 Review
Review

Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB-CSIC), 08036 Barcelona, Spain
2Liver Unit, Hospital Clinic I Provincial de Barcelona, Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
Affiliation:
1Department of Cell Death and Proliferation, Institute of Biomedical Research of Barcelona (IIBB-CSIC), 08036 Barcelona, Spain
2Liver Unit, Hospital Clinic I Provincial de Barcelona, Instituto de Investigaciones Biomédicas August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
3Center for the Study of Liver and Gastrointestinal Diseases (CIBERehd), Carlos III National Institute of Health, 28029 Madrid, Spain
Email: sandra.torres@iibb.csic.es
ORCID: https://orcid.org/0000-0002-2894-3188
Explor Dig Dis. 2024;3:474–503 DOI: https://doi.org/10.37349/edd.2024.00062
Received: October 11, 2024 Accepted: December 01, 2024 Published: December 10, 2024
Academic Editor: Ina Bergheim, University of Vienna, Austria
The article belongs to the special issue Mitochondria and Lipid Signalling in Liver Diseases
Mitochondria are essential organelles responsible for intracellular energy production and play crucial roles in cellular metabolism, inflammation, and apoptosis. Reactive oxygen species (ROS) are primarily produced in the mitochondria and endoplasmic reticulum of hepatocytes due to the activity of cytochrome P450 enzymes. Under ideal conditions, cells have specific molecular mechanisms that manage oxidative stress levels, thus ensuring a balance between oxidants and antioxidants. The interplay between ROS-induced mitochondrial dysfunction and the activation of the NLRP3 (nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3) inflammasome in the context of liver diseases has been extensively studied. However, the exact mechanisms by which mitochondria promote the activation of the NLRP3 inflammasome and contribute to the onset of liver disease remain unclear. This review aims to elucidate the recently discovered mitochondrial regulation of the NLRP3 inflammasome in liver disorders, including alcohol-related liver disease (ALD), metabolic-associated steatotic liver disease (MASLD), and hepatocellular carcinoma (HCC). Finally, it summarizes various natural and pharmaceutical agents that can mitigate liver damage by modulating the activation of the NLRP3 inflammasome through mitochondrial pathways. This work serves as an important resource for identifying new therapeutic approaches and provides further support for advancing the understanding of liver diseases.
Oxidative stress refers to a condition characterized by an imbalance between the generation of free radicals, known as reactive oxygen species (ROS), and the capacity of the body to counteract their effects through antioxidants. Free radicals are molecules that contain oxygen and are highly reactive due to the presence of unpaired electrons. While they are a natural byproduct of cellular metabolism, especially during processes like energy production in the mitochondria, excessive amounts can lead to cellular damage, with oxidative stress damaging proteins, lipids, and DNA [1]. Mitochondria are a major source of ROS during oxidative phosphorylation (OXPHOS) in the electron transport chain (ETC). Key ROS include superoxide anion (O2•−), hydrogen peroxide (H2O2), and hydroxyl radical (•OH) production. Excess ROS can trigger inflammation and activate the NLRP3 (nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3) inflammasome, linking mitochondrial dysfunction and increased ROS to immune responses. Disturbances in the balance of ROS production lead to oxidative stress, which is implicated in the progression of liver diseases, where mitochondrial dysfunction and increased ROS are prominent features [2]. Effective regulation of ROS, mitochondrial function, and related pathways is key to maintaining cellular health and preventing liver damage.
This review summarizes the current knowledge on mitochondrial ROS and the interplay with the NLRP3 inflammasome complex in the context of liver diseases.
ROS are by-products of oxygen metabolism of cell aerobic respiration. They are a family of free radicals, including O2•−, H2O2, •OH, and nitric oxide (NO). NO also belongs to the reactive nitrogen species (RNS) category [3].
ROS can come from exogenous sources (such as radiation, anticancer therapy, smoking, consumption of alcohol, and certain drugs) or endogenous sources. Mitochondria are one of the main endogenous sources of ROS, and specifically, it is produced during OXPHOS in the mitochondrial ETC, where ATP synthesis occurs [4]. The ETC is composed of four protein/cytochrome complexes in the mitochondrial inner membrane: complex I (NADH oxidase) which receives electrons from NADH, complex II (Succinate dehydrogenase) which oxidizes succinate to fumarate in the TCA cycle, complex III (Q-cytochrome c oxidoreductase), and complex IV (cytochrome c oxidase). In addition to these complexes, ubiquinone (UQ) and cytochrome c also participate in electron transport. Ultimately, complex V (ATP synthetase) produces ATP using ADP and inorganic phosphate (Pi) as substrates. Complexes I and III of the ETC are the primary sites where O2•− is formed, and then it is dismutated to H2O2 by superoxide dismutase (SOD) and then to water and oxygen by catalase or glutathione peroxidase (Gpx) [5]. ATP synthesis depends on the presence of oxygen (O2), which facilitates electron transfer reactions through the ETC. Two free radicals (O2•− and H2O2) are formed during the ETC, and they can interact with important biomolecules including DNA, proteins, and membranes [6] (Figure 1).
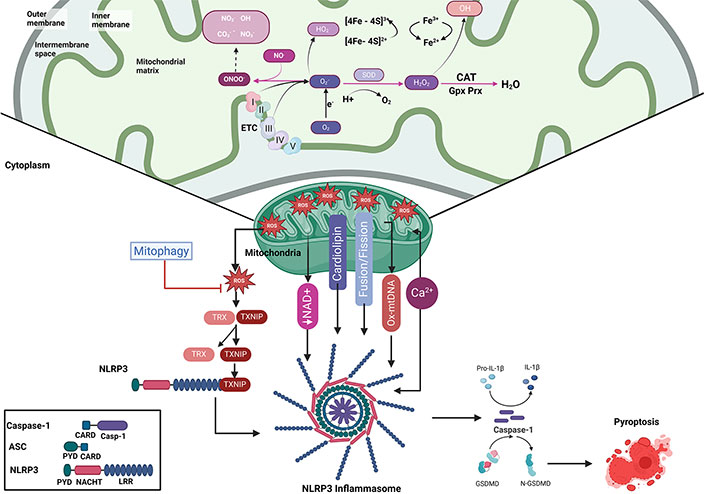
Mitochondrial ROS-induced dysfunction and inflammasome activation leading to pyroptosis. This figure illustrates the role of mitochondrial ROS in promoting mitochondrial dysfunction, inflammasome activation, and eventual pyroptosis. Within the mitochondria, the ETC generates ROS, such as O2•− and H2O2, which can lead to oxidative damage of mitochondrial components, including proteins and lipids. ROS-mediated damage impairs mitochondrial dynamics, reduces NAD+ levels, and disrupts fusion and fission balance, contributing to the release of mtDNA. Furthermore, Ca2+ dysregulation and cardiolipin translocation amplify inflammasome activation, reinforcing mitochondrial dysfunction and promoting cell death. Mitophagy is key in mitigating ROS-induced damage. Excessive ROS promotes the activation of TXNIP, which binds to the NLRP3 inflammasome complex, composed of NLRP3, ASC, and caspase-1. This activation triggers caspase-1 cleavage, leading to the maturation of pro-inflammatory cytokines, such as IL-1β, initiating pyroptosis, a form of inflammatory cell death. ASC: apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; Ca2+: calcium; CARD: caspase activation and recruitment domain; CAT: catalase; CO3−: carbonate radicals; ETC: electron transport chain; Gpx: glutathione peroxidase; GSDMD: gasdermin D; H2O2: hydrogen peroxide; IL-1β: interleukin-1β; LRR: leucine-rich repeat; mtDNA: mitochondrial DNA; NAD+: nicotinamide adenine dinucleotide; PYD: pyrin domain; NLRP3: NACHT-, LRR-, and PYD-containing protein 3; NO•: nitric oxide; O2: oxygen; O2•−: superoxide anion; •OH: hydroxyl radical; ONOO–: peroxynitrite; Ox-mtDNA: oxidized mtDNA; Prx: peroxiredoxin; ROS: reactive oxygen species; SOD: superoxide dismutase; TXNIP: thioredoxin-interacting protein. Created in BioRender. Torres, S. (2024) https://BioRender.com/b36d741
Besides mitochondria, other intracellular compartments are capable of ROS generation. These include the endoplasmic reticulum (ER), which produces ROS during protein folding [7]; peroxisomes, which generates ROS during the β-oxidation of fatty acids [8]; the plasma membrane, by membrane-bound NADPH oxidases (NOXs) during phagocytosis; in the cytosol, by monooxygenases of cytochrome P450 (CYP) superfamily, or other cytosolic enzymes, such as lipoxygenases and cyclooxygenases; or in the extracellular space, via xanthine oxidase.
