
Abstract
Cancer remains the second leading cause of death globally, posing an ongoing threat to public health. A hallmark of cancer cells is their capacity to invade adjacent tissues and evolve into malignant forms, often resulting in aggressive tumors resistant to conventional treatments. At the heart of this therapeutic challenge are cancer stem cells (CSCs), which possess distinctive capabilities for self-renewal, differentiation, and generation of diverse tumor cell populations. These CSCs have been identified across multiple tissue types, including lung, colon, breast, pancreas, and ovary. Research has demonstrated that CSC subpopulations contribute significantly to therapeutic resistance, tumor recurrence, and metastasis by regulating multiple signaling pathways, making them compelling targets for cancer therapy. Notably, emerging evidence suggests that natural products may offer protective benefits against cancer development while potentially targeting CSCs. This review synthesizes current knowledge of CSCs, examining their identifying markers, isolation techniques, study methods, and associated signaling pathways. Additionally, we explore various natural products that specifically target CSCs across different cancer types, presenting potential strategies to address the persistent challenges of drug resistance and cancer relapse.
Keywords
Phytochemicals, stem cell markers, signaling pathwaysIntroduction
Cancer represents one of the leading causes of death globally, with GLOBOCAN data reporting over 20 million new cases and 9.2 million deaths worldwide in 2022 [1]. These numbers are projected to increase dramatically, with estimates suggesting approximately 30 million cases and 16 million deaths by 2040 [1]. In the United States alone, projections indicate approximately 1,958,310 new cancer cases and 609,820 deaths annually, equivalent to about 1,670 deaths per day [2]. The hallmark of cancer is uncontrolled cell proliferation and invasion into surrounding tissues, with metastasis representing its most aggressive form. Despite significant advances in treatment modalities, including surgery, chemotherapy, radiotherapy, targeted therapy, and immunotherapy, therapeutic success remains limited due to metastasis, recurrence, therapy resistance, tumor heterogeneity, and immune evasion [3]—characteristics largely attributed to cancer stem cells (CSCs).
CSCs represent distinct subpopulations within tumors that share functional characteristics and cellular properties with normal stem cells [4]. Just as normal stem cells can self-renew and differentiate to generate specialized cells within a tissue, the CSCs have self-renewal capacity and the ability to differentiate into multiple lineages, contributing to tumor heterogeneity [5]. However, one important difference between normal stem cells and CSCs is that metastatic potential is a characteristic specific to CSCs and not shared by normal stem cells. Bonnet and Dick [6] first identified leukemia stem cells in acute myeloid leukemia (AML), demonstrating that CD34+CD38− cells could differentiate and proliferate in severe combined immunodeficiency disease (SCID) mice. Since then, CSCs have been recognized in several solid tumors, including breast, colorectal, lung, pancreatic, and brain cancers [7–11]. These cells can be identified through their expression of specific stemness-related markers, though some markers may be intracellular and vary in specificity across tumor types [11, 12]. Given their central role in cancer progression and metastasis, CSCs have emerged as crucial therapeutic targets in efforts to improve patient survival, enhance quality of life, and prevent cancer recurrence.
The role of CSCs in chemoresistance/relapse
Chemoresistance remains a critical challenge in cancer treatment, particularly due to intrinsic resistance stemming from intratumoral heterogeneity. At the heart of this resistance are CSCs, which comprise less than 1% of the tumor cell population but possess crucial abilities to self-renew and differentiate [13, 14]. While conventional treatments such as radiotherapy and chemotherapy can damage proliferating cells and suppress tumor growth, their effectiveness is limited, especially in advanced-stage patients, due to the presence of CSCs [15, 16]. Although these therapies successfully eliminate most tumor cells, they often fail to eradicate the CSC population [17].
