 Original Article
Original Article

Affiliation:
Department of Research and Development, Bio-Synthesis, Inc., Lewisville, TX 75057, United States
Email: frank_hong@biosyn.com
ORCID: https://orcid.org/0000-0002-6814-4671
Affiliation:
Department of Research and Development, Bio-Synthesis, Inc., Lewisville, TX 75057, United States
ORCID: https://orcid.org/0000-0002-5499-5061
Affiliation:
Department of Research and Development, Bio-Synthesis, Inc., Lewisville, TX 75057, United States
ORCID: https://orcid.org/0000-0003-0408-9391
Explor Digit Health Technol. 2025;3:101142 DOI: https://doi.org/10.37349/edht.2025.101142
Received: October 19, 2024 Accepted: February 26, 2025 Published: March 20, 2025
Academic Editor: Zhaohui Gong, Ningbo University, China
Aim: Genetic instability represents the hallmark of carcinogenesis. For cancer, the retinoblastoma (RB) gene defect allowing genetic instability was successfully exploited to eliminate cancer. Similarly, this study aims to assess the genetic instability of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike protein’s S1/S2 furin cleavage site in hopes of applying oligonucleotide-based therapeutics to suppress infectivity by exploiting hypermutability.
Methods: The Basic Local Alignment Search Tool was used to search for homology. Protein or nucleotide sequences were obtained from the National Center for Biotechnology Information database. BioEdit was used for multiple sequence alignment. Python-enhanced molecular graphics program was used for molecular modeling.
Results: To assess feasibility, comparative sequence alignment was performed on S1/S2 site plus juxtaposing residues of SARS-CoV-2 and avian infectious bronchitis virus (IBV) isolate AL/7052/97 that belongs to distinct genus. IBV amino acids correlating to 678-TNSPRRARSVASQS of SARS-CoV-2 spike protein were deciphered (nine identical, two conserved, two displaced, and one unconserved). The encoding nucleotides exhibited 14 identities, three transitions (C>U or U>C, two; G>A or A>G, one), and 15 transversions (U>A or A>U, eight; C>G or G>C, six; G>U or U>G, one) with mostly complementary base (14/15) for transversion. Analysis of SARS-CoV-2 variants corroborates that S1/S2 site continues to evolve. The overall data portrays an evolutionarily dynamic nature of S1/S2 site. The potential role of intragenomic ‘microhomology-mediated template switching’ by RNA-dependent RNA polymerase is described.
Conclusions: To apply virolytic pressure, peptide-guided oligonucleotides targeting S1/S2 site-encoding sequences may be deployed to trigger genomic RNA degradation. A potential consequence is that resistant variants (if emerge) may carry mutation(s) in S1/S2 site-encoding sequence to abrogate hybridization, which (by default) may encode defective substrate for furin. Thus, through ‘targeting oligonucleotides directed devolution’ of S1/S2 site, the infectivity of SARS-CoV-2 may be attenuated. An alternative strategy of oligonucleotide-based therapeutic editing by adenosine deaminases acting on RNA (ADAR) is mentioned.
Genetic instability constitutes the key driving force underlying tumoral evolution. The finding that multiple tumor suppressor genes function in DNA damage checkpoint to prevent mutagenesis confirmed this view. Therapeutically, the defect in checkpoint allowing genetic instability also represents vulnerability as it could be exploited to trigger genome disintegration, resulting in tumor cell death. This point has been successfully demonstrated in the case of a tumor suppressor gene defect (see below). Here, in a similar vein, we explored whether genetic instability underlying viral evolution could be exploited for the therapeutic purpose of suppressing infectivity.
Within cells, progression through the G1 phase is governed by the prototypic tumor suppressor retinoblastoma (RB) protein, a component of the DNA damage checkpoint [1]. RB also controls cell cycle progression across the S phase; after the cloning of the human RB gene by F. Hong (a.k.a. Frank Un, Frank D. Hong) et al. [2, 3] at the University of California in San Diego (United States), RB protein was found to be phosphorylated in vivo. His determination of the RB gene sequence was pivotal in identifying the cyclin-dependent kinase (CDK) complex as the enzyme catalyzing RB phosphorylation [2, 3]. The cyclic phosphorylation pattern of RB is maintained by distinct cyclin-CDK complexes, with the least phosphorylated species found in the G1 phase [4]. It led to the development of various inhibitors, which include palbociclib (Pfizer Pharmaceutical, United States), ribociclib (Novartis Pharmaceutical, Switzerland), and abermaciclib (Eli Lilly Pharmaceutical, United States) approved by the U.S. Food and Drug Administration (FDA) for treating metastatic breast cancer.
RB protein’s role in regulating transcription was inferred from F. Hong et al. [3] finding of its ability to bind to DNA, which was subsequently confirmed [5]. This property was corroborated via observing the RB-DNA complex formed by purified RB protein and double-stranded DNA [6]. Its atypical electrophoretic migration pattern led to his subsequent finding of RB’s oligomerizing property [7–9]. RB protein’s interaction with E2F confirmed RB gene’s role in transcriptional regulation. Partial proteolysis of RB by C. E. Hensey (University College Dublin, Ireland) et al. [7] revealed the existence of distinct domains for interaction with associating proteins and a separate module at the N-terminus. The prototypic antimetabolite drug hydroxyurea is FDA-approved for treating chronic myelogenous leukemia, and head and neck squamous cell carcinoma. [10]. Through the analysis of RB-associating protein, F. Hong discovered that the sensitivity to hydroxyurea could be restored to resistant human cancer cells at the City of Hope National Medical Center and Beckman Research Institute (United States) in collaboration with C. Bronner (Institut national de la santé et de la recherche médicale, France) et al. [11].
RB gene’s role in chromosome organization and architecture was indicated by F. Hong and W. H. Lee found [12]; that RB protein exhibits similarity in sequence with neurofilament L-type subunit, which was corroborated by R. F. Doolittle (University of California at San Diego, United States) [12]. Consistently, he observed RB oligomers and detected the polymerization of RB protein [7]. The association of RB with nuclear matrix was documented by S. Penman (University of Texas Health Science Center at San Antonio) et al. [13]. The finding that RB protein may self-interact to form higher-ordered structures was instrumental in extending RB gene’s function to DNA replication, repair, recombination, condensation, heterochromatinization, epigenetics, etc. [1]. The prototypic DNA crosslinking drug cisplatin is FDA-approved for treating ovarian cancer, testicular cancer, and bladder cancer. F. Hong [14] discovered that G1 arrest mediated by RB represents a critical determinant of cisplatin cytotoxicity in RB-positive human cancers retaining DNA damage checkpoint.
The inactivation of RB through mutation causes genetic instability [1, 15]. F. Hong observed that ectopic expression of RB driven by SV40 large T promoter causes transfected cells to arrest as enlarged cells (unpublished data). Retrovirally expressed RB suppressed the growth of human RB, prostate cancer, and breast cancer [16–18]. Further, purified human recombinant RB protein induced G1 arrest in microinjected cells [19]. These findings revealed RB gene’s function in DNA damage checkpoint in the G1 or S phase [20]. The prototypic antimicrotubule drug Taxol is FDA-approved for treating breast cancer, non-small cell lung cancer, and ovarian cancer. F. Hong [15] discovered that by exploiting the defective checkpoint rendered by inactive RB (or p53), Taxol (paclitaxel) induces lethality via triggering chromosome fragmentation, i.e., ‘mitotic catastrophe’. It led to the elucidation of the tumor-specific lytic path ‘hyperploid progression mediated death’ targeting RB or p53-mutant human cancer cells at the University of Texas Southwestern Medical Center (United States), establishing a framework for developing cytotoxic cancer therapy devoid of side effects [15].
