 Review
Review

Affiliation:
1Medical Oncology Department, Hospital Clínic of Barcelona, Translational Genomics and Targeted Therapeutics in Solid Tumors Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, 08036 Barcelona, Spain
ORCID: https://orcid.org/0009-0001-7837-020X
Affiliation:
1Medical Oncology Department, Hospital Clínic of Barcelona, Translational Genomics and Targeted Therapeutics in Solid Tumors Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, 08036 Barcelona, Spain
Affiliation:
1Medical Oncology Department, Hospital Clínic of Barcelona, Translational Genomics and Targeted Therapeutics in Solid Tumors Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, 08036 Barcelona, Spain
Affiliation:
2Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), 28029 Madrid, Spain
3Department of Biochemistry and Molecular Biomedicine and Institute of BIomedicine (IBUB), Faculty of Biology, University of Barcelona, 08028 Barcelona, Spain
4Gastrointestinal and Pancreatic Oncology Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
Email: martacascante@ub.edu
ORCID: https://orcid.org/0000-0002-2062-4633
Affiliation:
1Medical Oncology Department, Hospital Clínic of Barcelona, Translational Genomics and Targeted Therapeutics in Solid Tumors Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), University of Barcelona, 08036 Barcelona, Spain
2Centro de Investigación Biomédica en Red de Enfermedades Hepáticas y Digestivas (CIBEREHD), Instituto de Salud Carlos III (ISCIII), 28029 Madrid, Spain
4Gastrointestinal and Pancreatic Oncology Group, Institut D’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), 08036 Barcelona, Spain
Email: jmaurel@clinic.cat
ORCID: https://orcid.org/0000-0002-9413-5592
Explor Dig Dis. 2025;4:100564 DOI: https://doi.org/10.37349/edd.2025.100564
Received: December 06, 2024 Accepted: February 08, 2025 Published: February 18, 2025
Academic Editor: Evgeny Imyanitov, Saint Petersburg State Pediatric Medical University, Russian Federation
The article belongs to the special issue Immunotherapy for Cancer of Digestive System
Microsatellite unstable (MSI) colorectal cancer (CRC) tumors have a high mutational load (particularly frame-shift mutations) that creates numerous neoantigens that are presented to major histocompatibility complex molecules and recognized by T cells. Consequently, MSI tumors have a higher presence of tumor-infiltrating lymphocytes than mismatch repair-proficient tumors. Colorectal cancer patients with MSI constitute a rare group of immune checkpoint inhibitor (ICI)-responsive patients. Nonetheless, complete radiological responders comprise between 3% and 16% of MSI advanced CRC patients, which compares poorly with the 45% to 87% rate of pathological complete response in early MSI CRC patients treated with ICIs. In this review, we address the efficacy of current ICIs and the biological differences between early and advanced MSI CRC to potentially increase the efficacy of ICIs in both settings.
Early-stage microsatellite unstable (MSI) colorectal cancer (CRC) has traditionally been managed in the same way as mismatch repair-proficient/microsatellite stable (MSS) tumors, with surgery and chemotherapy for colon cancer and chemoradiotherapy followed by surgery for rectal tumors [1, 2]. However, evidence from a seminal paper published by Llosa et al. [3] demonstrated higher CD8+ cytotoxic infiltration in MSI tumors compared with MSS tumors and a significantly higher upregulation of multiple immune checkpoints, including PD-1 (programmed death-1), PD-L1 (programmed death-ligand 1), CTLA4 (cytotoxic T-lymphocyte associated protein 4), LAG3 (lymphocyte-activation gene 3), and IDO (indoleamine 2,3-dioxygenase). These findings identified this subset of CRC patients as optimal candidates for immune checkpoint inhibitor (ICI) therapy.
Although differences in neoantigen presentation can somewhat explain the efficacy of ICI therapy in MSI with respect to MSS tumors, at least 40% of MSS tumors present bona fide neoantigens that can be recognized by CD8+ cytotoxic cells [4]. Therefore, the differences in neoantigen presentation and T cell infiltration cannot exclusively explain the differences in ICI activity between MSI and MSS tumors. The type of T cell infiltration also differs between MSI and MSS tumors, with MSS tumors showing increased expression of regulatory T cells (Tregs) and exhausted CD8+ cells. Finally, metabolic differences have been found between MSI and MSS tumors, with a broad increase in glycolytic enzymes in MSI subtypes [5].
