 Review
Review

Affiliation:
1Internal Diseases Department of Stomatological Faculty, Pavlov First St-Petersburg State Medical University, 197022 St. Petersburg, Russia
2Laboratory of Medico-Social Problems in Podiatry, St. Petersburg State Pediatric Medical University, 194100 St. Petersburg, Russia
3Department of Molecular Microbiology, Institute of Experimental Medicine, 190000 St. Petersburg, Russia
Email: baryshnikova_nv@mail.ru
ORCID: https://orcid.org/0000-0001-7429-0336
Affiliation:
3Department of Molecular Microbiology, Institute of Experimental Medicine, 190000 St. Petersburg, Russia
ORCID: https://orcid.org/0000-0002-2569-6660
Affiliation:
3Department of Molecular Microbiology, Institute of Experimental Medicine, 190000 St. Petersburg, Russia
ORCID: https://orcid.org/0000-0002-9876-6594
Affiliation:
1Internal Diseases Department of Stomatological Faculty, Pavlov First St-Petersburg State Medical University, 197022 St. Petersburg, Russia
4Department of Faculty Therapy Named After V.A. Valdman, St. Petersburg State Pediatric Medical University, 194100 St. Petersburg, Russia
ORCID: https://orcid.org/0000-0001-6434-1267
Affiliation:
3Department of Molecular Microbiology, Institute of Experimental Medicine, 190000 St. Petersburg, Russia
ORCID: https://orcid.org/0000-0003-2312-5589
Explor Dig Dis. 2024;3:414–427 DOI: https://doi.org/10.37349/edd.2024.00058
Received: May 06, 2024 Accepted: July 17, 2024 Published: September 10, 2024
Academic Editor: Tzi-Bun Ng, The Chinese University of Hong Kong, China
The article belongs to the special issue Helicobacter Pylori and Infection: Genomics, Diagnosis, Pathogenesis, Antibiotic Resistance, Microbiota, Cancer, Prevention and Therapeutics
The formation of metabolic changes is based on many factors. In particular, the infectious theory of the development of metabolic “breakdowns” has not lost its relevance. In this regard, many scientists are investigating the role of various microorganisms in the pathogenesis of metabolic syndrome. The review provides the results of current research on the role of Helicobacter pylori (as one of the most well-known and widespread bacterial pathogens) in the pathogenesis of metabolic syndrome. However, the results of scientific work are sometimes contradictory, which dictates the need for novel further research to clarify the characteristics of the influence Helicobacter pylori on the formation of various components of the metabolic syndrome.
The formation of metabolic changes is based on many factors. In particular, the infectious theory of the development of metabolic “breakdowns” has not lost its relevance. In this regard, many scientists are investigating the role of various microorganisms in the pathogenesis of metabolic syndrome. Much attention is paid to the study of the role of Helicobacter pylori (H. pylori) as one of the most well-known and widespread bacterial agents [1–3].
Abdominal obesity serves as a key indicator of metabolic disorders. The potential involvement of an infectious agent in obesity formation warrants examination. Adipose tissue is recognized as an organ that secretes various adipokines, including leptin, angiotensinogen, adiponectin, and resistin, which regulate blood pressure, carbohydrate and lipid metabolism, and gastrointestinal motility. Some researchers suggest that adipose tissue, with its significant volume, synthesizes diverse hormones and cytokines that may exacerbate inflammatory processes, framing obesity as a chronic inflammatory response [4, 5].
Some of the agents that regulate appetite and, consequently, body weight are directly produced along the intestinal tract, including ghrelin and several incretins. The synthesis of these hormones is closely tied to the gastrointestinal tract’s condition. This has prompted interest in the influence of peptides controlling adipostasis on the course of gastroenterological diseases, which can be manifested as either dystrophy or obesity, complicating diagnosis, treatment, and prognosis.
In this context, the impact of H. pylori infection on blood ghrelin levels is particularly significant. Ghrelin, an orexigenic hormone secreted in the stomach, exerts a wide range of effects: It stimulates the secretion of growth hormone, prolactin, and adrenocorticotropic hormone, influences sleep and behavior, boosts appetite, and elevates blood glucose levels [6]. Ghrelin activates neurons in the hypothalamus and arcuate nuclei, promoting a positive energy balance by stimulating food intake and reducing fat utilization. Its effects are mediated through the growth hormone secretagogue receptor, which is widely distributed in tissues.