RB gene’s role as a therapeutic biomarker originates from F. Hong et al. [21] discovery of an RB mutant affecting its promoter activity [22]. Earlier, he identified and characterized the promoter of the human RB gene [23]. It represents the earliest RB mutant to be identified in human prostate cancer [21]. The mutational status of RB dictates the optimal strategy for treating advanced-stage cancer patients, e.g., hormone therapy [example (ex.), Enzalutamide] versus antimicrotubule chemotherapy (ex., Taxol) for prostate cancer.
Increasingly, modern medicine relies on targeting upstream genetic components such as mRNA or chromosomal DNA. Notable among them is genetic medicine using oligonucleotides to suppress translation by hybridizing to mRNA or degrading mRNA through RNA interference or RNase H1. The first clinical trial of siRNA employed siRNA targeting human ribonucleotide reductase M2 subunit contained in nanoparticles designed by M. E. Davis (California Institute of Technology, United States) et al. [24] to treat various types of human cancer. Nevertheless, oligonucleotide drugs, by themselves, cannot translocate across the cell membrane for delivery in a targeted manner, contributing to side effects. To facilitate delivery, the siRNA was conjugated to the tumor specifically internalizing peptide HN-1 for targeted delivery into the head and neck or solid breast tumor. HN-1 peptide was isolated via biopanning of M13 bacteriophage-based random peptide display library using human head and neck squamous carcinoma cells by F. Hong and G. L. Clayman [25] at the University of Texas M. D. Anderson Cancer Center (United States). HN-1 has been conjugated to various agents for cancer therapy (ex., siRNA oligonucleotide, Taxol, protein kinase C inhibitory peptide, diphtheria toxin, doxorubicin) or tumor imaging (ex., near-infrared dye, radioisotope) [26–34]. His delineation of discoidin domain tyrosine kinase receptor 1 (DDR1) as the putative receptor for HN-1 and its therapeutic application to breast, lung, and other cancer types was recently reviewed [35]. Its ability to penetrate tumor mass may be useful in delivering immunotherapeutics to the interior of solid human cancers. For prostate cancer, he described the role of monoamine oxidase A in modulating anticancer immunity at the University of Southern California (United States) [36, 37]. FDA has approved immunotherapeutics targeting hypermutated tumors [38].
The recent coronavirus disease 2019 (COVID-19) pandemic has affected many with underlying conditions [39, 40]. Among the impacted were those who have cancer, whose fatality rate increased markedly following the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection. Various preventive measures have been developed, including mRNA vaccines expressing the modified spike protein [41]. These countermeasures significantly reduced COVID-19-associated mortality. Nevertheless, subsequently emerged variants were able to impact host immunity via acquiring a mutation in epitopes [42]. Adaptation by SARS-CoV-2 was not unexpected, given its high level of mutability [43, 44]. The genetic instability of SARS-CoV-2 gives rise to point mutation, insertion, deletion, and complex mutation [45]. The virally encoded RNA-dependent RNA polymerase (RDRP) incurs a high error rate during nucleotide incorporation despite the proofreading capacity (3’-to-5’ exoribonuclease) of nonstructural protein 14 (NSP14). Furthermore, genetic recombination may occur due to template switching between homologous RNA strands by RDRP [45].
Modulating the cell cycle via targeting its regulators represents a key replicative strategy amongst viruses [46]. Virally encoded oncogenic proteins that interact with RB protein include large T antigen (simian vacuolating virus SV40), E1A (adenovirus), and E7 (human papillomavirus) [47]. For SARS-CoV-1, structural protein 3a inhibits RB phosphorylation, causing G1 arrest [48]. In the case of SARS-CoV-2, NSP1 was shown to suppress host cell protein synthesis, resulting in G1 arrest, which is regulated by RB [49]. SARS-CoV-2 was also shown to cause S arrest (which is controlled by RB) via depleting deoxynucleotides [50].
The acquisition of S1/S2 furin cleavage site in spike protein significantly expanded the tropism of SARS-CoV-2 (see below). The increased dissemination despite reduced virulence is a distinguishing feature amongst subsequently arisen variants. Here, to address this issue, the possibility of exploiting the hypermutability of SARS-CoV-2 to suppress its infectivity is explored. To apply virolytic pressure, oligonucleotides targeting the S1/S2 site-encoding sequence may be deployed to trigger genomic RNA degradation. A potential consequence of the above approach is that resistant variants (if they emerge) may carry a mutation in S1/S2 site-encoding sequence to block oligonucleotide hybridization, which may encode defective substrate for furin by default. Hence, the prospect of applying targeting oligonucleotides to direct the devolution of the S1/S2 site, delimiting tropism to attenuate the infectivity of SARS-CoV-2, is described. The precedent for the above comes from previous reports documenting reduced viral fitness conferred by resistance mutations. The approach is reminiscent, albeit distinct from the ‘directed evolution’ described by P. A. Romero and F. H. Arnold (California Institute of Technology, United States; Nobel Prize 2018) [51].
The protein-protein Basic Local Alignment Search Tool (BLASTp) was used to evaluate linear sequence similarity between two proteins (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). To avoid algorithmic bias that may prioritize longer-sequenced homologies in identifying the ‘best fit’, the search was conducted using the peptide sequence ‘PRRAR’ (Pro-Arg-Arg-Ala-Arg) of SARS-CoV-2 spike protein as the query. The screening was performed using BLASTp software (version BLAST+ 2.12.0) against the database of avian coronavirus IBV, which belongs to genus Gammacoronavirus (https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins). The database comprised of sequences from SwissProt, Protein Data Bank, Protein Information Resource, GenBank non-redundant coding sequence translations, and Protein Research Foundation (excluding environmental samples from Whole Genome Shotgun projects). For computing, the algorithm was set with the following parameters to execute the search for alignment. General parameters: max target sequences, 100; expect thresholds, 0.05; word size, 5; max matches in a query range, 0. Scoring parameters: matrix, BLOSUM62; gap costs, existence (11) extension (1); compositional adjustments, conditional composition score matrix adjustment. Filters and masking: none. Protein sequences and the corresponding nucleotide sequences in GenBank format were retrieved from the National Center for Biotechnology Information (NCBI) archived and publicly available through the PubMed (Public Medline) database (for protein sequence, https://www.ncbi.nlm.nih.gov/protein/; for nucleotide or protein sequence, https://www.ncbi.nlm.nih.gov/nucleotide/).
To align multiple polypeptide sequences, the BioEdit program was employed, which performs pair-wise alignment to identify the best matches of statistical significance. Alternatively, sequences were parceled into oligopeptides prior to performing alignment to identify residues that may match in a 3-dimensional context, albeit not in a 2-dimensional setting due to physical separation.