The present review seeks to inform clinicians and researchers about the latest advancements in the immunotherapy of MSI CRC tumors and to offer insights into future directions for optimizing patient care and outcomes. The studies included were published in peer-reviewed journals, focusing on clinical trials testing the safety and efficacy of immunotherapy in the neoadjuvant/adjuvant setting in early-stage and advanced MSI CRC. Non-English language studies, observational studies, case reports, and reviews without original data on MSI CRC were excluded. Key data extracted included study design, patient characteristics, intervention details (e.g., type of immunotherapy regimen), and outcomes (e.g., response rate, disease-free survival, progression-free survival). Data were narratively synthesized to summarize findings across studies, focusing on efficacy outcomes and implications for clinical practice. Key data were appropriately tabulated to facilitate comparison across studies and clearly present results. Potential bias toward published studies with positive results may limit the generalizability of the findings, while heterogeneity in study designs and methodologies may impact the robustness of the conclusions drawn. For biomarker evaluation, we revised the REMARK criteria [6, 7] and categorized the quality of evidence as A (high recommendation), B (low recommendation), and C (very low recommendation) (see Table 1). The following three criteria were used to evaluate the quality of the biomarkers for recommendation: a) whether a prospective sample size was defined for the biomarker, with prespecified differences in progression-free survival and/or overall survival (e.g., hazard ratio), after adjustment for other variables in advanced disease, or prespecified differences in the pathological complete response (pCR) in the neoadjuvant setting, b) the presence of a control arm or a comparison group exposed to another therapy to evaluate whether the effect is prognostic or predictive, and c) the inclusion of a validation set involving the use of at least one additional dataset using the same biomarker and cut-off.
Proposed adapted REMARK criteria for evaluating biomarker efficacy
| Classification | Recommendation | Prospectively design* | Control arm** | Validation set*** |
|---|---|---|---|---|
| A | High | YES | YES | YES |
| B | Low | NO | YES | YES |
| C | Very low | NO | NO | NO |
* Includes a prospective sample size for the biomarker, with pre-defined differences in PFS and/or OS (HR) after adjusting with other variables in advanced disease or pre-defined differences on pathological complete response in neoadjuvant setting. ** Control arm. Exposure to other therapies to evaluate if the effect is prognostic or predictive. *** Validation set. Use of at least one additional set with the same biomarker and cut-off. PFS: progression-free survival; OS: overall survival; HR: hazard ratio
The aim of neoadjuvant immunotherapy (nIT) with ICIs is to enhance T cell activity against tumor neoantigens in a preserved primary tumor microenvironment. In the adjuvant setting, ICI therapy may enhance T cell efficacy against neoantigens in a micrometastatic environment in which the primary tumor is no longer present. It is currently unclear whether the microenvironment and metabolism differ between early-stage disease and micrometastatic disease. In a colon cancer liver model, micrometastases exhibited increased T cell infiltration compared with macrometastases [8]. Nonetheless, other metastatic models (e.g., brain metastases from HER2+ breast cancer) showed that latent metastases have distinct metabolic fitness from synchronous and metachronous bulk metastases [9, 10]. Nevertheless, better efficacy with the neoadjuvant strategy has recently been shown in phase III trials in non-small cell lung cancer, melanoma, and triple-negative breast cancer [11–14].
One randomized clinical trial of note has completed recruitment in the adjuvant setting (NCT02912559) [15]. This study was initiated in 2017 and is comparing FOLFOX + atezolizumab to FOLFOX alone (n = 700). Recruitment closed in January 2023, and the final results are expected to be reported in April 2025. Although the results are eagerly awaited, because this will be the first randomized data from a neoadjuvant setting, several caveats should be noted. First, the duration (12 months therapy in the atezolizumab arm) compares negatively with the 6-week neoadjuvant period in the NICHE-II trial [16]. Second, the expected toxicity profile will also compare negatively with that of NICHE-II. Finally, even if the experimental arm is superior to the control arm, it would be difficult to match the efficacy of the NICHE-II study, with 100% disease-free survival at 3 years with neoadjuvant therapy with nivolumab-ipilimumab.
We have included in the revision all the published or non-published (but presented at ESMO or ASCO Meetings) with ICI neoadjuvant therapy (see Figure 1). The NICHE-I trial pioneered the evaluation of nIT with nivolumab and ipilimumab in patients with locally advanced colon cancer [17]. The study demonstrated a pCR rate of up to 60% and a favorable safety profile. These findings have prompted further investigations into the safety and efficacy of nIT. Phase II studies consistently demonstrate that nIT in early MSI patients yields significantly higher rates of a pCR (45%–80%) and major pathological response (< 10% residual tumor) (90%–100%), corresponding to Mandard tumor regression grade 1 (complete response) or 2 (near-complete response), compared to 6-week neoadjuvant chemotherapy with FOLFOX, in which only 4% of such patients achieved a pCR and 5% achieved a Mandard tumor regression grade 1 or 2 [18] (see Table 2). However, several questions remain, necessitating further research to obtain robust scientific evidence that could potentially influence standard clinical practice.