Low plasma ghrelin levels have been linked to insulin resistance, hypertension, and an increased prevalence of type 2 diabetes. There is also evidence suggesting that ghrelin may impact blood pressure levels. Additionally, ghrelin influences appetite, energy balance, gastric motility, and secretion. Plasma ghrelin levels typically rise approximately twofold before a meal and decrease within an hour afterward, earning it the moniker “empty stomach hormone”. Ghrelin also enhances motility and gastric emptying [7].
Leptin, a hormone primarily secreted by adipocytes and encoded by the ob gene, plays a pivotal role in regulating appetite and maintaining energy balance. Its receptor, known as obR, is expressed in the gastric epithelium. Research has shown that leptin administration can prevent gastric ulcer formation in rats and promote cell growth and reparative processes. Consequently, gastric leptin is believed to contribute to intestinal epithelial homeostasis and cell proliferation. Studies on experimental animals have revealed that leptin not only inhibits ulcer formation but also stimulates the growth of esophageal adenocarcinoma cells, indicating its involvement in maintaining the mucosal homeostasis of the stomach and esophagus, as well as pathological proliferation [8, 9].
Given that the stomach is a major source of circulating ghrelin and plasma leptin, there is growing interest in understanding the impact of H. pylori infection on endogenous peptide levels. Roper et al. [9] investigated leptin and ghrelin levels in the gastric juice of H. pylori-negative and H. pylori-positive patients, suggesting that H. pylori colonization may reduce circulating hormone levels [10]. Other studies have shown differences in ghrelin levels between H. pylori-negative and H. pylori-positive patients, with the lowest concentrations observed in patients with chronic gastritis associated with H. pylori infection [11, 12]. Additionally, patients infected with CagA-negative strains of H. pylori tend to exhibit higher ghrelin levels [10]. These findings support the notion that persistent H. pylori infection, through chronic inflammation and neutrophil activity, damages ghrelin-synthesizing cells, leading to reduced ghrelin levels and appetite loss [13]. Moreover, ghrelin levels have been found to positively correlate with plasma pepsinogen 1 levels and the pepsinogen 1/pepsinogen 2 ratio and inversely correlate with gastritis prevalence, suggesting that atrophic changes in the gastric mucosa and H. pylori infection contribute to these conditions. This decrease in ghrelin levels may be attributed to H. pylori infection and its relationship with the somatostatin-gastrin-acid modulation axis [14].
Several studies have investigated the impact of H. pylori eradication therapy on ghrelin levels. Researchers in Japan found that eradicating H. pylori infection resulted in a higher prevalence of hyperlipidemia and obesity. Kamada and colleagues [15] studied 50 patients with peptic ulcers confirmed to have H. pylori infection histologically, serologically, and via urease breath test. All patients received triple eradication therapy. The control group comprised individuals without peptic ulcer disease with a similar gender distribution. One year after eradication therapy, the patient group exhibited significant increases in body weight, body mass index (BMI), systolic and diastolic blood pressure compared to pre-eradication levels. Additionally, all patients showed significant elevations in total serum cholesterol, fasting glucose, leptin, and uric acid levels post-eradication. However, glycated hemoglobin levels did not change significantly. The incidence of hypercholesterolemia increased from nearly 30% to 58% post-eradication, hypertriglyceridemia from 28% to 44%, and obesity from 12% to 22%, while these parameters remained unchanged in the control group [15].
Other studies have indicated an inverse correlation between serum ghrelin concentrations and the severity of H. pylori-associated gastritis. Long-term eradication therapy (with observation periods ranging from 6 to 12 months) has been linked to a significant increase in BMI and fat mass alongside reductions in circulating ghrelin levels and increases in serum leptin. Thus, eradicating H. pylori infection may enhance appetite and elevate BMI. However, no statistically significant association was established between serum ghrelin levels in H. pylori-positive patients and obesity [11].
Currently, there is no definitive evidence suggesting that H. pylori directly influences obesity development. However, it’s noteworthy that countries with successful H. pylori eradication programs also tend to have higher rates of obesity. Although both obesity and H. pylori infection are prevalent pathologies, some studies suggest that they could be risk factors for cancer development, particularly gastric cancer.