The 3-dimensional structure of furin bound to a hexapeptide inhibitor was generated using the Python-enhanced molecular graphics (PyMOL) program. After downloading the structural coordinates from the National Institutes of Health (NIH) protein database (https://www.ncbi.nlm.nih.gov/Structure/index.shtml), PyMOL software was employed to re-enter the image of the enzyme-inhibitor complex. Structure-associated data was obtained from the National Library of Medicine (https://www.nlm.nih.gov/). The 3-dimensional structure of Arg-Arg-Arg-Val-Arg-Amba (aminomethylbenzoic acid) peptide competitively bound to the furin proprotein convertase enzyme determined through X-diffraction analysis was obtained via searching the structural database using ‘6EQX’ as the query.
Various types of organic chemistry underlying oligonucleotide synthesis and distinct types of chemical modification are used to resolve clinical issues associated with oligonucleotide therapeutics. These include the previously employed phosphodiester method or phosphotriester approach to synthesize oligonucleotides chemically. The current widely adopted solid-support-based approach utilizes nucleoside phosphoramidites as a precursor for oligonucleotide synthesis. For internucleosidic (backbone) modification, phosphorothioate may be used to reduce degradation. Alternatively, peptide nucleic acids or morpholino oligonucleotides may be utilized. To enhance nuclease stability or augment affinity for hybridization, bases may be modified (ex., methylation), or bridged nucleic acids (or locked nucleic acids) may be incorporated into oligonucleotides during the synthesis. Further, C2’ position of the sugar moiety may be modified to contain 2’-O-methyl, 2’-O-methoxyethyl, 2’-fluoro group, etc. To synthesize good manufacturing practice (GMP)-grade oligonucleotides for clinical application, additional works regarding molecular mass (as analyzed by mass spectroscopy), purity (as determined by high-performance liquid chromatography), sterility (as synthesized under a sterile condition), etc. will be necessary.
To assess the feasibility of the above therapeutic approach (see Introduction), the stability (or instability) of the amino acid sequence comprising the S1/S2 site was examined. Peptide sequences may be key to understanding the fitness-defining forces or requirements for protein design or evolutionary stability [52]. The discipline of molecular evolution was pioneered by R. F. Doolittle and B. Blombäck (Karolinska Institute, Sweden) [53]. However, obtaining insight into the evolutionary trajectory of the S1/S2 site of SARS-CoV-2 is difficult as proximally related coronaviruses lack the site in toto. Instead, we sought to gain insight by performing comparative sequence alignment with an avian infectious bronchitis virus (IBV) isolate, which belongs to a distinct genus.
IBV is the earliest coronavirus to be isolated [54–56]. IBV remains challenging to the commercial chicken industry globally due to the little cross-protection offered by vaccines against variants, resulting in a high rate of mortality. Following the IBV infection, symptoms appear within 24–48 hours [57]. The isolation of IBV was instrumental in identifying the causative agent for highly contagious respiratory disease afflicting chicks in North Dakota in 1931 [58]. The IBV strain Beaudette (IBV-Beaudette) was isolated by cultivating the IBV strain Massachusetts 41 (IBV-M41; isolated in the United States) in embryonated chicken eggs or cultured cells (primary chicken embryo kidney cells) [54, 59–61]. IBV-Beaudette has served as an in vitro infection model and can infect Vero monkey kidney cells, baby hamster kidney-21 (BHK-21) baby hamster kidney cells, or human cells due to adaptability [60, 62–67]. Unlike the virulent IBV-M41, IBV-Beaudette is nonpathogenic to chickens, and its attenuated virulence is associated with a reduced capacity of the spike protein to bind to target or altered replicase [58, 59, 68, 69].
The prototypic coronavirus IBV belongs to the genus Gammacoronaviridae and infects chickens’ respiratory, gastrointestinal, reproductive (female), and renal tissues [70]. The genomic structure of IBV consists of nonsegmented positive-sense single-stranded RNA (~27.6 kb) (Figure 1A). Occupying 5’ two-thirds of the IBV genome are the genes encoding polyproteins 1a and 1b, which generate 15 nonstructural proteins involved in transcription or RNA replication. The remaining 3’ portion of the IBV genome contains the genes encoding structural proteins (spike protein, envelope protein, membrane protein, nucleocapsid protein) and accessory proteins (3a, 3b, 5a, 5b) [69]. IBV delimits interferon production upon infection by suppressing host protein synthesis (via accessory protein 5b) [71]. Its spike protein is comprised of the S1 subunit, which contains the receptor binding domain, and S2 subunit containing two heptad repeat regions that undergo a conformational change to mediate the fusion of the viral envelope with the cell membrane. Though the S1 subunit binds to alpha-2,3 linked sialic acids, an additional determinant for IBV infection may exist [59, 63].
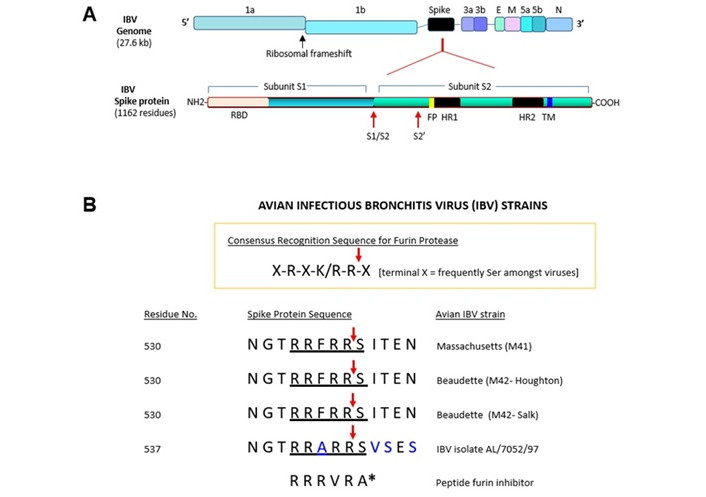
The S1/S2 furin cleavage site of spike protein in IBV isolate AL/7052/97. (A) A schematic showing structural and nonstructural genes of avian IBV genome (not to scale). 1a or 1b: open reading frame 1a or 1b; E: envelope protein; M: membrane protein; N: nucleocapsid protein; 3a, 3b, 5a, 5b: accessory proteins; SP: signal peptide; S1/S2: first furin cleavage site; S2’: second furin cleavage site; RBD: receptor binding domain; FP: fusion peptide; TM: transmembrane domain; HR1: heptad repeat 1; HR2: heptad repeat 2. The key domains of IBV spike protein are also shown. (B) The consensus sequence for the motif cleaved by furin is shown. The amino acid sequences of the S1/S2 site and the flanking regions of selected IBV strains and IBV isolate AL/7052/97 are listed. The residues that differ between the strains are indicated in blue letters. Red arrow denotes the predicted cleavage site by furin. Residue number corresponds to first amino acid of the peptide segment shown. The sources for the sequences are indicated in the text. Single letter codes are used to designate amino acids. For comparison, a noncovalent peptide furin inhibitor is shown, whose molecular structure is depicted in Figure 3B. A* denotes Amba (aminomethylbenzoic acid). IBV: infectious bronchitis virus; M41: Massachusetts 41; M42: Massachusetts 42
In the IBV-Beaudette spike protein, subunits S1 and S2 are generated through the proteolytic cleavage of the precursor protein (1,162 residues, including the signal peptide) [61]. In IBV-M41 spike protein, the N-terminal 253 residues mediate the binding of the coronavirus to the chicken respiratory tract [55]. Partial amino acid sequencing determined that the S2 subunit begins at residue Ser-538 [62]. The interconnecting peptide of Arg-Arg-Phe-Arg-Arg represents the first proteolytic cleavage site (S1/S2) in IBV-M41 and IBV-Beaudette [62]. Arg-Arg-Phe-Arg-Arg-Ser (residues 533–538) cleavage by furin was biochemically confirmed [60]. A second proteolytic cleavage site (i.e., ‘S2’) comprised of Arg-Arg-Arg-Arg-Ser or Arg-Arg-Lys-Arg-Ser (residues 687–691) that is cleavable by furin was found in IBV-Beaudette and other related strains [60]. The S2’ site arose due to the conversion of Pro-687 (IBV-M41) to Arg-687 through mutation (in IBV-Beaudette). The S1/S2 site promotes the infectivity of IBV-Beaudette in Vero cells or syncytium formation; the S2’ site is obligatory for furin-dependence [60]. The subsequent acquisition of the S2’ furin-cleavage site has expanded the cell and tissue tropism of IBV-Beaudette [67]. In the QX-type IBV strain, its acquiring of the S2’ site led to a significantly increased mortality in chickens [72].