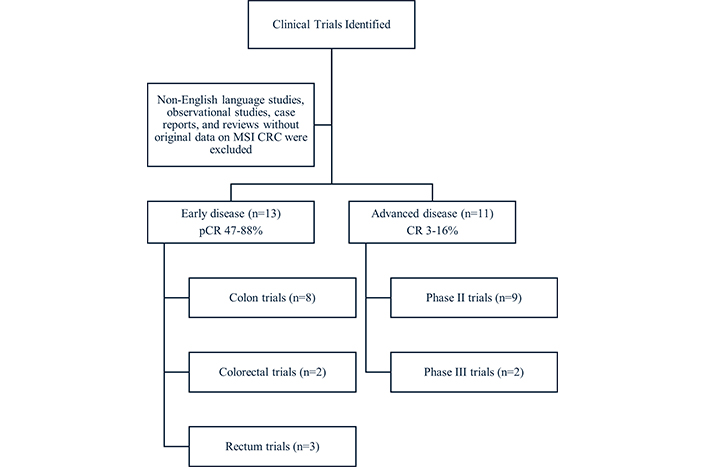
Flow diagram of clinical trials included in the review, categorized by disease stage (early or advanced) and type of study. Non-English studies, observational studies, case reports, and reviews without original data were excluded. MSI CRC: microsatellite unstable colorectal cancer; pCR: pathological complete response; CR: complete response
Clinical trials of ICIs in early-stage MSI colon and rectal cancer
| Studies | NCT | Drug (NA/A) | N | C/R | pCR (%) | Duration (w) | WW (%) | DFR (%)3-year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Chalabi, 2020 | NCT03026140 | Nivolumab/ipilimumab (NA) | 20 | C | 60 | 6 | - | - | [17] |
| Chalabi, 2024 | NCT03026140 | Nivolumab/ipilimumab (NA) | 119 | C | 67 | 6 | - | 100 | [16] |
| de Gooyer, 2024 | NCT03026140 | Nivolumab/relatlimab (NA) | 59 | C | 67 | 6 | - | - | [23] |
| Hu, 2022 | NCT03926338 | Toripalimab +/– celecoxib (NA) | 34 | C | 88 vs 65 | 12 | - | - | [25] |
| Xu, 2024 | NCT05890742 | IBI301 + sintilimabvs sintilimab (NA) | 101 | C | 80 vs 47 | 6 | - | - | [22] |
| Shiu, 2024 | NCT05197322 | Pembrolizumab (NA) | 32 | C | 59 | 6 | - | - | [27] |
| Ludford, 2023 | NCT04082572 | Pembrolizumab (NA) | 27 | C | 66 | 24 | - | - | [28] |
| AZUR2, 2024 | NCT05855200 | Dostarlimab vs SOC (NA/A) | 711 | C | - | 12 | - | - | |
| Yu, 2024 | NCT04715633 | Camrelizumab + afatinib (NA) | 52 | C/R | 61 | 12–24 | 54 | - | [24] |
| de la Fouchardiere, 2024 | NCT04795661 | Pembrolizumab (NA/A) | 87 | C/R | 47 vs 68 | 3–6 | - | - | [26] |
| Cercek, 2022 | NCT04165772 | Dostarlimab (NA) | 41* | R | - | 24 | 100 | - | [29] |
| Chen, 2023 | NCT04304209 | Sintilimab +/– CHT (NA/A) | 17 | R | - | 12 | 53 | - | [31] |
| AZUR1, 2024 | NCT05723562 | Dostarlimab (NA) | 100 | R | - | 24 | - | - |
ICIs: immune checkpoint inhibitors; MSI: microsatellite unstable; nivolumab: anti-PD-1; ipilimumab: anti-CTLA4; relatlimab: anti-LAG3; toripalimab: anti-PD-1; celecoxib: COX2 inhibitor; pembrolizumab: anti-PD-1; camrelizumab: anti-PD-1; afatinib: VEGF inhibitor; IBI301: anti-CTLA4; sintilimab: anti-PD-1; dostarlimab: anti-PD-1. NCT: National Clinical Trial; NA: neoadjuvant; A: adjuvant; CHT: chemotherapy; pCR: pathological complete response; WW: watch and wait; DFS: disease-free survival; C/R: colon/rectum; SOC: standard of care; Ref.: reference. * Updated information
First, although radiological staging is well-defined with pelvic magnetic resonance imaging in rectal cancer, the sensitivity and specificity of computed tomography are less clear in colon cancer [19]. This is important because MSI pT1-3N1 tumors, which represent roughly 50% of patients, have an excellent prognosis, with recurrence rates of less than 10%. In contrast, pT4N2 has a poor prognosis and 40% of these patients develop metastasis [20].