It is known, that high carbohydrate diet, soft drink, and sugar-sweetened beverage consumption are associated with an increased risk of metabolic syndrome and glucose metabolism disorders. But infection agents also can influence metabolism. From a pathogenetic perspective, H. pylori and increased insulin resistance can be seen as two independent factors contributing to metabolic disorders through chronic inflammation, oxidative stress, adipokine dysregulation, and hormonal imbalance (glucagon, catecholamines, glucocorticosteroids). Polyzos et al. [16] proposed an association between H. pylori and insulin resistance in a systematic review. Successful H. pylori eradication was associated with reductions in fasting insulin, total cholesterol, triglycerides, low-density lipoprotein (LDL), and C-reactive protein levels. Dogan et al. [17] reported that successful H. pylori eradication positively impacted the homeostatic model assessment of insulin resistance (HOMA index) in patients with normal blood glucose levels. Chinese researchers identified H. pylori as an independent predictor of hyperglycemia and insulin resistance. Several studies suggested a potential role of H. pylori in type 2 diabetes mellitus development, indicating that H. pylori persistence may exacerbate insulin resistance through inflammasome cytokine activation and significantly worsen glycemic control in diabetic patients [18–22].
A significant trend emerged indicating a higher prevalence of H. pylori infection in patients with type 2 diabetes mellitus compared to non-diabetic individuals. Studies conducted by American and Spanish researchers yielded similar findings, demonstrating a significant association between H. pylori presence and the development of coronary artery disease in individuals with type 2 diabetes mellitus. Moreover, another study suggested that H. pylori infection might contribute to atherosclerosis and ischemic stroke in type 2 diabetes patients, possibly due to elevated cytokine levels. However, there are conflicting studies that have not found a significant link between H. pylori infection and diabetes mellitus [23, 24].
Recent research has highlighted diabetes mellitus as a potential risk factor for H. pylori infection and reduced eradication efficacy of the microorganism. In a study comparing anti-Helicobacter therapy outcomes in patients with and without type 2 diabetes mellitus, eradication success rates were notably lower in diabetic individuals (50%) compared to the control group (85%) (P < 0.001) [25]. Longo-Mbenza et al. [26] also observed that eradication therapy led to a significant reduction in blood serum glucose levels. The diminished efficacy of anti-Helicobacter therapy in diabetic patients may stem from alterations in the gastric mucosa microvasculature, leading to impaired antibiotic absorption. Additionally, frequent antibiotic use in diabetic patients may promote the resistant of H. pylori strains to standard treatment [25]. An alternative treatment approaches (probiotics, autoprobiotics) and vaccination against H. pylori may be warranted in this patient population.
The research results on the relationship between H. pylori persistence and the development of diabetes complications are conflicting. According to some authors, there is no association between H. pylori infection and diabetes complications, such as nephropathy, retinopathy, and microangiopathy. However, other authors have found that the persistence of virulent H. pylori strains, particularly CagA+ strains, is associated with the development of macroangiopathy, neuropathy, and microalbuminuria in type 2 diabetes patients. This could be due to immunologically mediated endothelial damage leading to a systemic immune response to infection, resulting in albumin loss [27, 28]. Based on available data, it can be concluded that the role of H. pylori infection in the pathogenesis of diabetes is not yet fully understood.
A significant association was observed between H. pylori presence and increased systolic blood pressure, decreased high-density lipoprotein (HDL) levels and elevated LDL levels [29, 30]. The impact of H. pylori infection on lipid profiles, with results showing significantly higher levels of triglycerides and LDL in H. pylori-infected patients was investigated in several studies. These findings support the hypothesis of chronic infection influencing the development of atherogenic dyslipidemia, a risk factor for atherosclerosis [31].
In research implemented by other authors, it was obtained that H. pylori infection can induce dyslipidemia characterized by elevated levels of total cholesterol, LDL, apolipoprotein apo-B, and triglycerides, along with decreased levels of HDL and apolipoprotein apo-A1 [32]. Moreover, plasma cholesterol and LDL levels were found to be significantly higher in H. pylori-positive patients with ischemic stroke compared to H. pylori-positive patients without stroke. Thus, chronic H. pylori infection can shift the lipid profile towards an atherogenic direction by influencing proinflammatory cytokines, such as interleukin-1, interleukin-6, interferon-α, and TNF-α, which, in turn, affect lipid metabolism through various mechanisms, including the activation of lipoprotein lipase in adipose tissue and stimulation of free fatty acid synthesis in the liver.