Among the functions of mitochondrial ROS in physiological processes, they are important players in metabolic adaptation, differentiation, autophagy activation under starvation, and inflammatory cytokine production under pathogen infection. In terms of adaptive signaling, mitochondrial ROS-driven prolyl hydroxylase enzyme (PHD) inhibition stabilizes hypoxia-inducible factor 1 alpha (HIF1α) [9–12] and activates hypoxia by activation of HIF1/2α. Mitochondrial ROS activates cell survival mechanisms as well, such as phosphatase and tensin homolog deleted on chromosome 10 (PTEN), PI3K, protein kinase B (Akt) [13–16], and extracellular signal-regulated kinase (ERK1/2) [17–20]. Interestingly, mitochondrial ROS have also been shown to regulate cellular differentiation and chromatin structure remodeling via the oxidation of architectural chromatin components [21–22].
The equilibrium between ROS production and elimination is critical for maintaining redox homeostasis. Disruption of the redox equilibrium contributes to the pathogenesis of chronic diseases and aging [23, 24]. To counterbalance the excessive ROS production, cells have evolved a complex antioxidant system, which comprises both enzymatic and nonenzymatic mechanisms. The major enzymatic antioxidants are SODs, catalase (CAT) and Gpxs, glutathione-reductase (GR), and superoxide reductases (SOR). Among the most important low molecular weight antioxidants are endogenous, such as glutathione, thioredoxin (Trx), and melatonin, and exogenous antioxidants, such as vitamin C, vitamin E, carotenoids, and flavonoids.
Mitochondria dysfunction has been shown to contribute to the development and progression of many diseases, including metabolic and neurodegenerative diseases. As mentioned before, mitochondria perform multiple functions, such as the regulation of cell death signaling, inflammation, ROS production, and mitochondrial DNA (mtDNA) damage, which are factors associated with the progression of liver diseases, such as alcohol-related liver disease (ALD), metabolic-dysfunction associated steatotic liver disease (MASLD), previously known as non-alcoholic fatty liver disease (NAFLD), and hepatocellular carcinoma (HCC) [25, 26].
ALD is a condition caused by long-term, excessive alcohol consumption that leads to liver damage [27]. It is a major cause of chronic liver disease worldwide. The worldwide prevalence of alcohol use disorder (AUD) stands at 5.1%, affecting approximately 283 million individuals. The highest rates are observed in the European region, where the prevalence is 14.8% for men and 3.5% for women, followed by the Americas, with rates of 11.5% for men and 5.1% for women [28]. ALD encompasses a spectrum of liver conditions, ranging from simple fatty liver (steatosis) to more severe forms such as alcoholic hepatitis, fibrosis, cirrhosis, and even HCC [29]. The progression and severity of ALD depend on the duration and amount of alcohol intake, as well as individual factors like genetics and underlying health conditions.
Alcohol is metabolized partly in the mitochondria, first in the cytosol by the alcohol dehydrogenase (ADH) to acetaldehyde, and later by aldehyde dehydrogenase 2 (ALDH2) in the mitochondria. A part of ADH, in the cytosol ethanol is metabolized by CYP2E1 in the smooth ER [30]. Because of the alcohol metabolism, there is an increase in oxygen consumption and a decrease in ATP production, causing mitochondrial dysfunction. Alcohol-induced mitochondrial ROS formation [31] damages mtDNA and disrupts the ETC, releasing mtDNA [32, 33]. For example, mice fed with an alcohol diet and treated with manganese SOD (MnSOD), a mitochondrial antioxidant enzyme, were able to prevent mtDNA damage [34]. Furthermore, patients with alcohol-related hepatitis and cirrhosis presented mtDNA deletion and fragmentation [35, 36]. Research indicates that both chronic and acute ethanol exposure reduces intracellular antioxidant levels, particularly within mitochondria, leading to an increased susceptibility to liver damage caused by various apoptotic triggers [37, 38].
In terms of mitochondrial respiration, experimental models of alcohol intake are shown to be species-dependent. Several studies in rats fed alcohol orally with Lieber–DeCarli liquid diet containing 36% ethanol for 4 weeks showed decreased mitochondrial respiration in isolated liver mitochondria [39–42]. In contrast to these findings, mice fed alcohol orally in a pair-fed liquid diet [5.4% (w/v) ethanol] or by 4 weeks of intragastric alcohol feeding (alcohol accounted for ∼38.4% of the total caloric intake) exhibited increased respiration in isolated liver mitochondria [43–45] and a higher complex II relative to complex I-driven OCR [46, 47]. Furthermore, in the liver-humanized mouse models, chimeric mice xenotransplanted with human adult hepatocytes exhibited an increase in mitochondrial respiration after chronic and acute alcohol administration (5%, ethanol; 36% calories) [48].
Since the liver has a well-defined zonation, characterized by the perivenous (PV) and periportal (PP) areas, hypoxia is characteristic of the PV zone, and it is aggravated with alcohol intake that increases hepatic oxygen consumption. Therefore, the morphology and functional activity of mitochondria between PP and PV hepatocytes following alcohol feeding differs. Recent findings showed the presence of the mitochondrial cholesterol transporter, steroidogenic acute regulatory protein 1 (StARD1), predominantly in the PV zone, leading to cholesterol accumulation within the mitochondria that translates into a significant depletion of mitochondrial glutathione (mGSH) defense and increased ROS, lipid peroxidation and liver injury characteristic of ALD [49]. In this line, previous findings demonstrated an increased expression of StARD1 in patients with ALD [50]. Interestingly, the increase in mitochondrial function from alcohol-fed mice was predominantly observed in PV compared with PP hepatocytes. Chronic alcohol-fed rats presented reduced mGSH concentration, as happens in HepG2 treated with acetaldehyde, the primary product of ethanol oxidation [51, 52].
Chronic alcohol exposure also alters the mitochondrial structure and dynamics, producing mitochondrial swelling, cristae formation, and hyper-fragmentation of mitochondria in murine models. Research has demonstrated that exposure to ethanol disturbs the mitochondrial fusion-fission equilibrium, resulting in rapid changes to their structure in both in vivo and in vitro models. Conversely, a pioneering work described alterations in the structure of hepatic mitochondria from patients with ALD who develop megamitochondria as an adaptive response in alcohol-induced hepatotoxicity [53–55].
Alcohol exposure disrupts mitochondrial calcium homeostasis, leading to increased calcium uptake, which is linked to elevated mitochondrial ROS production [56]. To address dysfunctional mitochondria, cells typically rely on mitophagy for turnover. However, in alcohol-fed mice, mitophagy is impaired through a Parkin-p53 pathway, exacerbating liver injury [57, 58].