IBV is the earliest coronavirus to be reported with a furin cleavage site [73]. In 1986, F. Hong et al. [74] determined the genomic sequence of IBV encoding peplomer (spike protein) at the Salk Institute (San Diego, United States). The institute was founded by J. Salk (University of Pittsburgh, United States) in consultation with F. Crick (Nobel Prize 1962, double helix). In 1955, J. Salk developed a vaccine against poliovirus (distributed by Eli Lilly & Company, Kiehl’s Pharmacy, etc.) along with A. Sabin (University of Cincinnati, United States). Potential immunogenic epitopes were identified by comparing IBV-M41 (used for vaccinating chickens) with IBV-Beaudette strains M42-Houghton and M42-Salk. This work was done with W. J. Spaan (University of Utrecht, The Netherlands) [74], who was visiting B. M. Sefton (Salk Institute; discovered tyrosine kinase with T. Hunter, which led to its inhibitor Gleevec, which is FDA-approved for treating chronic myelogenous leukemia, acute lymphoblastic leukemia, gastrointestinal stromal tumor) [61, 75]. In 2021, most vaccines developed against SARS-CoV-2 also targeted spike protein [76].
Previous comparison of SARS-CoV-2 and IBV (i.e., IBV-Beaudette) did not show homology at the S1/S2 site despite the aligning at the S2’ site [77]. This result may have been due to algorithmic bias that prioritizes longer homologies (at the expense of shorter homologies) to arrive at the ‘best fit’. In a report where the S1/S2 site of the above two viruses have been compared side by side, only two residues matched, i.e., Arg at P4 and Arg at P1 (residues are denoted as P5-P4-P3-P2-P1-P1’ with cleavage occurring at P1-P1’) of SARS-CoV-2 (Pro-Arg-Arg-Ala-Arg-Ser) and IBV-Beaudette (‘Beaudette US’ strain; Arg-Arg-Phe-Arg-Arg-Ser) [78].
To gain insight into the evolutionary trajectory of the S1/S2 site in SARS-CoV-2 spike protein, a novel search was conducted. To this end, the search was confined to the database of distantly related avian coronavirus IBV, which belongs to genus Gammacoronavirus (whereas SARS-CoV-2 belongs to genus Betacoronavirus). The IBV database consists of numerous genotypic and serotypic variants, resulting from a high mutation rate of RDRP and recombination [79, 80]. To avoid the algorithmic bias mentioned above, query consisted of merely the pentapeptide PRRAR (Pro-Arg-Arg-Ala-Arg) of SARS-CoV-2’s spike protein. The query sequence was screened against databases, which included non-redundant GenBank coding sequence translations, Protein Data Bank, SwissProt, Protein Information Resource, and Protein Research Foundation (excluding environmental samples from Whole Genome Shotgun projects) using BLASTp program.
It yielded 5 homologues that completely matched ‘RRAR’ of the query sequence. Further analysis revealed that the top 5 homologues, represented 3 distinct entities in essence. (1) Nucleocapsid protein (Infectious bronchitis virus; Seq. ID ACN82401.1) with a duplicate entry. (2) Polyprotein 1ab (Infectious bronchitis virus; Seq. ID QDQ69132.1), which is identical to polyprotein 1a (Infectious bronchitis virus; Seq. ID QDQ69120.1). These two homologues were not studied further (as they do not represent spike protein). (3) Spike glycoprotein S1 subunit partial (Infectious bronchitis virus; Seq ID: AAM34741.1). GenBank entry AAM34741.1 contained a partial coding sequence of spike glycoprotein S1 subunit of avian IBV isolate AL/7052/97.
Previous works have identified ‘X-R-X-K/R-R-X’ (R, arginine) as the consensus recognition motif for furin proteases. In ‘XBXBBX’, B and X represent basic and hydrophobic residues, respectively [81]. Furin proteolytically cleaves between the terminally located Arg and X residue (terminal X is serine in many viruses). In Figure 1B, the amino acid sequence of the S1/S2 site in spike protein of IBV-M41, IBV-Beaudette M42-Houghton [82], and IBV-Beaudette M42-Salk are shown [69, 74, 83]. The corresponding amino acid sequence of IBV isolate AL/7052/97 (GenBank entry AAM34741.1), which conforms to the consensus recognition motif of furin, is also shown.
The results from the BLAST search revealed that Arg-Arg-Ala-Arg is identical between the spike proteins of SARS-CoV-2 and IBV isolate AL/7052/97. This information was used as the starting point to align peptide sequence containing the S1/S2 furin cleavage site of both viruses bidirectionally. The identity of amino acid at a given point in the protein sequence is dictated by the corresponding codon in mRNA (B. Holley, Salk Institute; H. G. Khorana, Massachusetts Institute of Technology; M. Nirenberg, National Institute of Health, United States, Nobel Prize 1968, genetic code). Hence, comparisons were made at both the nucleotide and amino acid levels. The nucleotide sequence comprising codons was instrumental in aligning amino acid sequences.
The amino acid sequence and nucleotide sequence of SARS-CoV-2 were obtained from GenBank entry MZ027645.1 [Severe acute respiratory syndrome coronavirus 2 isolate SARS-CoV-2/human/USA/CR0060_spike/2020 surface glycoprotein (S) gene, complete cds]. For IBV isolate AL/7052/97, GenBank entry AF509583.1 (Avian infectious bronchitis virus isolate AL/7052/97 spike glycoprotein S1 subunit gene, partial cds) provided both nucleotide sequence and amino acid sequence while GenBank entry AAM34741.1 [spike glycoprotein S1 subunit, partial (infectious bronchitis virus)] provided amino acid sequence. In the 14-residue segment shown, Arg-Arg-Ala of the above tetrapeptide is encoded by CGG-CGG-GCA (note: bold letters denote identical nucleotides throughout the text) in SARS-CoV-2 (Figure 2A). The identical residues in the IBV isolate AL/7052/97 are encoded by a closely matching sequence of CGG-CGU-GCU. Interestingly, the nucleotides that differ (i.e., A versus U) are complementary, a pattern frequently repeated amongst the rest of identical residues (see below).