Researchers from Korea, led by Kim et al. [33], also investigated the association between H. pylori infection and lipid profile in the elderly. According to their findings, the presence of H. pylori infection was significantly correlated with increased levels of total cholesterol and LDL (P < 0.05), although there was no significant correlation with triglyceride and HDL levels (P < 0.05) [33].
The impact of H. pylori eradication on the development of dyslipidemia remains controversial. A study by Scharnagl et al. [34] revealed that one year after eradication therapy in patients with duodenal ulcers, there was a significant increase in HDL, apo-A1, and apo-A2 levels. Moreover, eradication of H. pylori in healthy individuals led to elevated HDL levels and reduced LDL levels [34]. Furthermore, six months’ post-successful eradication, significantly lower plasma levels of total cholesterol and LDL were observed compared to H. pylori-positive patients in the control group.
The pathogenetic role of H. pylori in atherosclerosis development is supported by numerous studies [35–38]. In some studies, the microorganism DNA were even detected directly in atherosclerotic plaques [39].
A study by Nikolopoulou et al. [40] unveiled a link between the presence of IgG antibodies against H. pylori and the development of coronary artery atherosclerosis [40]. According to Figura et al. [41], the most plausible theory suggests that a high titer of antibodies to H. pylori CagA promotes atherogenesis. In experimental research, foreign scientists have demonstrated that successful eradication of H. pylori reduces the progression of the atherosclerotic process in H. pylori-infected mice on a high-fat diet [42]. However, some studies do not support an infectious etiology for atherosclerosis development [43], or an acceleration of age-related atherosclerotic changes due to H. pylori persistence [44] and postulate that atherosclerosis development related to lifestyle especially food selection [45].
Numerous studies confirm the pathogenetic role of H. pylori in coronary heart disease (CHD) development [46–49]. Several studies have indicated a significantly higher occurrence frequency of the bacterium in patients with various types of coronary artery disease compared to healthy individuals. For instance, Turkish scientists demonstrated that H. pylori was detected in 86.3% of patients with acute coronary syndrome, 74.6% with stable angina, and 54.5% of subjects in the control group [50]. Researchers from India concluded that elevated levels of C-reactive protein and IgA against H. pylori may serve as markers for assessing the risk of developing coronary artery disease [51]. Particularly high CHD risk is associated with infection by CagA+ strains of H. pylori [52]. Subsequent studies further support a significant direct correlation between H. pylori infection, especially CagA+ strains, and the presence of CHD. Successful eradication of H. pylori was shown to notably decelerate the progression of coronary artery lumen narrowing, possibly due to decreased chronic inflammation severity and reduced release of proinflammatory cytokines [53]. The meta-analysis includes of 9 studies demonstrated that the presence of antibodies against H. pylori CagA correlates with an increased risk of developing coronary acute disease [54]. Additionally, the study by Figura et al. [41] revealed a higher prevalence of antibodies against H. pylori CagA in patients with unstable angina compared to stable angina patients. Furthermore, the presence of antibodies to H. pylori CagA in the blood was associated with an increased incidence of acute coronary syndrome episodes [54]. According to our findings, screening examinations in patients with coronary artery disease revealed signs of gastritis, including atrophy, and H. pylori infection in 99% of patients in this category [55].
The persistence of the microorganism is linked to severe manifestations of coronary artery disease such as myocardial infarction. Italian scientists concluded that H. pylori is significantly more prevalent in patients with myocardial infarction than in the general population [56], a finding supported by other studies [57]. Moreover, research indicated that IgG levels against H. pylori are notably higher in individuals who died from heart disease compared to survivors of myocardial infarction, and in patients with myocardial infarction compared to the control group [58]. According to a meta-analysis by Liu et al. [59], H. pylori infection was associated with an increased risk of myocardial infarction (OR 1.73; 95% CI: 1.37–2.17).
Further investigations are necessary to precisely determine the role of H. pylori in the pathogenesis of ischemic heart disease (IHD), but the concept of the bacterium’s potential mechanisms as an etiopathogenetic factor in the development of this disease is reasonably clear (Figure 1).