CSCs employ multiple mechanisms to evade therapeutic interventions. First, they can regulate their cell cycle to maintain a quiescent state, making them less susceptible to conventional treatments that target actively dividing cells [18, 19]. Second, CSCs enhance their survival by deregulating apoptotic pathways, particularly through the overexpression of inhibitors of apoptotic proteins (IAP), which promote the degradation of pro-apoptotic proteins [20]. The complexity of targeting CSCs is further complicated by several factors. Single-target molecular therapies often prove ineffective, as blocking one signaling pathway can trigger compensatory survival pathways that promote CSC growth and chemoresistance [21, 22]. Additionally, CSCs express high levels of transporters of the ATP-binding cassette (ABC) family, including ABCB1, ABCG2, and ABCB5 [23, 24]. These transporters work to actively expel drugs from cells, reducing their intracellular concentration and effectiveness.
Perhaps most challenging is the inherent plasticity and heterogeneity of CSCs within tumors, allowing them to adapt to environmental changes and evade therapeutic effects. One critical adaptation mechanism is epithelial-mesenchymal transition (EMT), through which CSCs lose their cell-cell and cell-matrix adhesion properties while gaining mesenchymal characteristics [25, 26]. This transition enhances their invasive and metastatic potential, enabling migration to surrounding tissues and distant sites. Despite our understanding of these resistance mechanisms, conventional therapies remain ineffective at completely eradicating CSCs from tumor populations, leading to recurrence and treatment failure. This persistent challenge underscores the need for novel therapeutic strategies specifically targeting CSC populations and their resistance mechanisms.
CSC markers
A major challenge in CSC research is identifying which tumor cell subpopulations possess stem-like properties. While the gold standard for confirmation remains the ability to form tumors when injected at ultralow numbers into immunocompromised animals, this approach’s time-consuming and costly nature has driven researchers to seek alternative methods, particularly through the identification of stem cell markers [27].
These markers are specific proteins or molecules expressed on CSC surfaces that enable researchers to distinguish them from other cancer cells [27]. Through such markers, scientists have successfully identified CSCs across various tumor types [28]. However, a significant limitation exists: no universal marker is exclusive to CSCs, as many markers are shared with normal stem cells. These shared identifiers are termed “inclusive CSC markers”. This overlap in marker expression manifests in several ways. The Yamanaka factors (OCT4, SOX2, KLF4, and MYC) serve as stem cell markers but are expressed in both CSCs and normal stem cells [29]. Similarly, certain clusters of differentiation (CD) markers, including CD19, CD24, CD38, and CD44, originally identified as lymphocyte surface markers, are also present on the surface of CSCs. The range of stem cell markers extends to include epithelial cell adhesion molecules (EpCAM), CD133, aldehyde dehydrogenase (ALDH), CD44, CD24, and doublecortin-like kinase 1 (DCLK1), which can be expressed both on the cell surface and intracellularly across multiple cancer types [30].
CD44
CD44 is a type I transmembrane glycoprotein that serves multiple cellular functions, including cell signaling, division, differentiation, adhesion, and migration. This versatile protein appears in various cell types, including hematopoietic stem cells, mesodermal cells, neuroectodermal cells, fibroblasts, and epithelial cells [31]. As a widely recognized CSC marker, CD44 shows high expression across multiple cancers, including colorectal, breast, ovarian, glioblastoma, bladder, head and neck, pancreatic, and osteosarcoma [32–39]. Beyond its role as a marker, CD44 plays crucial roles in maintaining CSC properties, promoting metastasis, and conferring resistance to apoptosis [40].
While CD44 acts as a receptor for multiple ligands, including chondroitin, osteopontin (OPN), collagen, and fibronectin [41–43], its primary ligand is hyaluronic acid (HA). HA, a glycosaminoglycan ubiquitously present in tissues, constitutes a major component of the extracellular matrix (ECM). When HA binds to CD44, it triggers conformational changes that initiate various cellular signaling cascades and biological processes. These processes, which include cell proliferation, adhesion, migration, and invasion, involve pathways mediated by metalloproteinases (MMPs) and the cytoskeletal matrix, ultimately contributing to tumor progression [44].