In addition to these findings in ALD, mitochondrial dysfunction is considered a hallmark of MASLD. MASLD is characterized by abnormal fat accumulation in the liver, approximately 15% to 20% of MASLD patients develop metabolic dysfunction-associated steatohepatitis (MASH), characterized by the presence of liver damage, inflammation, and fibrosis, and eventually can progress to cirrhosis [59]. The worldwide prevalence of MASLD has increased from 25.3% from 1990 to 2006 to 38.2% between 2016 and 2019, reflecting nearly a 50% increase in its global prevalence over the last thirty years [60]. Hepatic mitochondria are crucial in the development of hepatic steatosis, as their metabolic processes generate acetyl-CoA, which serves as the fundamental component for the synthesis of lipids and cholesterol in the liver. The accumulation of free fatty acids (FFAs) in hepatocytes predisposes the liver to oxidative stress and cellular damage, leading to increased mitochondrial fatty acid import, β-oxidation to generate acetyl-CoA, and increased hepatic de novo lipogenesis [61].
Mitochondrial dysfunction precedes the development of insulin resistance and hepatic steatosis, playing a pivotal role in the progression of MASLD [62]. This dysfunction serves as a link between insulin resistance, steatosis, oxidative stress, and the dysregulation of gluconeogenesis [63, 64]. In the context of hepatic steatosis, there is a disruption in the ETC, leading to a reduction in the activity of mitochondrial complex III. This impairment results in inhibited ATP synthesis and a disturbance in the TCA cycle [65]. Additionally, there is an increase in mtDNA content and mitochondrial density [66], while the production of mitochondrial ROS is elevated. Targeting ROS inhibition is considered a crucial strategy for reducing lipid accumulation in hepatocytes [67].
Animal models of obesity (ob/ob mice) and obese patients with and without MASLD, showed increased hepatic mitochondrial β-oxidation [68, 69]. Studies in rats fed with a high-fat diet (HFD) or choline-deficient diet (CDD) resulted in increased levels of ROS and reduced antioxidant defenses [70–72]. In patients with MASH, increased ROS levels are linked to greater evidence of tissue oxidative damage, such as lipid peroxidation [35, 69, 73], and significantly reduced SOD activity and mGSH levels [73, 74].
Cardiolipin is one of the lipids from the mitochondrial membrane that changes in experimental models of MASH (HFD-fed mice or diabetes mice), but also in MASH patients [75–78]. Cardiolipin exhibits a significant susceptibility to oxidative damage caused by ROS [75]. In addition, cardiolipin is a hallmark of apoptotic cell death and mitochondrial fission [79]. The depletion of cardiolipin from the inner mitochondrial membrane (IMM) results in total disorganization of mitochondrial respiratory complexes, as cardiolipin is essential for the effective coupling of the respiratory chain with ATP synthase [76, 80]. In CDD-fed rats, the peroxidation of cardiolipin results in reduced complex I activity [81].
In addition to cardiolipin, cholesterol trafficking and its accumulation in the mitochondria also play a role in the pathogenesis of MASH [82, 83]. In genetic models, mice lacking niemann-pick type C1 (NPC1), a late endosomal cholesterol trafficking protein, as well as obese ob/ob mice, exhibit mitochondrial cholesterol accumulation, altered mitochondrial membrane fluidity, mGSH depletion, and increased susceptibility to inflammatory cytokines [84, 85]. In this regard, levels of STARD1 are also increased in human MASH patients, which aligns with the reported increase in mitochondrial cholesterol levels and GSH depletion [73, 86, 87].
As occurs in ALD, murine models of MASLD show increased oxidative mtDNA damage with impaired mtDNA repair mechanisms and disrupted ETC [88]. In MASLD patients have an increase in oxidative mitochondrial damage, which leads to defects in the mitochondrial ETC (complexes I, III, IV, and V), OXPHOS, mitochondrial uncoupling, and ATP production [69, 89–91]. HFD-fed mice have reduced complex IV activity and CDD-fed mice have a significant reduction in subunits of complexes I and IV and ATP content [81, 92, 93].
In addition to changes in OXPHOS, mitochondria from MASH patients present alterations in structure, showing megamitochondria [35, 94–97]. Ultrastructural studies have shown that megamitochondria appear swollen, with a loss of cristae, multilamellar membranes, and paracrystalline inclusions [82, 83]. MASH mice models showed megamitochondria formation and liver damage. Recently, a study performed in mice fed with a methionine-choline-deficient (MCD) diet showed that the MCD diet produces megamitochondria in hepatocytes, and the depletion of OPA1, a mitochondrial dynamin-related GTPase that mediates mitochondrial fusion, decreases mitochondrial size in this megamitochondria-associated MASH mouse model [98].
Another critical mitochondrial process involved in the progression into MASH, is mitophagy, the removal of damaged mitochondria through mitochondrial fission, which is impaired in both patients and experimental dietary models [99, 100], such as HFD-fed mice. Recently, it was shown that the formation of megamitochondria in MASH involves arrested mitophagy intermediates [101]. In addition, HFD-fed mice show increased pro-apoptotic proteins and apoptotic cells that correlate to steatosis and inflammation [100]. In the same line, patients with MASH show increased frequency of apoptotic cells as well as increased pro-apoptotic proteins and Fas expression [102].
HCC is a common form of primary liver cancer and ranks among the foremost causes of cancer-related mortality globally. The prognosis for patients with HCC is poor, with the 5-year relative survival rate being only 18% [103]. In 2018, there were 840,000 cases reported [104]. Asia and Africa have the highest incidence rates worldwide. China has the largest number of cases due to its large population (18.3 per 100,000) and largest population (1.4 billion people), while Mongolia has the highest incidence (93.7 per 100,000) [105]. As a result, the diagnosis and treatment of HCC have become critical focal points for research. Mitochondria are central to HCC progression, affecting energy metabolism, maintaining redox balance, and controlling autophagy and apoptosis.
Aerobic glycolysis is the process by which cancer cells produce more lactate regardless of oxygen availability. Most cancer cells prefer aerobic glycolysis to OXPHOS for glucose metabolism, which is different from normal proliferating cells [106]. The Warburg effect is the name given to this phenomenon [107]. According to Warburg, mitochondrial dysfunction in cancer cells impairs aerobic respiration and increases reliance on glycolytic pathways. The presence of oxygen in healthy cells increases the production of NADH through the TCA cycle, in which NADH acts as an electron donor in the mitochondria, starting the process of electron transfer and ATP synthesis [108]. On the other hand, this electron transfer mechanism is impeded in the anaerobic conditions that are commonly found in tumor cells because oxygen is not present as an electron acceptor [109]. The excessive generation of ROS from this aberrant electron flow within the ETC can lead to oxidative stress, lipid peroxidation, mtDNA damage and mutations, proapoptotic cytokine activation, and a reduction in the effectiveness of OXPHOS. As a result, HCC has fewer copies of mtDNA than normal tissues, which causes mitochondrial dysfunction and tumor growth. Additionally, mtDNA copy number variations and mutations lead to impairments in OXPHOS-mediated ATP production [110]. Nevertheless, many cancer cells have competent OXPHOS activity that can produce ATP, even though the glycolytic phenotype in cancer cells was thought to be caused by defective mitochondrial OXPHOS [111–114]. Moreover, the unfolded protein response (UPR) and worsening of mitochondrial dysfunction can be caused by mtDNA mutation and deletion. Defects in the mitochondrial respiratory system, mtDNA depletion, and mtDNA transcription suppression can therefore promote the development of HCC [115].
Concerning the biology of cancer, aberrant cholesterol metabolism in cancer cells encourages uncontrolled cell division. One molecular factor that specifically coordinates some of these metabolic changes in cancer cells is the accumulation of cholesterol in the mitochondria, which disrupts mitochondrial function [116]. Therefore, the Warburg effect may be partially attributed to mitochondrial cholesterol loading in cancer cells. Paradoxically mitochondrial cholesterol accumulation in cancer cells does not result in mGSH depletion because of compensated overexpression of the SLC25A11 carrier, which counteracts the inhibitory effect of cholesterol, as observed in HCC [117]. Because cholesterol prevents cancer cells from undergoing apoptosis, the adaptive response of HCC cancer cells’ mitochondria to show increased mitochondrial cholesterol loading while maintaining unrestricted mGSH is advantageous for tumor growth.