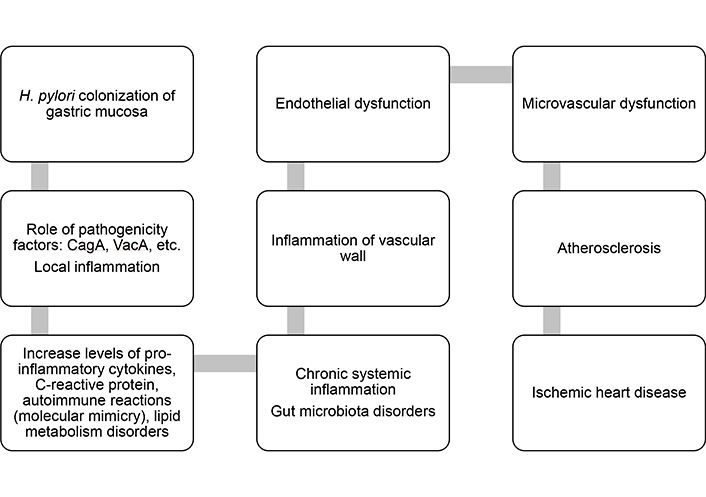
Pathogenetic mechanisms of the influence of Helicobacter pylori (H. pylori) infection on the development of coronary heart disease
Some studies have noted that patients with non-alcoholic fatty liver disease (NAFLD) have a higher titer of antibodies to H. pylori (IgG class), as well as a decrease in adiponectin, an increase in the level of TNF-α, compared with individuals without NAFLD [60]. Subsequently, Polyzos et al. [61] found that in individuals with histologically confirmed non-alcoholic steatohepatitis, against the background of successful eradication of H. pylori, there was a clear tendency to reduce the index of steatohepatitis and fibrosis, as well as the level of homocysteine, transaminases, and erythrocyte sedimentation rate (ESR). H. pylori infection can influence negatively on glucose metabolism, lipid metabolism, and inflammatory cytokines in nonalcoholic fatty liver disease patients [62]. Meta-analysis of Xu et al. [63] shows that H. pylori infection is associated with NAFLD, especially moderate and severe NAFLD.
Quite often, invasion of stomach mucosa with H. pylori infection can be associated with change in the gut microbiota. This can worsen lipid metabolism disorders and accelerate the development of NAFLD (Figure 2).
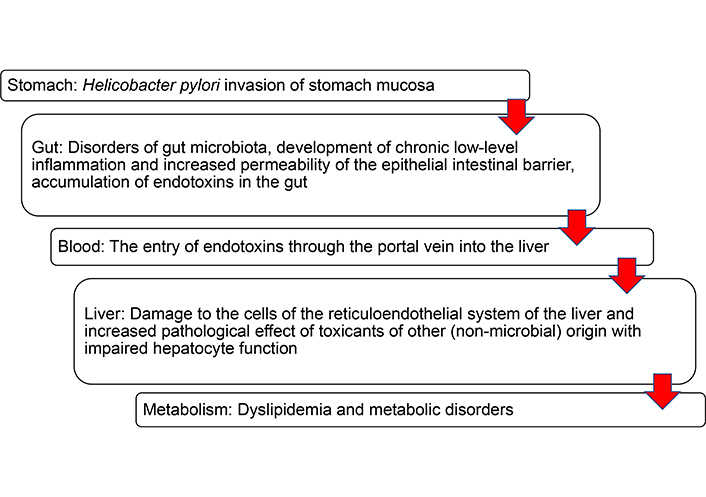
“Steps” in the development of lipid metabolism disorders in Helicobacter pylori-positive individuals with changes in the gut microbiota
The main goal when conducting eradication therapy in patients with metabolic syndrome is high efficiency (the desire to achieve successful eradication using first-line therapy) and safety (taking into account the multimorbidity of this category of patients). In accordance with the Maastricht Consensus VI [64], to increase the effectiveness and safety of treatment, it is recommended to include probiotics, bismuth preparations, and cytoprotectors in treatment regimens together with standard eradication regimens, as well as after their completion.
The inclusion of probiotics in eradication therapy regimens helps to increase the effectiveness of eradication, reduce the frequency and severity of side effects of antibiotics and increase patient adherence to treatment. Potential mechanisms of action of probiotics against H. pylori include not only correction of the intestinal microbiota, but also immunological effects (increasing humoral and cellular immunity, reducing oxidative stress), direct antagonistic effects against H. pylori (such as colonization resistance and bacteriocin synthesis) and stimulation of local immunological protection (strengthening the protective barrier of the mucous membrane and reducing inflammation of the gastric mucosa) [65, 66]. The use of indigenous enterococci (autoprobiotic) orally 50 mL 2 times a day for a course of 20 days in 22 patients infected with H. pylori provided to complete relief of dyspepsia and shown the eradication rate of 80% according to the results of the determination of the antigen of the microorganism in the feces [67]. In addition, the use of autoprobiotic enterococci improved many of metabolic parameters: a decrease in body weight, a decrease in elevated blood sugar levels, an improvement in lipidogram parameters, as well as a decrease in the level of C-reactive protein and proinflammatory interleukin-6 [67].