Second, it is further unclear whether adjuvant therapy will add clinical value after nIT. Findings from the NICHE-II trial with only 3-week therapy with nivolumab (two cycles) and ipilimumab (one cycle) [16, 21] indicated a 67% pCR and 95% major pathological response and an outstanding 100% disease-free survival rate at 3 years, suggesting that adjuvant therapy will likely not increase the efficacy of nIT.
The remaining question is if monotherapy with anti-PD-1 alone can achieve the same efficacy as combination therapy with anti-CTLA4 [17, 21, 22]. Recently, the NICHE-III trial, involving patients treated with nIT comprising two cycles of nivolumab and one cycle of relatlimab (anti-LAG3), showed similar efficacy (67% pCR) and toxicity as anti-CTLA4 therapy [23]. Most phase II studies suggest that monotherapy with anti-PD-1 is sufficient to achieve a similar pCR (46%–66%) as combination regimens. Only a small phase II randomized study has compared the efficacy of anti-PD-1 alone with that of anti-PD-1 combined with anti-CTLA4. That study showed a significant reduction in the pCR with anti-PD-1 alone (47.7% vs 80%) [22]. The benefit of adding other strategies to anti-PD-1 therapy, such as anti-angiogenic therapy with afatinib [24] and anti-COX2 with celecoxib [25], is currently unknown.
The duration of nIT may also influence outcomes. In the NICHE trials, combination therapies were administered during a 6-week nIT period, whereas other trials involving monotherapy with anti-PD-1 extend the nIT to 12 or even 24 weeks. For instance, a pCR was achieved in 46% and 68% of patients with pembrolizumab alone in 3- or 6-week nIT therapy, respectively, in the IMHOTEP study [26], in 59% of patients with 6-week therapy [27], and in 66% of patients with 24-week therapy [28]. Therefore, it is currently unclear if the number of cycles or the nIT duration improves the pathological response and 3-year disease-free survival. Because of the absence of phase III data compared with conventional therapy (surgery followed by chemotherapy), the results of the ongoing AZUR2 study (NCT05855200), a phase 3 clinical trial comparing nIT with dostarlimab vs standard treatment that is currently enrolling patients, are awaited with interest.
In early rectal cancer, anti-PD-1 alone with dostarlimab improves the strategy of non-operative management (NOM) to almost 100% [29, 30]. Other studies with other anti-PD-1 agents, such as camrelizumab [24] and sintilimab [31], also showed impressive results in NOM. Recently, the AZUR1 study (NCT05723562) has recruited 100 patients with early-stage MSI rectal cancer treated with dostarlimab. The primary objective is a 65%–90% complete clinical response at 12 months. The results are highly expected, particularly because the appropriate radiological evaluation for selecting patients for NOM and nIT is currently unclear.
The clinical characteristics of MSI metastatic patients (only 5% of metastatic patients are MSI) differ in some aspects from those of MSS individuals. Compared with MSS tumors, the primary tumors are typically located on the right side (65%–70% vs 30%–35%), are more prone to the development of BRAF mutations (25%–30% vs 5%–10%), have more peritoneal metastasis (35%–40% vs 10%–15%) and less liver metastasis (35%–40% vs > 70%), and exhibit less abnormally high lactate dehydrogenase levels (5%–10% vs 30%–35%).
In Table 3, we included all the studies that have been published with ICI in advanced MSI tumors. We included phase II trials of first-line therapy with nivolumab-ipilimumab [32] and of pre-treated patients with nivolumab [33], nivolumab-ipilimumab [34, 35], nivolumab-relatlimab [36], avelumab [37, 38], durvalumab [39] and pembrolizumab [40]. Overall, these studies showed a high response rate (33%–65%) and 3-year progression-free rate (38%–60%). Although the results compare favorably with chemotherapy, roughly 25%–40% of patients are ICI refractory and less than 50% remain progression free at 3 years.