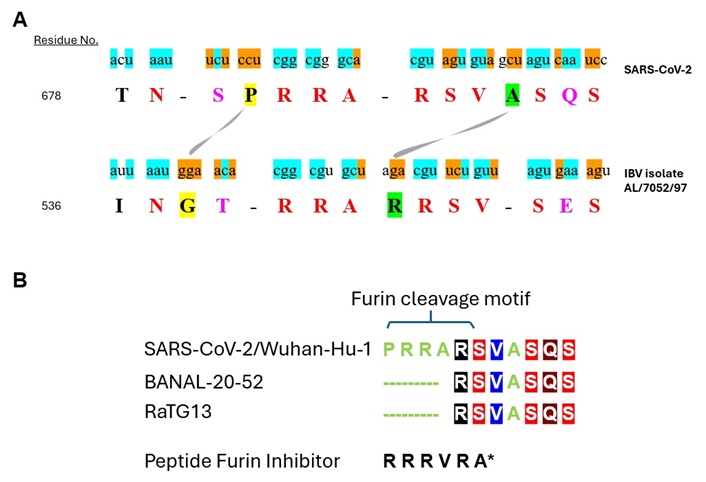
Comparative sequence alignment of the peptide segment spanning the S1/S2 furin cleavage site in spike protein of SARS-CoV-2 and IBV isolate AL/7052/97. (A) IBV database was searched using the pentapeptide PRRAR (Pro-Arg-Arg-Ala-Arg) in spike protein of SARS-CoV-2 as the query. Arg-Arg-Ala-Arg of the query completely matched the spike protein of IBV isolate AL/7052/97. The aligned region extended to flanking residues. The aligned amino acid sequence (capital letter) of SARS-CoV-2 and IBV isolate AL/7052/97 are shown. The residue number refers to the initial amino acid in the peptide sequence. Identical amino acids are indicated in red letter while conserved residues are designated in purple letter. Residues being compared are highlighted yellow or green and connected by a curved line. The nucleotide sequence (lower key letter) encoding the peptide sequence is also shown. Identical nucleotides are highlighted blue while complementary nucleotides are highlighted orange. (B) Multiple sequence alignment of the juxtaposed residues located downstream of the S1/S2 site in spike protein of SARS-CoV-2, RaTG13, and BANAL-20-52. The sources for the sequences listed are described in the text. Amino acids are designated in single letter codes. For therapeutic consideration, a noncovalent peptide furin inhibitor is shown, whose molecular structure is depicted in Figure 3B. A* denotes Amba (aminomethylbenzoic acid). IBV: infectious bronchitis virus
In assessing the evolutionary relationship between distinct proteins based on the homology, a meaningful comparison may require aligning of the amino acid sequences comprising a common secondary structure of concern. Such endeavor, however, may be difficult for spike protein as its S1/S2 furin cleavage site is thought to assume a random coil conformation in vivo as implied from the inability to detect a defined structure using cryoelectron microscopy or X-ray diffraction crystallography [84]. As an alternative, alignment was performed using the parceled sequences to account for potential oligopeptides that may match in a three-dimensional context but may be separated by intervening amino acids in the linear peptide (thus difficult to match in a two-dimensional context).
Though not apparent at the outset, we then discovered that the alignment extends to the juxtaposed residues located downstream. As described above, Arg (which succeeded Arg-Arg-Ala described above) is also identical between the two viruses. However, the codon (CGU) for Arg-685 in SARS-CoV-2 does not match the codon (AGA) for Arg-543 located at the equivalent position in IBV isolate AL/7052/97. Instead, it matches the codon (CGU) for the subsequent residue Arg-544 in IBV isolate AL/7052/97 (Figure 2A). It unexpectedly led to the recognition that Arg-Ser-Val that succeeds Arg-Arg-Ala is also identical between the two viruses. Aside from identical nucleotides, the nucleotides that differ between the respective codons are also complementary: CGU-AGU-GUA in SARS-CoV-2 versus CGU-UCU-GUU in IBV isolate AL/7052/97.
Further downstream, Ser-Gln-Ser in SARS-CoV-2 nearly matched Ser-Glu-Ser of the IBV isolate AL/7052/97. Gln-690 of SARS-CoV-2 and Glu-548 of IBV isolate AL/7052/97 represent two amino acids with structurally similar side chains. In addition to identical nucleotides, complementary nucleotides are also found between their respective codons where sequences differed: AGU-CAA-UCC in SARS-CoV-2 versus AGU-GAA-AGU in IBV isolate AL/7052/97.
Intriguingly, the above alignment uncovered that SARS-CoV-2 is missing a residue at the equivalent position for Arg-543, which is located between Arg-Arg-Ala and Arg-Ser-Val in IBV isolate AL/7052/97. Instead, SARS-CoV-2 contains Ala-688 located between Arg-Ser-Val and Ser-Gln-Ser (see green highlighted residues connected by a curved line in Figure 2A). Potentially, the presence of Ala-688 may have compensated for the missing residue to keep the spatial gap between Arg-Arg-Ala and Ser-Gln-Ser in SARS-CoV-2 in synchrony with that between Arg-Arg-Ala and Ser-Glu-Ser in the IBV isolate AL/7052/97. Thus, the disruptive effect on the local conformation caused by the missing residue may have been compensated by the presence of Ala-688 to restore the corresponding structural information. Previously, it was shown that compensatory mutations are often local and the second mutation undoes the insult on 3-dimensional structure caused by the first mutation [85]. Out of three nucleotides comprising their codons, two are complementary (i.e., AGA for Arg-543 in IBV isolate AL/7052/97 versus GCU for Ala-688 in SARS-CoV-2), with the remaining nucleotide being purine (adenine or guanine) in both viruses. A notable consequence of the above is its negative impact on the furin cleavage site. Hence, the displacement of the residue may have led to the evolution of a suboptimal substrate for furin in SARS-CoV-2. Consistently, Pro-Arg-Arg-Ala-Arg-Ser at the S1/S2 boundary of SARS-CoV-2 poorly adheres to the consensus recognition motif of ‘X-R-X-K/R-R-X’ (R, arginine) for furin.
A similar scenario unfolds in the sequence upstream of Arg-Arg-Ala-Arg. Of 4 residues located upstream, Asn and the corresponding codon AAU are identical in both viruses. The preceding residue, i.e., Thr (codon ACU) in SARS-CoV-2 or Ile (codon AUU) in IBV isolate AL/7052/97, differ by a single nucleotide (pyrimidine in both viruses) in their respective codons. The subsequent residue, i.e., Ser (codon UCU) in SARS-CoV-2 or Thr (codon ACA) in IBV isolate AL/7052/97, represent conserved residues, whose codons differ by two nucleotides, which are complementary. Interestingly, the remaining residue Pro (codon CCU) in SARS-CoV-2 or Gly (GGA) in IBV isolate AL/7052/97 (yellow highlighted residues connected by a curved line in Figure 2A) also appears displaced, i.e., positioned on the opposite sides of the conserved residue Ser/Thr, with their codons entirely encoded by complementary nucleotides.
In the aligned region, the nucleotides in the coding sequence of SARS-CoV-2 and IBV isolate AL/7052/97 displayed 14 identities, three transitions (C>U or U>C, two; G>A or A>G, one), and 15 transversions (U>A or A>U, eight; C>G or G>C, six; G>U or U>G, one). Transversion occurred more commonly than transition (15/3) for substitutions, and complementary base occurred frequently (14/15) for transversions. Presumably, the region may have undergone a series of complex genetic events during its ontogeny (see below).