The use of bismuth tripotassium dicitrate is due to the increasing resistance of H. pylori to clarithromycin. Bismuth drugs are increasingly being included in first-line treatment regimens to improve therapeutic efficacy. They note the absence of primary and secondary resistance of H. pylori to them, as well as a reduced risk of side effects caused by antibiotics, such as diarrhea and intestinal dysbiosis [68]. Bismuth preparations protect cells of the gastric mucosa from the damaging effects of inflammatory products, preventing the progression of gastritis, including post-eradication gastritis [69]. An important positive effect of bismuth preparations is the suppression of lipid peroxidation processes while simultaneously protecting DNA, i.e., bismuth preparations can be considered as “traps” of free radicals [70].
An example of a cytoprotector is rebamipide, which protects the gastrointestinal mucous membrane by stimulating prostaglandin synthesis and inhibiting peroxidation products, proinflammatory cytokines, and chemokines, crucial in metabolic disorders. The drug enhances gastric mucosal blood supply, glycoprotein and bicarbonate synthesis, and gastric epithelial cell proliferation [71, 72]. Rebamipide was demonstrated in 1998 to reduce H. pylori adhesion to gastric epithelial cells in cell culture [73], with subsequent studies confirming its anti-Helicobacter activity [74–76]. Achieving significant regression of gastritis morphological signs with reduced neutrophil and monocyte infiltration due to H. pylori and/or post-infection eradication requires long-term (6–12 months) rebamipide treatment [77, 78].
Even after successful H. pylori eradication in patients with chronic atrophic gastritis, atrophy regression may not occur universally. The Maastricht Consensus III introduced the concept of a “point of no return”, beyond which H. pylori eradication no longer significantly prevents gastric cancer development [79]. Severe atrophy and intestinal metaplasia mark this “point of no return”. According to the Maastricht Consensus VI, patients with high-stage (OLGA/OLGIM III–IV) gastritis and/or extensive endoscopic atrophy remain at risk of gastric cancer development post-H. pylori eradication [80]. Additionally, visceral obesity serves not only as a principal component of metabolic syndrome but also as an independent risk factor for stomach cancer development.
In patients with metabolic syndrome and H. pylori infection, it is crucial to promptly identify atrophy and prescribe medications that promote its regression while aiding in the restoration of gastric mucosal structure and function. Alpha-glutamyl-tryptophan, a stimulator of repair, has showed high efficacy in regressing inflammatory and atrophic changes in H. pylori associated gastritis. Experimental evidence by Ohashi et al. [81] supports its regenerative and anti-inflammatory properties, showing its capacity to activate metabotropic glutamate receptors (mGlu-R-Ia and mGlu-R-III), thereby initiating intracellular regulatory cascades that accelerate cell proliferation and differentiation. Moreover, alpha-glutamyl-tryptophan was observed to expedite intercellular matrix restoration by modulating matrix metalloproteinase activity [82].
A double-blind, placebo-controlled, multicenter study assessing the effectiveness and safety of alpha-glutamyl-tryptophan in chronic atrophic gastritis treatment demonstrated its regenerative effects. Morphometric analysis revealed a 26.1% increase in the number of glands per 1 mm2 of gastric mucosa (Figure 3), restoration of stomach acid production, and normalization of the pepsinogen I/pepsinogen II ratio, indicative of restored gastric mucosal functional activity. Endoscopic and histological assessments further evidenced its anti-inflammatory effects, with reductions observed in inflammatory cell infiltration per 1 mm2 of gastric mucosa (eosinophilic granulocytes: 3 times, neutrophilic leukocytes: 4 times, macrophages: 1.5 times, lymphocytes by 28.2%, plasma cells by 29.6%). Gastroscopic findings revealed a statistically significant decrease in mucosal edema severity (P = 0.008), total acute inflammatory process signs (P = 0.006), and a reduction in negative inflammatory process outcomes (P = 0.038) [83]. Statistically significant correlations (P < 0.05) were also observed between endoscopic inflammation and the number of neutrophilic, eosinophilic granulocytes, macrophages, and lymphocytes per 1 mm2 [83].