Mitophagy, the deliberate elimination of mitochondria, is probably a contributing component to the drug resistance of cancer cells. Numerous studies that examined the effect of mitophagy deficiency on carcinogenesis have found that inhibiting mitophagy promotes tumor formation [118]. According to recent research, resistant hypoxia-induced HCC cells have reduced expression of the main mitophagy regulatory gene ATPase family AAA domain-containing 3A (ATAD3A), and resistance to sorafenib necessitates hyperactivated mitophagy [119]. For patients with HCC, restoring these cells’ susceptibility to sorafenib through mitophagy inhibition may provide a novel therapeutic strategy.
Immunity is the body’s sophisticated defense network, designed to identify and neutralize harmful invaders like bacteria, viruses, and damaged cells. At its core, the immune system is divided into two main branches: innate and adaptive immunity [120]. The innate immune system acts as the first responder, rapidly detecting common threats through pattern recognition and initiating an immediate response. In contrast, the adaptive immune system provides a more tailored defense, learning to recognize and remember specific pathogens for future protection [121]. This intricate balance between rapid action and long-term memory keeps the body resilient against external infections and internal disruptions.
Inflammasomes are critical components of the cytoplasmic architecture of immune cells, including macrophages and dendritic cells. These complexes play a pivotal role in the innate immune system by recognizing and responding to cellular stress and danger signals [122]. Central to this process are pattern recognition receptors (PRRs), which mediate the recognition of conserved molecular signatures, known as pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PRRs are categorized based on their location; membrane-bound PRRs include toll-like receptors (TLRs) and C-type lectin receptors (CLRs), and cytoplasmic PRRs include NACHT-like receptors (NLRs) and RIG-I-like receptors (RLRs) [123].
The NLRs, a type of intracellular PRR, share a conserved structure consisting of three key domains. At the C-terminus, the leucine-rich repeat (LRR) domain is responsible for ligand recognition and autoregulation. This domain is linked to a central NACHT domain, which mediates the assembly of the inflammasome, which facilitates nucleotide binding and oligomerization, essential for receptor activation [124]. Phylogenetic analyses of the NACHT domain allowed the classification of NLRs into five distinct subfamilies based on their N-terminal effector domain. These subfamilies include NLRCs (nucleotide-binding oligomerization domain-like receptors caspase activation and recruitment domain-containing proteins), which possess a caspase activation and recruitment domain (CARD); NLRPs, which comprise a pyrin domain (PYD); and NLRPA, NLRPB, NLRPX1, which lack an effector domain. The specific effector domain determines the functional properties and immune response mechanisms of each NLR subfamily [125].
The NLRP proteins, also known as NALPs, are critical components of the innate immune system. The NLRP subfamily is composed of 14 proteins [123, 126]. Among them, NLRP3 was first found in the human brain specifically in microglia, and has been the most extensively studied for its role in responding to different stimuli of TLR, pathogens, proinflammatory cytokines, and intracellular components [127]. The NLRP3 inflammasome is the most extensively characterized member of the NLR family and has been the subject of considerable research in the context of liver disease. Within the liver, NLRP3 is primarily expressed in macrophages and monocytes, while hepatocytes and stellate cells demonstrate comparatively lower levels of NLRP3 expression [128]. The N-terminal PYD of NLRP3 is responsible for initiating immune responses by facilitating the formation of inflammasomes [129].
The NLRP3 inflammasome plays a critical role in orchestrating inflammatory responses. This multiprotein complex is involved in detecting a broad array of PAMPs and DAMPs, leading to the activation of inflammatory cascades and pyroptotic cell death. It is typically composed of a cytosolic NLR sensor, the adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD), and the effector enzyme pro-caspase-1 [127].
The cytosolic sensor NLR, specifically from the NLRP3, detects danger signals (PAMPs and DAMPs) and recruits downstream signaling molecules. Its PYD domain is important for protein-protein interactions, particularly with the adaptor protein ASC, which helps to form the inflammasome complex [127]. Pro-caspase-1 and NLRP3 are connected by ASC, which consists of two domains: CARD, which interacts with pro-caspase-1, and PYD, which binds to NLRP3. Upon the activation of NLRP3 as an inflammasome sensor molecule, ASC oligomerizes to form a large cytoplasmic structure known as the “ASC speck”, which acts as a bridge that connects NLRP3 to caspase-1 through CARD-CARD, PYD-PYD interactions for pro-caspase-1 recruitment and activation. Caspase-1 is a cysteine effector protease synthesized from its inactive precursor (pro-caspase-1). Upon recruitment to the inflammasome complex by ASC, pro-caspase-1 undergoes autocatalytic cleavage to become active [130].
Inflammasomes are activated upon receiving the initial signal, converting caspase-1 to its cleaved form. This activated caspase-1 is responsible for processing pro-inflammatory cytokines, such as pro-IL-1β and pro-IL-18, into their mature forms (IL-1β and IL-18) [123]. Additionally, it activates the cytoplasmic protein gasdermin D (GSDMD), which facilitates pore formation in the plasma membrane. The formation of these pores by GSDMD can lead to pyroptosis, a type of cell death characterized by DNA fragmentation and increased plasma membrane permeability, allowing for the release of IL-1β and IL-18 into the extracellular space [131] (Figure 1).
Inflammasomes can be broadly categorized into canonical and non-canonical inflammasomes based on their activation mechanisms and downstream signaling pathways [132]. The canonical inflammasome involves a sensor protein, an adaptor protein (ASC), and caspase-1, with a well-defined priming and activation pathway. The sensor protein is upregulated through signaling pathways like those involving TLRs and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB). The non-canonical inflammasome includes components like NLRC4 and Pyrin. These are characterized by the direct activation of caspase-11 (or caspase-4/5 in humans), without the need for the ASC adaptor. The sensor protein in this case can sense bacterial flagellin or other specific stimuli. The NLRP3, NLRP1, and absent in melanoma 2 (AIM2) inflammasomes fall into the canonical category, with a well-characterized pathway involving priming and subsequent activation leading to inflammation and pyroptosis [123].
The activation of the NLRP3 inflammasome is a highly regulated, multi-step process that incorporates various cellular stress signals, resulting in the formation of the inflammasome complex and the commencement of an inflammatory response. In response to these triggers, a downstream inflammatory pathway is activated to eliminate microbial infection and repair damaged tissues [133]. This pathway requires two steps: the first step, priming, consists of a transcriptional process that positively regulates NLRP3 and proinflammatory mediators, preparing the cell for the second step, which is the activation of the inflammasome [134].
The priming phase of NLRP3 inflammasome activation, also known as signal 1, is a critical step that prepares immune cells, such as macrophages, for subsequent inflammasome assembly. This phase is initiated by PAMPs, such as bacterial lipopolysaccharide (LPS), or DAMPs released by damaged cells. These signals activate PRRs such as TLRs or NLRs, leading to the activation of the transcription factor NF-κB. NF-κB drives the transcriptional upregulation of NLRP3, pro-IL-1β, and pro-IL-18, the inactive precursors of inflammatory cytokines [133]. Importantly, these components are not constitutively expressed at high levels, so priming ensures their availability in preparation for inflammasome assembly. This step can also involve post-translational modifications like NLRP3 phosphorylation, which further prepares NLRP3 for activation.
While the first stage involves cell priming, a second phase in the inflammasome activation process takes place leading to full activation and inflammasome formation once an NLRP3 activator is recognized [134]. The activation of the NLRP3 inflammasome is triggered by a range of stimuli that induce common intracellular signals, including ionic flux (K+, Cl−, Ca2+), lysosomal damage, and ROS production by damaged mitochondria. These processes collectively lead to the activation of NLRP3, which is crucial for initiating the inflammatory response [133].