Clinical trials of ICIs in advanced MSI colorectal cancer
| Studies | NCT | Phase | N | Drug | BOR (%) | CR (%) | DFR (%)2-year | DFR (%)3-year | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Lenz, 2022 | NCT04008030 | II/1L | 45 | Ipilimumab/nivolumab | 65 | 13 | 73 (57–84.5) | - | [32] |
| André, 2022 | NCT02060188 | II/≥ 2L | 119 | Ipilimumab/nivolumab | 55 | 3 | 63 (53–71) | 60 (50–68) | [35] |
| Overman, 2018 | NCT02060188 | II/≥ 2L | 74 | Nivolumab | 51 | 3 | 50 (38–61) | - | [34] |
| Overman, 2024 | NCT02060188 | II/> 2L | 50 | Relatlimab/nivolumab | 48 | 16 | 51 (37–64) | 38 (24–52) | [36] |
| Le, 2020 | NCT02460198 | II/2L | 61 | Pembrolizumab | 33 | 3 | 31 | - | [40] |
| Le, 2020 | NCT02460198 | II/> 2L | 63 | Pembrolizumab | 33 | 8 | 37 | - | [40] |
| Diaz, 2022 | NCT02563002 | III/1L | 307 | Pembrolizumab vs CHT | 45 vs 33 | 13 vs 4 | 48.3 (39.9–56.2) | 42.3 (34–50.4) vs 11.1 (6.1–17.9) | [42] |
| Andre, 2024 | NCT04008030 | III/1L | 505 | Ipilimumab/nivolumab vs nivolumab vs CHT | - | - | 72 vs 14 | - | [43] |
| Kim, 2020 | NCT03150706 | II/> 2L | 30 | Avelumab | 24 | 14 | - | - | [37] |
| Taïeb, 2023 | NCT03186326 | II/2L | 122 | Avelumab vs CHT | 30 vs 26 | 7 vs 5 | 25 vs 10 | - | [38] |
| Oh, 2022 | NCT03435107 | II/> 2L | 30 | Durvalumab | 42 | - | - | - | [39] |
ICIs: immune checkpoint inhibitors; MSI: microsatellite unstable; ipilimumab: anti-CTLA4; nivolumab: anti-PD-1; relatimab: anti-LAG3; pembrolizumab: anti-PD-1; CHT: chemotherapy; avelumab: anti-PD-L1; durvalumab: anti-PD-L1; BOR: best overall response; CR: complete response; DFR: disease-free rate; NCT: National Clinical Trial; Ref.: reference
Two large phase III trials have compared ICI to standard chemotherapy. The KEYNOTE-177 trial demonstrated superior progression-free survival of pembrolizumab over chemotherapy as frontline treatment for patients with advanced MSI disease [41, 42]. The CheckMate 8HW has been recently published [43]. The dual primary endpoints were progression-free survival, assessed by blinded independent central review, for nivolumab + ipilimumab vs nivolumab across all lines and for nivolumab + ipilimumab vs chemotherapy in the first-line setting in patients with centrally confirmed MSI-H metastatic CRC (mCRC). This study shows the superiority of nivolumab-ipilimumab vs chemotherapy. The comparison between nivolumab-ipilimumab and nivolumab is planned only in patients that received ≥ 2 prior lines of therapy and the study will therefore not clarify if nivolumab-ipilimumab is superior to nivolumab as first-line therapy. Because chemotherapy is currently not an adequate control arm for advanced MSI disease, it is unclear whether nivolumab-ipilimumab should be considered a new standard of care.
Only a few studies have analyzed biomarkers to address ICI therapy efficacy in early MSI disease. In the NICHE-I study, although the tumor mutational burden (TMB) varied widely between MSI (median, 1,438 mutations; range, 648–4,458) and MSS (median, 111 mutations; range, 48–261) tumors, no differences in TMB were found between MSS responders (median, 108; interquartile range, 62–153) and non-responders (median, 117; interquartile range, 93–143). In addition, no major differences were found between MSI and MSS in CD3+, FOXP3+, PD-L1, and interferon scores, tertiary lymphoid structure presence, and CXCL13 expression. The sole biomarker related to the MSS ICI response was the presence of T cell co-expression of CD8 and PD-1 [17]. In the PICC study, early-stage MSI patients were treated with toripalimab ± celecoxib. The authors found no differences in activity based on TMB, but more responders were seen in the group of patients with HLA-DQA1 and HLA-DQB1 expression and in tumors that showed CD8+ T cells with low PD-1 expression [44]. Finally, the same group used single-cell RNA-seq analysis to show that patients who achieved a pCR in post-treatment samples exhibited decreased proinflammatory features, with fewer CD8+ TRM-mitotic cells (with exhausted phenotype markers, e.g., PDCD1, LAG3, TIGIT, HAVCR2, TOX, and ENTPD1), CD4+ Tregs, proinflammatory IL1B+ monocytes, and CCL2 fibroblasts [45]. Although these findings suggest a polarization to an exhausted ICI refractory phenotype after therapy, these studies do not clarify in pre-treated biopsies the intrinsic causes driving this polarization.
There is extensive literature regarding the use of potential biomarkers to assess ICI efficacy in advanced MSI patients (see Table 4). Nonetheless, using our proposed quality biomarker analysis (modified REMARK), none of the studies fulfill all of the strict criteria for their recommended use in clinical practice (Table 1). This means that none of these biomarkers can replace immunohistochemistry (loss of expression of any of four proteins [MLH1, PMS2, MSH2, MSH6]) or determination of mismatch repair deficiency (dMMR) defined by microsatellite analysis (at least two of five microsatellites with different length variations). These methods are cheap, highly reproducible, and, most importantly, have been used prospectively to select patients in clinical trials of MSI patients undergoing ICI. The fact that none of the published MSI predictive biomarkers, are used in clinical practice, reflect the poor credibility of REMARK criteria.
