysterols and BAs, which can contribute to the progression of MASLD to HCC development. In addition, alterations in hepatic mitochondrial lipid composition may also contribute to liver metastatic colonization by invasive cancer cells [213]. Hence, future developments aimed to prevent the increase in mChol to maintain appropriate mitochondrial function may be promising for the treatments/preventions of these prevalent chronic liver diseases.
ARLD: alcohol-related liver disease
BAs: bile acids
CL: cardiolipin
Cyt C: cytochrome c
DAMPs: danger-associated molecular patterns
Drp-1: dynamin-related protein 1
ER: endoplasmic reticulum
ETC: electron transport chain
FA: fatty acid
FFA: free fatty acids
GPX: glutathione peroxidase
GSH: glutathione
H2O2: hydrogen peroxide
HCC: hepatocellular carcinoma
HSCs: hepatic stellate cells
IL: interleukin
IMM: inner mitochondrial membrane
IRF: interferon regulatory factor
KCs: Kupffer cells
MAMs: mitochondrial-associated membranes
MASH: metabolic-associated steatohepatitis
MASLD: metabolic dysfunction-associated steatotic liver disease
MAT1A: methionine adenosyltransferase 1A
mChol: mitochondrial cholesterol
mGSH: mitochondrial glutathione
mtDNA: mitochondrial DNA
NLRP3: NLR family pyrin domain containing 3
O2: molecular oxygen
O2•−: superoxide anion
OMM: outer mitochondrial membrane
OXPHOS: oxidative phosphorylation
PC: phosphatidylcholine
PE: phosphatidylethanolamine
PGC-1β: peroxisome proliferator-activated receptor gamma co-activator 1β
Prx: peroxiredoxins
PS: phosphatidylserine
ROS: reactive oxygen species
SAM: S-adenosylmethionine carrier
STARD1: steroidogenic acute regulatory protein
TG: triglycerides
Trx: thioredoxin
Cartoons in Figures were created with BioRender.com. Carmen Garcia-Ruiz and José C. Fernández-Checa acknowledge the support of the Spanish National Research Council’s Cancer Hub.
LF and LCdlR: Conceptualization, Writing—original draft, Writing—review & editing. JCFC and CGR: Conceptualization, Writing—original draft, Writing—review & editing, Funding acquisition, Supervision. All authors have read and agreed to the published version of the manuscript.
Prof. José C. Fernández-Checa is the Editor-in-Chief of Exploration of Digestive Diseases, and Prof. Carmen Garcia-Ruiz is a member of the Editorial Board and a Guest Editor of Exploration of Digestive Diseases. However, neither was involved in the decision-making or review process for this manuscript.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
We are grateful for the support from grants [PID2020-115055RB-I00], [PID2022-1429560B-I00] and [2023AEP068] from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and the Fondo Europeo de Desarrollo Regional (FEDER) and from the CIBEREHD; as well as support from AGAUR of the Generalitat de Catalunya [SGR-2021-00491], European Cooperation in Science & Technology (COST) [ACTION CA17112], Prospective European Drug-Induced Liver Injury Network, the 2018-102799-T “Enfermedades Metabólicas y Cancer” from the Red Nacional of the Spanish Health Ministry and the COVID grant from the Spanish Association for the Study of the Liver (AEEH). In addition, this project has received funding from the European Horizon’s research and innovation program HORIZON-HLTH-2022-STAYHLTH-02 under agreement No [101095679]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Sanda Win ... Filbert Win Min Aung
Daniel L. Pouliquen
Vicent Ribas