The release of K⁺ and Ca2+ mobilization is prompted by stimuli such as ATP binding to P2X7 receptors, nigericin (a K⁺ ionophore), and certain microbial agents [129]. K⁺ outflux from the cytoplasm to the extracellular media occurs through specific ion channels or transporters, resulting in a decrease in intracellular K⁺ concentration that is critical for inflammasome activation, as lower K⁺ levels facilitate the assembly and functional activation of the inflammasome complex by mediating IL-1 maturation of macrophages and monocytes, thereby driving the inflammatory response. On the other hand, Ca2+ mobilization occurs through voltage-dependent anion channels (VDAC) at the mitochondria-associated membranes (MAMs). This mobilization of Ca2+ is achieved by opening the plasma membrane channels or releasing intracellular Ca2+ from the ER. The efflux of K+, which acts as a counter ion, helps the entry of Ca2+ through the plasma membrane channels and promotes the release of Ca2+ stores linked to the ER. Therefore, the release of K⁺ and Ca2+ mobilization are linked to achieving the same goal.
In addition to ion flux, lysosomal disruption acts as a signal for NLRP3 activation. Lysosomes are essential intracellular organelles responsible for degrading biomolecules and maintaining cellular homeostasis by breaking down cellular waste, damaged organelles, and pathogens. Under pathological conditions, such as the phagocytosis of crystals, lysosomal membrane integrity can be compromised, leading to lysosomal membrane permeabilization (LMP). This process results in the release of lysosomal contents, including cathepsins, into the cytoplasm, which serve as critical signals for activating the NLRP3 inflammasome. This activation relies on potassium efflux, suggesting that molecules released during lysosomal damage may interact with the cellular membrane to trigger this process [130].
Mitochondrial dysfunction, often producing ROS, is another key signal for NLRP3 inflammasome activation. ROS can directly or indirectly activate NLRP3 by disrupting mitochondrial integrity. Mitochondrial damage, caused by stressors such as ROS or toxins, can lead to the release of mtDNA into the cytosol. This release is particularly concerning when the mtDNA is oxidized (Ox-mtDNA), as its proximity to the ETC makes it highly susceptible to oxidative damage. The presence of mtDNA outside the mitochondria is recognized as a danger signal by the immune system, triggering the activation of the NLRP3 inflammasome [135]. Additionally, recent studies indicate that priming triggers the synthesis of mtDNA via interferon regulatory factor 1 (IRF1) activation, which is required for full NLRP3 activation [136].
As mentioned before, cardiolipin is a phospholipid crucial for mitochondrial function and highly sensitive to ROS-induced oxidative damage [75, 76]. Moreover, Ca²⁺ influx promotes the translocation of cardiolipin to the outer membrane during mitochondrial stress, especially at MAMs, where cardiolipin’s movement facilitates NLRP3 inflammasome assembly by influencing mitochondrial membrane integrity and signaling [137, 138] (Figure 1). Thus, cardiolipin creates a favorable environment for inflammasome formation and amplifies NLRP3 activation.
Elevated ROS levels play a significant role in the pathogenesis of various liver diseases. The liver is particularly vulnerable to oxidative stress because of its central role in detoxification, metabolism, and exposure to environmental toxins. ROS-driven damage occurs through various mechanisms, such as lipid peroxidation, in which ROS attack cell membranes; DNA damage, leading to mutations; mitochondrial dysfunction, which impairs energy production and amplifies ROS generation; inflammation, by activating signaling pathways that promote inflammatory responses; and fibrosis, where ROS activate stellate cells, contributing to scar tissue formation and liver damage [139]. These processes collectively drive the progression of liver diseases, from initial injury to more advanced stages such as cirrhosis and cancer.
Acetaldehyde, the primary byproduct of ethanol metabolism, has pro-inflammatory and fibrogenic properties that significantly contribute to the development of steatohepatitis and fibrosis in ALD [140]. The progression of ALD involves the activation of a pro-inflammatory cascade that occurs both in the liver and systemically. In the liver, this inflammatory response is initiated by PAMPs from the gut and DAMPs released from injured hepatocytes, which subsequently activate the inflammasome [141]. When alcohol is consumed, it passes into the intestines, where it disrupts the intestinal barrier—a protective layer that blocks harmful substances. This disruption weakens the tight junctions between the epithelial cells, compromising the barrier’s integrity [142]. As a result, LPS, molecules found on the surface of certain bacteria, begin to pass into the bloodstream. The translocation of LPS triggers an immune response, as these bacterial components, recognized as DAMPs, are detected by TLRs. This activation initiates inflammatory signals in the liver [143, 144] (Figure 2).
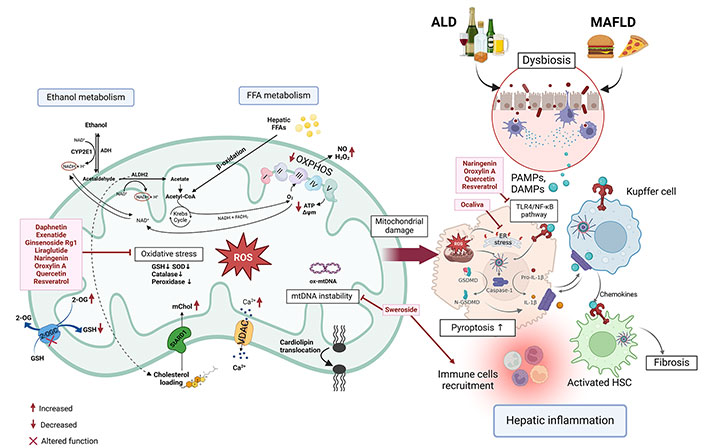
Targeting mitochondrial ROS-induced inflammasome in ALD and MASLD. Ethanol intake, along with diets high in FFA and cholesterol, can compromise the integrity of the intestinal barrier. This disruption allows PAMPs and DAMPs, such as FFA or microbial byproducts, to traverse the impaired tight junctions, thereby stimulating the activation of the NLRP3 inflammasome in hepatic cells. This process initiates the activation of caspase-1, which facilitates the conversion of pro-IL-1β and GSDMD into their active forms. Furthermore, the metabolism of ethanol and FFA in hepatocytes elevates ROS levels within the mitochondria, which subsequently triggers inflammasome activation. The intake of ethanol and fatty diets enhances the transport of cholesterol into mitochondria via the StARD1 protein. Cholesterol increases the membrane rigidity altering the membrane proteins and the antioxidants (GSH) transport into mitochondria to reduce ROS. Ethanol intake and fatty diets increase the cholesterol in mitochondria through StARD1 protein. PAMPs and DAMPs activate KCs and HSCs, leading to the recruitment of immune cells and the onset of hepatic inflammation, which may progress to advanced stages of ALD. CYP2E1: cytochrome P450 2E1; ADH: alcohol dehydrogenase; ALDH2: aldehyde dehydrogenase 2; 2-OG: 2-oxoglutarate; GSH: glutathione; NAD+/NADH: nicotinamide adenine dinucleotide; SOD: superoxide dismutase; mChol: mitochondrial cholesterol; StARD1: steroidogenic acute regulatory protein 1; FFA: free fatty acid; FADH2: flavin adenine dinucleotide; VDAC: voltage-dependent anion channel; ROS: reactive oxygen species; NO: nitric oxide; O2: oxygen; H2O2: hydrogen peroxide; ALD: alcohol-related liver disease; MASLD: metabolic-dysfunction associated steatotic liver disease; PAMPs: pathogen-associated molecular patterns; DAMPs: damage-associated molecular patterns; TLR4: toll-like receptor 4; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; ER: endoplasmic reticulum; IL-1β: interleukin-1β; HSC: hepatic stellate cell; GSDMD: gasdermin D; KCs: Kupffer cells; mtDNA: mitochondrial DNA; Ox-mtDNA: oxidized mtDNA; OXPHOS: oxidative phosphorylation. Created in BioRender. Torres, S. (2024) https://BioRender.com/g11w167
Meanwhile, ethanol is metabolized in hepatocytes by ADH and the enzyme CYP2E1. During this process, CYP2E1 utilizes oxygen to convert ethanol into acetaldehyde, which generates free radicals and ROS as byproducts. The excessive production of ROS, along with inducible nitric oxide synthase (iNOS), induces ER stress and activates inflammatory pathways through the TLR4/MyD88/NF-κB signaling axis, which significantly enhances NLRP3 inflammasome activation [145, 146]. Studies employing the Lieber-DeCarli model demonstrated that mice expressing human CYP2E1 exhibited increased susceptibility to the progression of ALD, displaying more severe liver damage, oxidative stress, inflammation, and mild fibrosis compared to wild-type mice [147].