Studies evaluating biomarkers in patients treated with ICIs in MSI cancer, with adapted REMARK criteria
| Studies | N | Biomarker | Prospective set | Control arm | Validation set | Recommendation | Ref. |
|---|---|---|---|---|---|---|---|
| Mandal, 2019 | 33 | MSI sensor | NO | NO | NO | C | [47] |
| Georgiadis, 2019 | 23 | MSI sensor | NO | NO | NO | C | [48] |
| Kawazu, 2022 | 114 | HLA-ABC | NO | YES | NO | B | [54] |
| Middha, 2019 | 13 | B2M | NO | NO | NO | C | [55] |
| Germano, 2021 | 38 | B2M | NO | NO | NO | C | [56] |
| Zhang, 2022 | 35 | B2M | NO | NO | NO | C | [57] |
| Schrock, 2019 | 22 | TMB | NO | NO | NO | C | [49] |
| Loupakis, 2020 | 80 | TMB/TILS | NO | NO | NO | C | [50] |
| Chida, 2021 | 45 | TMB | NO | NO | NO | C | [51] |
| Manca, 2023 | 110 | TMB | NO | NO | NO | C | [52] |
| Westcott, 2023 | 26 | Clonal neoantigen burden | NO | NO | NO | C | [53] |
| Quintanilha, 2023 | 320 | MSI (NGS) | NO | YES | YES | B | [46] |
| Pietrantonio, 2021 | 305 | Nomogram | NO | NO | YES | C | [60] |
| Sui, 2022 | 66 | Inflammatory signature | NO | NO | YES | C | [58] |
| Corti, 2021 | 163 | Inflammatory signature | NO | NO | NO | C | [59] |
| Saberzadeh-Ardestani, 2023 | 33 | Inflammatory signature | NO | NO | NO | C | [61] |
| Gallois, 2023 | 138 | Stromal signature | NO | NO | YES | C | [62] |
| Chida, 2022 | 36 | CMS | NO | NO | NO | C | [63] |
| Sui, 2021 | 43 | DKK1 | NO | NO | NO | C | [64] |
| Bortolomeazzi, 2021 | 29 | WNT/TMB/B2M | NO | NO | YES | C | [65] |
| Ratovomanana, 2023 | 129 | Multiplex MSI signature/TGF-β signature | NO | NO | YES | C | [71] |
ICIs: immune checkpoint inhibitors; MSI: microsatellite unstable; HLA: human leukocyte antigen; B2M: beta 2 microglobulin; TMB: tumor mutational burden; TILS: tumor infiltrating lymphocytes; NGS: next generation sequencing; CMS: consensus molecular subtype; DKK1: Dickkopf 1; TGF-β: transforming growth factor beta
The only study that fulfills at least two modified REMARK criteria was recently published by Quintanilha et al. [46]. They used next-generation sequencing to assess MSI and compared the results with standard methods (immunohistochemistry of dMMR) and showed that next-generation sequencing better discriminates the benefit of ICI therapy.
MSI sensor software analyzes aligned sequencing data to assess available microsatellite regions with sufficient coverage in a tumor/normal tissue pair and thereby identify variations in deletion lengths indicative of MSI. Current evidence does not justify its use in clinical practice [47, 48]. In addition, TMB [49–52], which uses different cut-offs or clonal neoantigen burdens, does not fulfill any of our adapted REMARK criteria [53]. Loss of the expression of beta 2 microglobulin (β2M), which is usually associated with β2M mutations (occurring in 24%–28% of MSI tumors), has been shown pre-clinically [54] to preclude ICI efficacy due to poor major histocompatibility complex (MHC)-I presentation, although the clinical data do not support the pre-clinical findings [55–57]. Importantly, none of these studies fulfill our modified REMARK criteria and therefore cannot be used in clinical practice.