The alignment of the downstream juxtaposed sequence of the S1/S2 furin cleavage site despite the evolutionary distance between SARS-CoV-2 and IBV isolate AL/7052/97 prompted us to examine coronaviruses proximally related to SARS-CoV-2 [86]. In Figure 2B, the presence of Arg-Ser-Val and Ser-Gln-Ser in the spike protein from SARS-CoV-2 isolate Wuhan-Hu-1 is shown. The corresponding amino acid sequence was obtained from GenBank entry MN908947.3 (Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome). Other SARS-CoV-2 strains also retain the identical residues, ex., SARS-CoV-2-6XR8, SARS-CoV-2-7C2L, SARS-CoV-2-6XCN [77]. Arg-Ser-Val and Ser-Gln-Ser are also present in subsequently emerged SARS-CoV-2 variants, ex., Alpha (B.1.1.7), Delta (B.1.617.2), and Omicron (B.1.1.529) [87]. The genomic sequence of RaTG13 bat coronavirus shares 96.1% identity with SARS-CoV-2 [88–90]. Arg-Ser-Val and Ser-Gln-Ser are found in the spike protein from RaTG13 (Figure 2B). The amino acid sequence of RaTG13 was retrieved from GenBank entry MN996532.2 (Bat coronavirus RaTG13, complete genome). The genomic sequence of BANAL-20-52 bat coronavirus (isolated in Laos) shares 96.8% identity with SARS-CoV-2. At the protein level, 16 out of 17 residues interacting with human angiotensin converting enzyme-2 receptor are conserved between BANAL-20-52 and SARS-CoV-2, significantly higher than RaTG13 (11 out of 17). Arg-Ser-Val and Ser-Gln-Ser are present in the spike protein from BANAL-20-52 (Figure 2B) as well as BANAL-20-103 and BANAL-20-236. Their amino acid sequences were obtained from the report by S. Temmam et al. [90]. Taken together, the above data, which are consistent with previous reports [77, 78], portray an evolutionarily dynamic nature of the S1/S2 site.
This view is supported by a high degree of recombination observed around the spike protein [91]. Of potential relevance is ‘microhomology-mediated template switching’ by RDRP, which is facilitated by the complementarity between donor and acceptor sites occurring intragenomically [45]. The S1/S2 site of SARS-CoV-2 spike protein exhibits a high degree of variant frequencies [45]. RDRP may cause insertion or multiple template-switching events followed by realignment to correct sequence, which may explain triplet substitution (GGG to AAC) associated with Omicron lineage. SARS-CoV-2 variant Alpha from the 20B clade is associated with three nucleotide substitutions (CTA), which are complementary. SARS-CoV-2 variant Gamma from the 20B clade features two nucleotide substitutions (TC), which are complementary [45]. Despite these alterations, the reading frame for translation remains intact. Additionally, RDRP may cause deletion via slippage at repeat sequences. Similarly, T7 RNA polymerase may undergo microhomology-mediated template switching to generate insertion or deletion and accrue a high level of transversions (C>A) in transcripts [45].
The medical community has recently been confronted with the latest SARS-CoV-2 coronavirus strain Omicron [92, 93]. A significant fraction of the mutations occurred in spike protein, with many of them in the receptor binding domain [94]. Mutations also affected RDRP [95]. Another report evaluated the efficacy of vaccines targeting spike protein against Omicron [96]. A similar study was conducted with antibody-based drugs targeting spike protein.
The results from the preceding sections (The evolutionary trajectory of the S1/S2 furin cleavage site in SARS-CoV-2 spike protein and Comparative sequence alignment of the S1/S2 site in SARS-CoV-2 and the IBV isolate AL/7052/97) prognosticate an evolutionary continuum at the S1/S2 site. To ascertain, mutations documented at the S1/S2 site in subsequently arisen SARS-CoV-2 variants were interrogated. For this, only the naturally occurring mutations associated with variants in published articles or archived databases were examined. Mutations that were introduced artificially were excluded from analysis—e.g., mutations inserted to stabilize spike protein for X-ray diffraction study (Figure 3A). For reference sequence, SARS-CoV-2 strain Wuhan-Hu-1 (GenBank entry NC_045512) was used [97]. Point mutations that occurred in the S1/S2 site include: P681H, P681R [98]; P681L [99]; R685S (Protein Data Bank entry QZR93666.1); A684S, A684T, S686G, R682W, R682Q, A684V [100]; P681S, R683P, R683Q, A684E [97]. Deletions found in the S1/S2 site include P681(-), A684(-), R685(-) and S686(-) [100]. The sequences listed may not represent the full repertoire of the S1/S2 site mutations documented to date.
The S1/S2 furin cleavage site mutations found in spike protein of SARS-CoV-2 variants. (A) Residue number corresponds to the mutated amino acid in SARS-CoV-2 spike protein. Only the mutations documented in naturally occurring SARS-CoV-2 variants are listed. Their order does not reflect the time point of their emergence. The S1/S2 furin cleavage site sequence of the reference strain Wuhan-Hu-1 is shown at top. The sources for the sequences listed are described in the text. Amino acids are designated in single letter codes. Mutated residues are highlighted in red. Deleted residues (denoted by hyphen) are highlighted in blue. The sequences listed may not represent the full repertoire of the S1/S2 site mutations accrued to date. (B) A 3-dimensional view of inhibitor-bound furin. The molecular structure of proprotein convertase furin bound to Arg-Arg-Arg-Val-Arg-Amba is shown. The 3-dimensional structure of furin bound to the hexapeptide inhibitor was generated by downloading the structural coordinates for PDB ID 6EQX and using PyMOL to re-enter the image of the enzyme-inhibitor complex. Space-filling model shows the structure of the hexapeptide
We also note that, in addition to the mutations described above, multiple other mutations have been documented at various regions outside the S1/S2 site in spike protein amongst SARS-CoV-2 variants. These include K417N, E484A, N501Y, and D614G, with some of them occurring in the binding domain for the receptor, angiotensin-converting enzyme 2 (ACE2). The evolutionary progression of SARS-CoV-2 continues, and JN.1 has dominated variants derived from the XBB lineage. Among the resultant subvariants KP.2 and KP.3, KP.3.1.1 represents the predominantly circulating strain in the U.S. The latter is associated with the F456L, R346T, and Q493E mutations. The variant XEC, which arose through recombination between variants KS.1.1 and KP.3.3, was initially detected in Germany. It harbors the mutations T22N and Q493E, and its transmission potential is being assessed.
Taken together, the overall data support the view that the S1/S2 furin cleavage site of spike protein continues to evolve in SARS-CoV-2.
SARS-CoV-2 is a positive-stranded RNA virus that belongs to the subgenus Sarbecoviruses (order Nidovirales; family Coronaviridae; subfamily Orthocoronavirinae; genus Betacoronavirus) [101, 102]. SARS-CoV-2 is internalized following the interaction of its spike protein with the surfacially expressed ACE2, which hydrolyzes the vasoconstrictor angiotensin II [103]. Internalization of SARS-CoV-2 may occur via distinct pathways [98]. Its spike protein contains two proteolytic cleavage sites, i.e., S1/S2 located at the junction between S1 and S2 subunits and S2’ located within the S2 subunit [98, 104]. Despite the cleavage at the S1/S2 site (priming), the S1 and S2 subunits are held together via noncovalent interactions whereas the cleavage at the S2’ site triggers structural rearrangement to expose the ‘fusion peptide’ to engage cell membrane for internalization [105]. The cleavage at the S1/S2 by furin may occur during the virus maturation in SARS-CoV-2-producing cells [98]. One entry route involves cleaving SARS-CoV-2 spike protein (internalized via clathrin-mediated endocytosis after binding to ACE2) at the S2’ site by cathepsin to enable membrane fusion necessary for cytosol entry. For direct entry at the cell surface, the S2’ site is cleaved by transmembrane protease, serine 2 (TMPRSS2) to allow fusion of the viral envelope with the cell membrane. Cleavage of the S2’ site by furin was also reported [106].