The development of ALD is driven by various factors, including an imbalance in mitochondrial homeostasis. CYP2E1, an enzyme highly active in hepatocytes, has a strong catalytic capacity for ethanol, leading to excessive lipid accumulation, increased ROS production, mitochondrial dysfunction, and eventually hepatocyte pyroptosis [148]. Moreover, ROS plays a significant role in regulating inflammatory pathways, making them key contributors to the inflammation and progression of ALD [149].
Numerous studies have demonstrated a notable correlation between ALD and increased concentrations of uric acid and ATP. These elevated levels can trigger the activation of the NLRP3 inflammasome within Kupffer cells (KCs), the macrophages that reside in the liver, leading to the activation of caspase-1 [150] (Figure 2). Within these immune cells, NLRP3 activation is associated with intracellular mechanisms involving ROS and spleen tyrosine kinase (SYK) [151]. This inflammatory response is closely linked to chronic alcohol consumption, as ethanol and its metabolites are major contributors to hepatocyte necrosis. When hepatocytes become necrotic, they release ATP, which binds to the P2X7 receptor on KCs, causing K+ efflux and Ca2+ influx. This cascade of events further promotes NLRP3 inflammasome activation, highlighting the intricate relationship between necrosis and inflammation in the context of ALD [152]. Furthermore, alcohol consumption results in the release of extracellular ATP and uric acid from injured liver cells, while simultaneously enhancing the transport of gut-derived LPS to the liver a phenomenon attributed to increased gut permeability, which is partially facilitated by IL-18 [153]. This intricate relationship between the gut and liver, exacerbated by alcohol intake, highlights how alcohol promotes an environment that fosters liver inflammation and damage. Such conditions can elevate levels of pro-inflammatory cytokines, such as tumor necrosis factor alpha (TNF-α) and IL-6, in both clinical patients and experimental models of alcohol exposure. This cascade subsequently triggers the activation of the NLRP3 inflammasome in liver cells, leading to increased production of inflammatory cytokines and further aggravating liver inflammation and injury [147].
Pyroptosis can be triggered by either canonical or noncanonical inflammasomes, primarily through activating proinflammatory caspases, specifically caspase-1 and caspase-11, via the cleavage of GSDMD [128]. This inflammatory response leads to pyroptosis in hepatocytes. Khanova et al. [154] utilized a hybrid-feeding model that combined a Western diet with intragastric ethanol administration to induce alcoholic steatohepatitis (ASH). Their findings indicated an upregulation of caspase-1 and increased processing of GSDMD, underscoring the significant role of pyroptosis in the pathogenesis of alcoholic hepatitis [154].
In the context of ALD, studies have demonstrated a link between pyroptosis in hepatocytes and the activation of the NLRP3 inflammasome. Recent experimental models of ALD have shown a notable decrease in the expression of microRNA 148a (miR-148a) in hepatocytes. This miRNA is regulated by FoxO1, a key metabolic regulator and a tumor suppressor in the liver. This reduction promotes the overexpression of Trx-interacting protein (TXNIP), further activating the NLRP3 inflammasome and leading to pyroptosis [155] (Figure 1).
A key characteristic of MASLD is chronic liver inflammation, which is closely associated to both mitochondrial dysfunction and activation of the NLRP3 inflammasome [156]. Understanding these interconnected processes is essential for unraveling the progression of MASLD and identifying potential therapeutic interventions.
The accumulation of FFAs and triglycerides within liver cells compromises mitochondrial integrity, leading to overproduction of ROS, primarily generated during mitochondrial β-oxidation of fatty acids [157], that consequently leads to inflammasome activation, cellular injury, and eventually fibrosis, hallmarks of MASH [2] (Figure 2). Furthermore, mitochondrial dysfunction initiates ER stress by activating the UPR, a protective mechanism that becomes maladaptive under prolonged stress [158]. This ER stress promotes de novo lipogenesis, worsening hepatic steatosis and accelerating the transition from simple fatty liver to the more inflammatory and fibrotic stages of MASH [159]. The tight relationship between mitochondrial health, oxidative stress, and lipid metabolism underscores mitochondria as a key driver in the pathogenesis of MASLD [160].
The NLRP3 inflammasome is another critical player in the inflammatory response associated with MASLD. Elevated levels of NLRP3 inflammasome components, such as caspase-1, have been detected in MASLD patients, and their presence is strongly correlated with the severity of liver damage [128]. In particular, therapies that modulate cholesterol metabolism, restore mitochondrial integrity, or inhibit receptors, such as the P2X7 receptor, which is a specific NLRP3 activator triggered by potassium efflux, may potentially delay or even reverse MASH progression [161]. Experimental models in mice demonstrate that deletion of the NLRP3 inflammasome reduces hepatic inflammation and fibrosis, highlighting its central role in disease progression. Pharmacological NLRP3 inhibitors like MCC950 have shown promising results in blocking NLRP3 activation, mitigating inflammation, and protecting against liver injury [162]. The intricate link between mitochondrial dysfunction and NLRP3 inflammasome activation represents a crucial pathway in MASLD progression. DAMPs, such as saturated FFAs, further enhance this activation in the early stages of MASH, making the liver more vulnerable to subsequent inflammatory insults, including LPS exposure [150]. Cholesterol overload adds another layer of complexity to this process. Excessive cholesterol, particularly when it accumulates within mitochondria, disrupts mitochondrial membrane integrity and amplifies oxidative stress [163]. In addition, cholesterol crystals can activate the NLRP3 inflammasome within KCs, accelerating the inflammatory process and contributing to fibrosis [164]. Studies have shown that reducing mitochondrial cholesterol or using cholesterol-lowering agents, like statins, can alleviate steatohepatitis in experimental models, further emphasizing the interplay between cholesterol metabolism, mitochondrial function, and NLRP3 activation [165]. However, further research is needed to assess their long-term effects on steatosis and cholesterol deposition.
In mouse models of MASLD induced by an HFD, mitochondrial damage emerges as a critical factor. This damage results in the release of mtDNA into the cytoplasm, which activates inflammasomes and worsens liver inflammation. Notably, mtDNA promotes activation of the NLRP3 and other inflammasome complexes, like AIM2 inflammasome, in liver cells, which, in turn, triggers pyroptosis—a highly inflammatory form of cell death—further aggravating liver pathology [166]. The AIM2 inflammasome, like NLRP3, responds to mitochondrial dysfunction by initiating the formation of inflammasomes, which leads to the maturation and release of pro-inflammatory cytokines, including IL-1β and IL-18 [167]. These cytokines amplify inflammation through pyroptosis, thereby advancing the disease state in MASLD [167].
Mitochondrial dysfunction and NLRP3 inflammasome activation are tightly interconnected processes that drive the progression of MASLD from simple steatosis to MASH [150]. These pathways contribute to inflammation, oxidative stress, and fibrosis—key aspects of the disease. Understanding these mechanisms not only enhances our knowledge of MASLD pathogenesis but also points to potential therapeutic targets. Future research focusing on therapies that improve mitochondrial health or inhibit inflammasome activation holds significant promise for more effective management of MASLD and its complications in clinical practice.
Chronic liver diseases, characterized by sustained inflammation—including those with NLRP3 inflammasome activation—drive the progression from liver damage to HCC. While hepatotropic viruses evade NLRP3 to maintain chronic infections with minimal immune activation, the established HCC environment exhibits active NLRP3 signaling, which paradoxically promotes tumor growth [168]. In HCC, NLRP3 activation drives a pro-inflammatory and pro-tumorigenic environment, supporting cancer cell survival, immune evasion, and angiogenesis [169].