Inflammation has been pre-clinically related to ICI resistance. The inflammation pattern can be measured in different ways. For instance, in blood samples, we can evaluate inflammation by using the ratio between neutrophils/myeloids over lymphocyte infiltration. The main concern of this biomarker is the difficulty in the clear definition of the optimal cut-off. Using our strict REMARK methodology, none of the published studies fulfill the A or B recommendation [58–61] suggest that a close proximity of CD8+ T cells expressing PD-1 with CD3, CD8, and CD68 macrophages expressing PD-L1 correlates with ICI efficacy. Finally, transcriptomics has also been used to evaluate the ratio of proliferation to stromal components [62]. Progression-free survival was shorter in tumors treated with ICIs and in stromal-high/proliferation-low tumors. Again, different cut-offs and different ways to evaluate inflammation limit its value as a biomarker. Other authors have focused on pathways that have been described in other tumors as potential biomarkers of ICI resistance. For example, the study by Chida et al. [63] reported that WNT activation, commonly observed in CMS2/3 subtypes, is associated with ICI resistance. Similarly, studies by Sui et al. [64] and Bortolomeazzi et al. [65] further support the link between WNT activation and ICI resistance. Consensus molecular subtype 2, characterized by MYC and WNT expression and reduced immune infiltration [66, 67], has been shown to be increased in CRC metastases, compared with primary tumors [68]. This CMS subtype has been also related to ICI-resistance [69]. Additionally, a transforming growth factor-β (TGF-β) signature, also enriched in colorectal metastases [70] has been correlated with reduced ICI efficacy [71].
Tumor cells undergo metabolic reprogramming to maintain malignant features and fitness in metastatic microenvironments. A seminal paper published by the Bartman group [72] showed recently that primary tumors rely on high levels of glycolysis compared with normal tissue but, contrary to what was expected, have a lower tricarboxylic acid (TCA) flux and ATP production. Compared with primary tumors and normal tissues, an increased TCA flux and elevated lactate consumption was noted in lung metastatic tissues from triple-negative breast cancer. Interestingly, the high glycolysis and low TCA is explained by a replacement of normal specific tissue functions (e.g., protein synthesis in pancreatic cancer) for increased proliferation. Similar conclusions were reached by the Bezwada group [73], which showed that acetate and glutamine increase TCA labeling in kidney cancer metastases compared with primary tumors. The Faubert group [74] elegantly propose that metastatic cells in circulating blood increase OXPHOS and reactive oxygen species (ROS) capacities [75, 76], coupled with antioxidant mechanisms to sustain cell viability under stressful, highly oxidative, conditions. Thus, it seems that metastatic seeding requires functional mitochondria [77].
Several antioxidant pathways have been reported to minimize the death of circulating metastatic cells. Most of the studies suggest that ferroptosis resistance associated with lipogenesis is one of the crucial antioxidant pathways [78, 79]. Other antioxidant pathways that enable cells to mitigate ROS in the metastatic process are increased NADPH in mitochondria through glutamine reductive carboxylation [80] and the folate pathway [81].
Metastases show distinct metabolic characteristics from primary tumors that potentially can confer more ICI resistance. For instance, atypical glycolysis [82, 83], hexosamine biosynthesis pathways [84–86] and epithelial-to-mesenchymal transition, all well-described mechanisms of ICI resistance, have been related to disease dissemination [87–91].
Tumors with increased OXPHOS, have also been reported to be associated with ICI resistance [92–94]. Interestingly increased OXPHOS alone or associated with high levels of glycolysis is a well-known mechanism of dissemination in a variety of tumors. Lung metastases from triple-negative breast cancer [72, 95, 96] and kidney cancer [73] are supported by increased TCA/OXPHOS function. Interestingly, liver metastases show higher glycolysis and low TCA/OXPHOS despite increased PDK1 expression [95]. In contrast, breast cancer cells with broad metastatic potential (4T1 cells: bone, lung, and liver) engage both OXPHOS and glycolysis. This can suggest that alternative energy supplies can activate both glycolysis and OXPHOS. Given that other studies of primary lung and pancreatic cancers show that lactate is used to fuel the TCA/OXPHOS pathway, we speculate that these highly metabolically flexible tumors use lactate instead of glucose to fuel the mitochondria [97, 98]. This pattern is also supported by antioxidant mechanisms such as the pentose phosphate pathway (PPP), as has been well described by other authors [99–101]. Despite PPP inhibition, alternative antioxidant pathways such as isocitrate dehydrogenase and malic enzyme upregulate functions through glutaminolysis [102–104]. Finally, glutamine in these tumors supports urea cycle dysregulation, high chromosomal instability [105] and polyamine synthesis [106]. It is currently unclear if glutaminase [107, 108] or polyamine inhibition [109, 110] can increase the clinical efficacy of ICIs.
The efficacy of cancer immunotherapy in MSI CRC differs between patients with early disease (roughly 60%–70% pCR) and advanced disease (mCRC) (3%–16% complete radiological response). Currently, there is no clear biological explanation for this difference. One of the key differences between early-stage and advanced MSI CRC lies in the tumor immune microenvironment (TIME). Early-stage tumors are characterized by a higher infiltration of cytotoxic CD8+ T cells and a lower proportion of Tregs, creating a more immunogenic environment. In contrast, advanced tumors exhibit an increase in immunosuppressive elements, such as Tregs and exhausted CD8+ T cells expressing PD-1, LAG3, and TIM-3. Additionally, advanced tumors harbor more tumor-associated macrophages (TAMs) with an M2 immunosuppressive phenotype, which further contributes to immune evasion [111–113]. These distinctions in TIME likely explain the reduced efficacy of ICIs in advanced-stage MSI tumors compared to early-stage tumors.