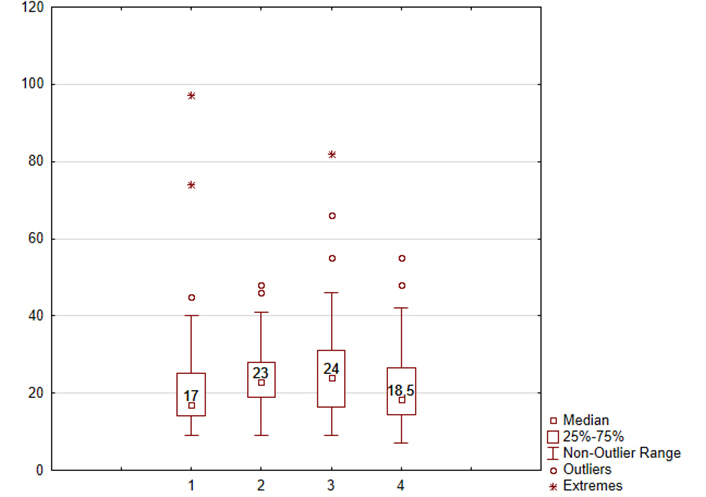
Dynamic of the number of glands per 1 mm2 of the gastric mucosa after intake of the alpha-glutamyl-tryptophan or placebo. Abscissa axis of the treatment group: 1—results of alpha-glutamyl-tryptophan group before treatment; 2—results of alpha-glutamyl-tryptophan group after treatment; 3—results of placebo group before treatment; 4—results of placebo group after treatment. Ordinate axis: the number of glands per 1 mm2 of the gastric mucosa in the atrophic zone. The medians, 25% and 75% quartiles, outliers of values, minimum and maximum values of the indicator are presented. Comparison 1 and 2 points: P = 0.028; comparison 2 and 4 points: P = 0.013
Note. Adapted from “Alpha-Glutamyl-Tryptophan in the Treatment of Chronic Atrophic Gastritis, Associated with Helicobacter pylori” by Uspensky YP, Baryshnikova NV, Fominykh YA, Krasnov AA, Petlenko SV, Apryatina VA. In: Lazăr DC, editor. Helicobacter pylori Infection. London: IntechOpen; 2022 (https://www.intechopen.com/chapters/84437). CC BY.
In conclusion, the evidence presented suggests a hypothesis regarding the involvement of H. pylori infection in the pathogenesis of metabolic syndrome. Furthermore, the role of gastrointestinal tract microbiota must be considered, as normal microbiota is recognized as a key factor in ensuring proper macroorganism function. Alterations in microbiota, particularly with prolonged H. pylori infection, exacerbate metabolic disturbances and disruptions in lipid and carbohydrate metabolism, initiating the development of metabolic syndrome. Numerous studies highlighting the systemic detrimental effects of H. pylori, including on metabolic syndrome target organs, underscore the importance of further investigation in this area. Additionally, research discussing the correlation between H. pylori invasion, gastric mucosa contamination, and intestinal candidal dysbiosis development suggests potential common pathogenetic mechanisms of these microorganisms’ adverse effects on the human body. However, large-scale studies are necessary to validate the infectious (microbial) theory of metabolic disorder onset.
BMI: body mass index
CHD: coronary heart disease
H. pylori: Helicobacter pylori
HDL: high-density lipoprotein
LDL: low-density lipoprotein
NAFLD: non-alcohol fatty liver disease
NVB: Conceptualization, Investigation, Writing—original draft. EIE: Conceptualization, Investigation, Writing—original draft, Validation. GFL: Writing—original draft, Writing—review & editing. YPU and ANS: Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The work was supported by the Ministry of Science and Higher Education of the Russian Federation, No. [075-15-2022-302] (20.04.2022), the Federal State Budgetary Scientific Institution “Institute of Experimental Medicine” (FSBSI “IEM”), and the Scientific and Educational Center “Molecular Bases of Interaction of Microorganisms and Human” of the world class research center “Center for Personalized Medicine” at FSBSI “IEM”. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Luiza Gilmanovna Bektemirova ... Vasiliy Ivanovich Reshetnyak
Elias Kouroumalis ... Argyro Voumvouraki
Rosabel Corrales ... Rafael Llanes
Tatyana Anatolievna Kuchmenko ... Arina Kopaeva
Surbhi Dumra, Abhishek Ray