The NLRP3 inflammasome plays a pivotal role in shaping the HCC tumor microenvironment (TME), which is heavily influenced by chronic liver inflammation. The HCC TME consists of diverse cell populations—cancer-associated fibroblasts (CAFs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and regulatory T cells (Tregs)—all of which contribute to immune evasion and tumor progression [169, 170]. Inflammatory cytokines released upon NLRP3 activation can recruit immune cells to the tumor site, where they might support tumor growth and metastasis by promoting an immunosuppressive TME. Elevated levels of IL-1β, for example, have been shown to promote the recruitment of MDSCs, which contribute to chemotherapy resistance.
Recent research has investigated how NLRP3 expression in HCC cells influences NK cell-mediated cancer surveillance, reducing their number and functionality. Findings indicate that knocking out NLRP3 in HCC cells enhances NK cell cytotoxic effects. In line with this data, in xenograft mouse models, NLRP3 knockout in HCC cells was associated with slower tumor progression and metastasis, as well as an increased susceptibility of the tumor to NK cell attacks. Altogether, this suggests that targeting NLRP3 in HCC may boost NK cell immunosurveillance, offering a promising strategy for NK cell-based immunotherapies in liver cancer [171]. In addition to this, NLRP3 activation impacts metabolic pathways within the TME. For instance, it promotes fatty acid oxidation (FAO) in TAMs, fostering an immunosuppressive M2 macrophage phenotype conducive to tumor progression [172]. Furthermore, ROS generated through FAO activate the NLRP3/IL-1β signaling pathway, which is crucial for shaping immune responses within the TME [173]. Further, recent studies indicate that receptor-interacting protein kinase 3 (RIPK3) influences fatty acid metabolism and HCC progression through NLRP3/caspase-1 signaling in TAMs.
On the other hand, other studies suggest that NLRP3 activation has anti-tumor potential in HCC, notably through inducing pyroptosis, which can suppress tumor progression by stimulating the recruitment of NK cells to the tumor site. In this line, in vitro studies indicate that NLRP3 inflammasome activation may inhibit tumor progression by reducing cell proliferation and migration. Specifically, in Bel-7402 and SMMC-7721 HCC cell lines, NLRP3 activation suppresses DNA replication, slows cell proliferation, and induces G1 cell cycle arrest. Combined LPS and ATP treatment significantly reduces the migration of these HCC cells without inducing apoptosis, suggesting that NLRP3 activation may inhibit tumor growth by limiting cancer cell proliferation and migration [173]. Similarly, research suggests that inflammasomes may initially act to minimize tumor growth in response to lactate buildup in the TME. However, tumor cells often adapt by increasing their production of TGF-β, a cytokine that promotes further immunosuppressive mechanisms, thereby counteracting this restriction [174].
Lastly, MiR-223-3p has been shown to regulate HCC by binding to the 3′-untranslated region of NLRP3, thereby suppressing its expression [175, 176]. This suppression of the NLRP3 inflammasome induces apoptosis and inhibits the proliferation of HCC cells [177]. However, this observation contrasts with previous studies suggesting that NLRP3 inflammasome inhibition promotes apoptosis in HCC cells [178].
In summary, the NLRP3 inflammasome plays a complex and dualistic role in HCC progression, acting as both a tumor suppressor and promoter depending on the context [179–181]. In early stages, NLRP3 activation may enhance anti-tumor immunity and tumor cell death, whereas, in established HCC, it can facilitate immune evasion and tumor growth through inflammatory pathways involving NF-κB and ATP. Targeting NLRP3 or its downstream pathways thus presents a promising therapeutic approach to modulate the TME in favor of anti-tumor responses. Context-dependent effects of NLRP3 require tailored therapeutic strategies that account for factors such as HCC stage, underlying liver disease (e.g., viral hepatitis, NAFLD), and the patient’s immune status [178]. Further research is essential to clarify NLRP3’s mechanisms across diverse cell types and disease stages, which could ultimately inform the development of treatments that enhance immune surveillance while minimizing tumor-promoting inflammation in HCC.
Since mitochondria are associated with the regulation of NLRP3 activation, this has led to the development of new methods and drugs that target mitochondria for the treatment of inflammation-associated human pathologies.
Numerous drug candidates have been discovered through chemical synthesis or the analysis of natural products and their derivatives. Several of these drug candidates have demonstrated potential efficacy in treating diseases in both in vitro and in vivo models, and a few have advanced to the clinical trial stage.
From a mechanistic perspective, numerous steps involved in NLRP3 inflammasome activation and signaling present viable targets for pharmacological intervention in NLRP3 inflammasome-associated liver diseases (Figure 2).
Liraglutide, an analog of glucagon-like peptide-1 (GLP-1), has recently been recognized as an effective first-line therapy for type 2 diabetes mellitus (T2DM) and has demonstrated the ability to reduce hepatic steatosis. In a specific mouse model, liraglutide was shown to improve MASLD experimental models and to alleviate hepatitis by inhibiting the activation of the hepatic NLRP3 inflammasome [182]. Additionally, recent findings suggested that liraglutide could mitigate MASLD by regulating the activation of the NLRP3 inflammasome and pyroptosis, thereby influencing mitophagy [183].
Clinical evidence also supports the notion that liraglutide can enhance liver function and promote histological improvement in MASLD patients [184, 185]. These findings suggest that liraglutide may be a viable option for early intervention in patients with T2DM and MASLD as a preventive measure.
Exenatide is classified as a GLP-1 receptor agonist that promotes insulin secretion and is utilized in the management of T2DM. Research has demonstrated that exenatide effectively reduces the severity of MASLD in both in vivo and in vitro settings. Exenatide treatment has demonstrated the ability to enhance the flux of the mitochondrial tricarboxylic acid cycle, while also significantly reducing insulin resistance, steatosis, and lipotoxicity in hepatocytes [186]. Additionally, exenatide mitigates oxidative stress-induced damage and regulates NLRP3 inflammasome activation by modulating mitophagy and autophagy pathways in the liver, providing protective effects in murine models of MASLD and diabetes [186]. In clinical trials, exenatide has been shown to mitigate the pathological symptoms of MASLD in patients with T2DM, promoting weight reduction, partially suppressing inflammation, and hindering the advancement of steatosis [187–189]. Therefore, exenatide may be integrated into personalized treatment strategies for MASLD.
Ocaliva (obeticholic acid) functions as a ligand for farnesoid X receptor (FXR), which is essential for maintaining liver homeostasis and is crucial for cellular and mitochondrial functionality [190]. The expression levels of hepatic FXR are inversely correlated with the activation of the NLRP3 inflammasome in MASLD [191], which can be attributed to hepatitis B virus infection or liver failure in patients, as well as liver injury in murine models [192–195]. FXR has been shown to inhibit the activation of the NLRP3 inflammasome induced by ER stress in hepatocytes, while also downregulating the expression of NLRP3 and TXNIP through the PERK–CHOP signaling pathway [196]. Ocaliva has indeed been utilized in various clinical trials for the treatment of MASLD, showing a significant reduction in fibrosis and an improvement in pathological symptoms among patients with MASH, indicating a potential clinical benefit [197].
In this regard, numerous natural products have been recognized for their ability to influence NLRP3 inflammasome activation, either directly or indirectly, by enhancing mitochondrial function. Among them, ginsenoside Rg1, recognized as the predominant component in ginseng saponin [99], demonstrates protective properties in both in vivo and in vitro experimental models against hepatic steatosis, inflammation, and fibrosis associated with various liver diseases [198–200]. The hepatoprotective mechanisms of Rg1 include the action of antioxidants, the reduction of ROS release, the inhibition of apoptosis-related proteins and inflammatory cytokines, and the suppression of NLRP3 inflammasome activation [201]. More specifically, Rg1 facilitates the blockade of steatosis in hepatocytes, inhibits fibrosis in hepatic stellate cells, and promotes mitophagy in KCs, thereby further inhibiting NLRP3 inflammasome activation [202–205].