Liquid biopsy (LB) analysis (determination of mutations in blood) soon after surgical resection (4–8 weeks) adds valuable prognostic clinical information in early colon cancer. Patients with LB positivity (LB+) (usually 10%–20% of patients) have a high risk of metastasis (> 50% risk and usually with a short follow-up). In contrast, LB negative (LB−) patients (which constitute 80%–90% of resected tumors) have less than a 10% risk of relapse [114]. Unfortunately, the intrinsic biological and metabolic characteristics of the LB+ and LB− patients that ultimately develop metastases are poorly understood.
We previously identified two major clusters with metabolic interactions among tumors, stromal cells, and immune cells across 11 different tumor types [115–117]. Specifically in colon cancer, cluster 1 (IMC1) has mesenchymal features, atypical glycolysis, and high stromal and immune infiltration and is represented by the CMS4 subtype, correlated with ICI-resistance and are increased in advanced disease compared with early CRC [86, 90, 118]. Cluster 3 (IMC3) is driven by high chromosomal instability, high OXPHOS with uptake of multiple nutrients (lactate, fatty acids, glucose, and glutamine), and high metabolic flexibility supported by multiple antioxidant mechanisms and is represented by the CMS2 and CMS3 subtypes [119]. This CMS can also induce immune-suppression through ammonia and polyamine accumulation [110, 120]. As mitochondrial respiration and antioxidant stress protection (in the IMC3 subtype) and atypical glycolysis (in the IMC1 subtype) play critical roles in metastatic spread, we hypothesize that adaptation of the metabolic microenvironment in metastatic sites can potentially explain ICI resistance in at least a subset of patients with advanced MSI CRC. Supporting it, our group [121], also showed, using 13C-labelled glucose, that the entry of glucose into the TCA cycle is higher in the metastatic colon cancer cell lines SW620 and LIM2 than in the paired primary cell line SW480 and that the levels of phosphorylated pyruvate dehydrogenase (pPDH) are consistently lower in metastatic cells.
Although early and advanced MSI diseases exhibit a similar T cell infiltration and TMB, they show high disparities in ICI efficacy. Due to marked differences in metabolism between limited and metastatic disease, potential metabolic vulnerabilities can also enhance ICI efficacy specially in advanced MSI. We strongly support to evaluate immune-suppressive metabolic profiles in MSI CRC [for example, mesenchymal metabolic tumors (IMC1) and high glycolytic/high OXPHOS (IMC3)] to identify those patients that can be potentially ICI resistant. In IMC1 profile, hexosamine biosynthesis inhibitors or MCT4 inhibitors can increase ICI efficacy. Alternatively, in IMC3 subtype, OXPHOS inhibitors, PPP inhibitors, or inhibitors of polyamine synthesis can increase ICI efficacy.
CRC: colorectal cancer
CTLA4: cytotoxic T-lymphocyte associated protein 4
dMMR: determination of mismatch repair deficiency
ICIs: immune checkpoint inhibitors
IMC1: cluster 1
LAG3: lymphocyte activation gene 3
LB: liquid biopsy
mCRC: metastatic colorectal cancer
MSI: microsatellite unstable
MSS: microsatellite stable
nIT: neoadjuvant immunotherapy
pCR: pathological complete response
PD-1: programmed death-1
PD-L1: programmed death-ligand 1
PPP: pentose phosphate pathway
ROS: reactive oxygen species
TCA: tricarboxylic acid
TIME: tumor immune microenvironment
TMB: tumor mutational burden
Tregs: regulatory T cells
MR: Funding acquisition, Writing—original draft, Writing—review & editing. CR and RM: Data curation, Writing—review & editing. MC and JM: Conceptualization, Data curation, Writing—original draft, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This research was funded by Spanish Ministry of Science and Innovation [PID2023-150539OB-I00], funded by MCIN/AEI/10.13039/501100011033 to MC, and the ICREA Academia Prize funded by ICREA Foundation to MC. This research was funded by Instituto de Salud Carlos III (ISCIII) [AC24/00028] to JM. Grants were from the Catalan Agency for the Management of University and Research Grants (AGAUR), specifically [2021-SGR-01328] to JM and [2021-SGR-00350] to MC. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 9211
Download: 30
Times Cited: 0
Anastasia Rays ... Аlexey Tryakin