Multiple coronavirus species, whose genomic RNA sequences align closely with that of SARS-CoV-2, have been identified [86]. SARS-CoV-2 spike protein contains S2’ furin cleavage site (PSKPSKRSFIEDLLFNKV: furin recognition motif in italics), which includes Arg-815 (GenBank entry MN908947.3, ‘Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1’) [107]. Likewise, the S2’ site is also present in the spike protein of closely related coronaviruses: bat coronavirus RaTG13 (PSKPSKRSFIEDLLFNKV, GenBank entry MN996532.2, ‘Bat coronavirus RaTG13’), human SARS-1 coronavirus (PLKPTKRSFIEDLLFNKV, GenBank entry AY278741.1, ‘SARS coronavirus Urbani’), bat coronavirus Rs4231 (PLKPTKRSFIEDLLFNKV, GenBank entry KY417146, ‘Bat SARS-like coronavirus isolate Rs4231’), and bat coronavirus BtRs-BetaCoV/YN2018B (PLKPTKRSFIEDLLFNKV, GenBank entry MK211376.1, ‘Coronavirus BtRs-BetaCoV/YN2018B’). Intriguingly, most proximally related coronaviruses lack the S1/S2 site despite the presence of the S2’ site.
The acquisition of the S1/S2 furin cleavage site in spike protein is critical for the expanded tropism (plus human transmission) of SARS-CoV-2 [108]. The presence of both the S1/S2 site and the S2’ site in spike protein of SARS-CoV-2 was confirmed using furin inhibitors [105]. Consistently, the absence of the S1/S2 site reduced the pathogenicity of SARS-CoV-2 [109]. Further, ectopically introducing the S1/S2 furin cleavage site to the SARS-CoV-1 spike protein (which lacks the S1/S2 site) increased fusogenicity [110, 111]. Thus, the S1/S2 site may represent a critical determinant for SARS-CoV-2 infectivity. Subsequent SARS-CoV-2 variants such as Omicron exhibited increased dissemination despite reduced virulence [73].
To delimit tropism, an optimal strategy for disabling the S1/S2 site was sought, which could exploit the hypermutability of SARS-CoV-2. Inhibiting furin may cause adverse side effects as the enzyme participates in various normal physiological functions. The structural analysis of furin revealed that its active site for catalysis consists of a groove for binding substrates, which is lined with numerous negatively charged amino acids—i.e., Asp and Glu. Consistently, peptides containing positively charged polyarginine represent potent inhibitors of furin. As prior screenings may have relied on assays that monitor the inhibition of furin-like activity in cell lysate, inhibitors with higher selectivity for furin were sought. The molecular structure of proprotein convertase furin bound to the hexapeptide Arg-Arg-Arg-Val-Arg-Amba is shown (Figure 3B).
Due to its random-coil secondary structure, sterically blocking access to furin by using antibodies to mask the S1/S2 site of spike protein may be difficult [112]. An alternative strategy is to utilize oligonucleotides to target the S1/S2 site for pharmacological inhibition at the nucleic acid level: (1) as the S1/S2 furin cleavage site is retained by SARS-CoV-2 for propagation, it represents a valid therapeutic target; (2) targeting RNA than protein is more efficacious as it lies upstream in genetic information flow; (3) oligonucleotides may confer targeting specificity at the nucleotide sequence level; (4) oligonucleotides could trigger genomic RNA degradation via RNA interference (ex., siRNA) or RNAse H (ex., gapmer); (5) antisense oligonucleotides (ex., morpholino) can interfere with translation or RNA processing; (6) antisense oligonucleotides targeting SARS-CoV-2 genome have been reported, indicative of its feasibility; (7) oligonucleotide drugs may provide virus-specificity at the genetic level; (8) unlike conventional structure-activity relationship-based chemical drug development, only mRNA sequence is necessary to design oligonucleotide drugs.
Briefly, developing the chemical method underlying oligonucleotide synthesis entailed several major advances. In 1955, A. Todd (University of Cambridge, England; Nobel Prize 1957, nucleotide, nucleotide coenzyme) and A. M. Michelson reported the synthesis of dithymidine dinucleotide, which involved condensing of thymidine 3’-(benzyl phosphorochloridate) 5’-(di-benzyl phosphate) with 3’-O-acetylthymidine [113]. In 1958, H. G Khorana and P. T. Gilham (British Columbia Research Council, Canada) [114] introduced the phosphodiester method for oligonucleotide synthesis, which utilized N,N’-dicyclohexylcarbodiimide as activating agent; however, branching at internucleosidic phosphate remained problematic. In 1969, R. L. Letsinger and K. K. Ogilvie (Northwestern University, United States) [115] described a phosphotriester approach to resolve the above issue by protecting the phosphate moiety and subsequently implemented a solid-phase synthesis method. In 1975, W. B. Lunsford (a former colleague of W. Letsinger; Baylor University, United States) and colleagues [116] reported phosphite triester methodology, which uses 3’-O-chlorophosphite containing more reactive phosphorus(III) to form internucleosidic linkage, with whom M. Castro and F. Hong worked on oligonucleotide synthesis. In 1981, F. Hong worked on cloning of E. coli genes encoding the phosphate transferase system with S. Roseman (discoverer of phosphate transferase system, Johns Hopkins University, United States; a former colleague of H. G. Khorana) [117]. In 1981, M. H. Caruther (a former colleague of W. Letsinger; University of Colorado, United States) and M. D. Matteucci [118] used nucleoside phosphoramidites as a precursor for oligonucleotide synthesis, which utilizes 2-cyanoethyl phosphite protecting group. Critical to current solid-phase oligonucleotide synthesis using deoxynucleoside phosphoramidites was the solid support-based peptide synthesis methodology developed by R. B. Merrifield (Rockefeller Institute, United States; Nobel Prize 1984) [119] in 1963.
Synthetic oligonucleotides have been used extensively in various molecular biological applications, i.e., gene cloning, sequencing, site-directed mutagenesis, analytical assay (ex., microarray), gene expression analysis (ex., fluorescent in situ hybridization), gene synthesis, etc. These include diagnostic assays (e.g., polymerase chain reaction to identify genetic mutants, DNA paternity testing). The use of oligonucleotides has also been extended to therapy. In 1978, M. L. Stephenson and P. C. Zamecnik (Harvard University, United States) [120] described suppressing the translation of Rous sarcoma virus 35S RNA using oligonucleotides. Rous sarcoma virus, which causes chicken cancer, was discovered by P. Rous (Rockefeller Institute for Medical Research, United States; Nobel Prize 1966) [121] in 1911. Following the discovery of RNA interference by A. Fire (Carnegie Institution of Washington, United States; Nobel Prize 2006) et al. [122], the potential utility of siRNA as a pharmaceutical agent was investigated. Its advantages include targeting mRNA rather than protein, exploiting the host cell’s RNA interference complex, its repeated use for mRNA degradation, etc. For clinical application, multiple issues need to be addressed, i.e., inciting of innate immunity, degradation by ribonucleases, off-target suppression, selective delivery to intracellular target, etc. Critical to resolving these issues is the ability to chemically modify oligonucleotides. For instance, to protect from nucleases, internucleosidic phosphodiester linkage may be modified to contain phosphorothioate. Alternatively, sugar moiety may be replaced with locked nucleic acid (i.e., bridged nucleic acid) or phosphoramidate morpholino oligomer [123]. Modified nucleobases (ex., pseudouridine) may be incorporated to attenuate immunogenicity [124]. To reduce side effects caused by off-targeting, specific modifications may be introduced to the sugar moiety [125].