Another natural product that protects against liver disease is the polyphenolic plant pigment quercetin. Numerous studies have indicated that quercetin plays a positive role in MASLD, ALD, and T2DM experimental models by modulating the expression of lipid metabolism genes and influencing inflammatory mediators’ release [206, 207]. Recent findings in alcohol-induced acute liver injury in rats showed that quercetin might protect against liver damage by upregulating the expression of HO-1, an Nrf2-dependent gene, thereby inhibiting NLRP3 inflammasome activation and inflammatory cytokine secretion [208]. In mice fed with an MCD diet, quercetin ameliorates liver pathology by facilitating mitophagy through the activation of AMPK [209]. This indicates that enhancing mitophagy through the upregulation of AMPK could serve as a potential therapeutic approach for combating MASLD.
Sweroside, a key ingredient in traditional Chinese medicine for the treatment of hepatitis, demonstrates a notable protective effect against liver injury and fibrosis induced by chemicals [210, 211]. In experimental models of MASLD, sweroside has been shown to enhance the infiltration of hepatic immune cells while also regulating lipid metabolism, liver fibrosis, and inflammatory responses. The mechanism of action for sweroside involves the inhibition of de novo synthesis of mtDNA in the liver, which plays a role in reducing the activation of the hepatic NLRP3 inflammasome and lowering levels of hepatic IL-1β and caspase-1 [212, 213].
Oroxylin A and daphnetin are additional natural compounds recognized for their hepatoprotective properties [214–216]. Both substances demonstrate the ability to mitigate oxidative stress and mitochondrial dysfunction, thereby preventing the activation of the NLRP3 inflammasome, albeit through distinct mechanisms. For instance, in models of ALD, oroxylin A has been found to inhibit the activation of pyroptosis mediated by mitochondrial ROS via the peroxisome proliferator-activated receptor gamma coactivator 1α (PGC-1α)/Mfn2 pathway [217]. Conversely, daphnetin exhibits protective effects in models of drug-induced liver injury by inhibiting the activation of the ROS-TXNIP/NLRP3 inflammasome pathway [218].
Resveratrol, a natural polyphenol with phytoalexin properties, exhibits beneficial effects on lipid accumulation and lipid profiles, enhances insulin sensitivity, and possesses significant anti-inflammatory and antioxidant characteristics, positioning it as a potential hepatoprotective agent for the treatment of MASLD. The administration of resveratrol in a rodent model of HFD-induced obesity has been shown to alleviate hepatic metaflammation and fatty liver, which is associated with changes in NLRP3 inflammasomes and a reduction in pro-inflammatory marker levels [219]. Resveratrol also improves the condition of ALD by reducing the expression of HIF1α and decreasing the production of mitochondrial ROS [220]. However, a meta-analysis involving 302 patients with MASLD across seven randomized clinical trials concluded that the current evidence is inadequate to confirm the effectiveness of resveratrol in managing and treating MASLD [221]. Further investigation in this field is warranted.
Naringenin is a flavonoid predominantly found in high levels within citrus fruits. It has demonstrated protective effects against liver damage, attributed to its anti-inflammatory, anti-fibrotic, antioxidant, and anticancer characteristics [222]. Studies involving naringenin administration have revealed its beneficial impact on the management of MASLD by affecting lipid metabolism, which encompasses the regulation of lipid and cholesterol oxidation, accumulation, and synthesis. Additionally, naringenin decreases the expression of NLRP3 and IL-1β, while also inhibiting the advancement of MASLD by targeting the NLRP3/NF-κB signaling pathway in hepatocytes and KCs [223]. As a result, naringenin is considered a promising candidate for the treatment of liver diseases.
NLRP3 inflammasome activation is acknowledged as a critical contributor to the initiation and advancement of numerous liver diseases. Mitochondria, which are among the most abundant organelles in the liver, are essential in modulating NLRP3 inflammasome activation in the context of liver injury. This modulation takes place through various mechanisms, such as enhancing the release of DAMPs, promoting the recruitment of NLRP3 to mitochondria, inducing mitochondrial calcium overload, increasing mitochondrial ROS production, and the inhibitory effects of mitophagy. The overproduction of ROS and disruptions in OXPHOS play a crucial role in mitochondrial dysfunction, along with oxidative damage that affects both mitochondrial respiratory chain complexes and mtDNA. This damage partially obstructs electron flow, leading to an increased production of mitochondrial ROS, which in turn creates a harmful cycle of escalating damage. Understanding the role of mitochondria in NLRP3 inflammasome activation during liver injury provides valuable insights for developing novel therapeutic strategies targeting NLRP3 inflammasome-related conditions. Both pharmacological derivatives and natural compounds have shown the potential to alleviate liver damage by influencing NLRP3 inflammasome activation through mitochondrial pathways. Studies conducted on human and animal models highlight the promise of new antioxidant strategies to mitigate mitochondrial ROS production, providing a promising approach for the prevention and treatment of liver diseases.
•OH: hydroxyl radical
ADH: alcohol dehydrogenase
AIM2: absent in melanoma 2
ALD: alcohol-related liver disease
ASC: apoptosis-associated speck-like protein containing a caspase activation and recruitment domain
CARD: caspase activation and recruitment domain
CDD: choline-deficient diet
CYP: cytochrome P450
DAMPs: damage-associated molecular patterns
ER: endoplasmic reticulum
ETC: electron transport chain
FAO: fatty acid oxidation
FFAs: free fatty acids
FXR: farnesoid X receptor
GLP-1: glucagon-like peptide-1
Gpx: glutathione peroxidase
GSDMD: gasdermin D
H2O2: hydrogen peroxide
HCC: hepatocellular carcinoma
HFD: high-fat diet
HIF1α: hypoxia-inducible factor 1 alpha
KCs: Kupffer cells
LPS: lipopolysaccharide
MAM: mitochondria-associated membranes
MASH: metabolic dysfunction-associated steatohepatitis
MASLD: metabolic-dysfunction associated steatotic liver disease
MCD: methionine-choline-deficient
MDSCs: myeloid-derived suppressor cells
mGSH: mitochondrial glutathione
mtDNA: mitochondrial DNA
NAFLD: non-alcoholic fatty liver disease
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NLRCs: nucleotide-binding oligomerization domain-like receptors caspase activation and recruitment domain-containing proteins
NLRP3: nucleotide-binding oligomerization domain-like receptor family, pyrin domain containing 3
NLRs: NACHT-like receptors
NO: nitric oxide
O2•−: superoxide anion
OXPHOS: oxidative phosphorylation
PAMPs: pathogen-associated molecular patterns
PP: periportal
PRRs: pattern recognition receptors
PV: perivenous
PYD: pyrin domain
ROS: reactive oxygen species
SOD: superoxide dismutase
StARD1: steroidogenic acute regulatory protein 1
T2DM: type 2 diabetes mellitus
TAMs: tumor-associated macrophages
TLRs: toll-like receptor
TME: tumor microenvironment
Trx: thioredoxin
TXNIP: thioredoxin-interacting protein
UPR: unfolded protein response
HSJ: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. ST: Validation, Writing—review & editing, Supervision. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
We acknowledge the support from the grants [PID2022-140169OB-C22] from Plan Nacional de I+D funded by the Agencia Estatal de Investigación (AEI) and from Beatriu de Pinós Post-doctoral grant [2021 BP00187]; as well as from the CIBER, the intramural project grant for junior researchers [CIBEREHD 2023] and the support from AGAUR of the Generalitat de Catalunya [SGR-2021-00491]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Sanda Win ... Filbert Win Min Aung
Daniel L. Pouliquen
Vicent Ribas
Laura Fàbrega ... Carmen Garcia-Ruiz