Various oligonucleotide-based therapeutics (ex., siRNA, gapmer, antisense oligonucleotides, aptamer, morpholino oligonucleotide) have been approved by FDA for clinical use. The siRNA drugs approved by FDA include patisiran (hereditary transthyretin-mediated amyloidosis), givosiran (acute hepatic porphyria), lumasiran (primary hyperoxaluria type 1). Gapmers represent single-stranded oligonucleotides composed of DNA and RNA, which rely on the intracellular enzyme RNase H to degrade target mRNA. Gapmers that have been FDA-approved include mipomersen (homozygous familial hypercholesterolemia), and inotersen (familial amyloid neuropathies). For targeted delivery to the liver, siRNA drugs were conjugated to N-acetylgalactosamine (GalNAc) [126]. For tumor-specific delivery, siRNA therapeutics targeting ribonucleotide reductase were conjugated to tumor-specifically internalizing peptide HN-1 [26, 35]. Several phosphoramidate morpholino oligomers have been FDA-approved to modulate dystrophin exon splicing defects associated with Duchenne’s X-linked genetic disorder’s muscular dystrophy [127].
Multiple oligonucleotide therapeutics targeting viral RNA have been reported. For SARS-CoV-1, siRNAs have been developed that target the genomic RNA sequence encoding ORF1b (NSP12) or spike protein [128]. To inhibit the replication of SARS-CoV-1, its ‘leader sequence’ has been targeted using siRNA [129]. For SARS-CoV-2, modified antisense oligonucleotides (ex., morpholino-based oligonucleotides, gapmers) have been devised to suppress translation or promote degradation [130, 131]. Knockdown of TPPRSS2 expression by using ‘cell penetrating peptide-conjugated phosphorodiamidate morpholino oligomer (PPMO)’ reduced the cytopathic effect of SARS-CoV-2 in vitro [78].
A potential consequence of applying the above-described virolytic pressure is that resistant variants (if emerge) may carry mutation in the S1/S2 site-encoding sequence to abrogate hybridizing to the oligonucleotides, which is facilitated by its hypermutability. The altered sequence, by default, may encode a degenerated S1/S2 site that is no longer recognizable to furin for proteolytic cleavage, thus delimiting the tropism of SARS-CoV-2. Previous reports documenting reduced viral fitness conferred by resistance mutations set the precedent for the above. Firstly, in the case of cancer, drug treatment may induce evolution in treated cells toward acquiring resistance [132]. For RNA viruses, resistance to siRNA drugs may be acquired via incurring mutation in the target sequence to yield a mismatched template that prevents hybridization. In the case of the human immunodeficiency virus (HIV), viral escape variants that arose following the treatment with Nef-targeting siRNA accumulated mutation in target sequence, i.e., partial or complete deletion, nucleotide substitution [133, 134]. Secondly, resistance mutation may reduce viral fitness. In the case of the influenza virus, mutations that caused resistance to Oseltamivir, which inhibits neuraminidase, reduced replicative fitness [135]. Decreased fitness may occur when mutations occur in highly conserved sequences in the viral genome [134]. The high mutation rate may cause RNA viruses to exist as a population of ‘quasi-species’, i.e., closely related mutants [135]. It, in turn, may elevate the extent of genetic variability, increasing the likelihood of yielding variants that are less competent in replication. Consistently, mutations occurring in functionally significant regions were detrimental to hepatitis C virus [136]. For highly evolving RNA viruses, antiviral drugs inhibiting critical viral proteins may induce mutations that impede viral replication or pathogenesis.
Significant advances have been made in our understanding of biological organisms at the genetic level. Nonetheless, the genetic information gained may need to be interpreted in the context of their evolving nature given its fluidity. Earlier, molecular evolution was examined via the analysis of fibrinogen [137]. Yet, our understanding of the nature of the forces driving evolution appears incomplete—as was apparent from the inability to predict the emergence of SARS-CoV-2 despite years of research. Presented in this report is a therapeutic strategy through which the genetic hypermutability of SARS-CoV-2 may be exploited to attenuate infectivity. ‘Targeting oligonucleotide directed devolution’ entails applying virolytic pressure by inducing genomic RNA degradation to drive molecular evolution towards selecting degenerative S1/S2 site mutations in naturally occurring resistant variants. The possibility of resistance mutations occurring at a distant site interfering with oligonucleotide hybridization at the S1/S2 site structurally cannot be excluded. Conceivably, this approach may target multiple critical domains within a single viral gene, may target multiple essential viral genes, or may be applied repeatedly to ensuing variants. As an alternative approach, oligonucleotides may be used for ‘therapeutic RNA editing’ to inactivate the S1/S2 site. Adenosine deaminases acting on RNA (ADAR) represent intracellular enzymes which catalyze the hydrolytic deamination of adenosine to inosine. To treat genetic disorders caused by base substitution, Woolf (Ribozyme Pharmaceuticals, Inc., United States) et al. [138] used complementary RNA oligonucleotide to hybridize to mutant dystrophin mRNA encoding a premature UAG stop codon. The endogenously expressed double-stranded RNA adenosine deaminase recognized the duplex and converted to inosine, allowing translation to resume [138]. All in all, we hope these novel approaches may lead to the development of a potential deterrence against rapidly evolving pathogens like SARS-CoV-2 that exacerbate the disease burden caused by cancer [139].
ACE2: angiotensin-converting enzyme 2
ADAR: adenosine deaminases acting on RNA
Amba: aminomethylbenzoic acid
BLASTp: protein-protein Basic Local Alignment Search Tool
CDK: cyclin-dependent kinase
COVID-19: coronavirus disease 2019
FDA: U.S. Food and Drug Administration
IBV: infectious bronchitis virus
IBV-Beaudette: infectious bronchitis virus strain Beaudette
IBV-M41: infectious bronchitis virus strain Massachusetts 41
PyMOL: Python-enhanced molecular graphics
RB: retinoblastoma
RDRP: RNA-dependent RNA polymerase
SARS-CoV-1: severe acute respiratory syndrome coronavirus 1
FUH: Conceptualization, Data curation, Investigation, Formal analysis, Methodology, Writing—original draft. KDL: Methodology, Writing—review & editing. MMC: Resources, Writing—review & editing. All authors have read and approved the submitted version.
All authors declare that they have no conflicts of interest.
Since all the original data in this study were de-identified and obtained from open databases, ethical approval was not required.
Since all the original data in this study were de-identified and obtained from open databases, consent to participate was not required.
Not applicable.
The URLs for database-sourced data can be found in the Materials and methods section. Other data are available from the corresponding author upon reasonable request.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3247
Download: 157
Times Cited: 0