The CD44/HA signaling axis is particularly significant in cancer development, with cancer cells typically expressing higher HA levels than normal tissues. This interaction activates protein kinase C, which subsequently phosphorylates the transcription factor Nanog. Nanog maintains stem cell characteristics by upregulating the expression of the drug efflux pump gene ABCB1, thereby promoting therapy resistance [45]. Additionally, the CD44/HA pathway interfaces with both Ras/MAPK (mitogen-activated protein kinases) and PI3K signaling pathways, creating a complex network of cellular signals.
The CD44 gene consists of 19 exons, with 10 constant exons present in all isoforms produced through alternative splicing, forming the standard CD44 (CD44s). The variant isoforms (CD44v) incorporate these 10 constant exons plus different combinations of the remaining 9 exons [46, 47]. These variant isoforms predominantly appear in epithelial cells and cancer cells, where their expression correlates with tumor progression and metastasis [48].
Specific CD44 variants play distinct roles in different cancer types. In head and neck cancers, CD44 variant 3 (CD44v3) promotes tumor growth, migration, and metastasis [49]. CD44v6 facilitates metastasis in colorectal, pancreatic, and breast cancers [35–37], while CD44v8 specifically enables metastatic colonization in lung cancer [50]. Given these variant-specific roles in cancer progression, researchers have proposed that CD44 could serve as a valuable diagnostic biomarker for predicting therapeutic response in various cancers [51, 52].
CD24
CD24 is a glycosylphosphatidylinositol (GPI)—anchored protein expressed on various cell types, particularly hematopoietic and neuronal stem cells, where it functions as a differentiation marker. This protein facilitates cell adhesion, signal transmission, and immune response regulation [53]. Through its interaction with P-selectin, CD24 promotes cell migration by enabling leukocyte adhesion to endothelial cells and activated platelets [54]. In cancer biology, CD24 plays a critical role in tumorigenesis and progression. Its expression correlates with aggressive tumor characteristics, disrupted oncogenic signaling pathways, and poor prognosis [55]. CD24 influences both cell-cell interactions and the tumor microenvironment, enhancing cancer cell malignancy. Researchers frequently use CD24 along with other markers, particularly CD44, to identify CSCs in various cancers, including breast, ovarian, lung, colorectal, head and neck, and leukemia [56–61].
CD24’s role in chemotherapy resistance has been demonstrated across multiple studies. In retinoblastoma cells, elevated CD24 reduces vincristine sensitivity by activating autophagy signaling pathways [62]. In endometrial carcinoma cells, increased CD24 expression promotes cell proliferation, invasion, and resistance to paclitaxel and doxorubicin [63]. High CD24 expression correlates with worse prognosis following chemoradiotherapy in glioblastoma patients [64]. Finally, CD24 overexpression contributes to cisplatin resistance in head and neck squamous cell carcinoma (HNSCC) cell lines [65]. Additionally, CD24 facilitates immune evasion through its interaction with sialic-acid-binding Ig-like lectin 10 (Siglec-10). Present on activated T-cells, B-cells, and macrophages, Siglec-10’s binding to CD24 disrupts macrophage signaling pathways, reducing phagocytic activity [66]. This mechanism suggests that targeting CD24 could potentially overcome both chemoresistance and enhance phagocytosis in cancer cells.
ALDH
ALDHs are enzymes that catalyze the oxidation of aldehydes to carboxylic acids in the body. These enzymes play crucial roles in cellular detoxification and protection from oxidative stress, promoting cell survival [67]. In normal stem cells, ALDH helps maintain the balance between stemness and differentiation. ALDH activity, first identified in normal hematopoietic stem cells, has emerged as a reliable CSC marker in various malignancies, including gastric, lung, head and neck, esophageal, breast, and ovarian cancers [68–73].
Humans express 19 ALDH protein isoforms, most of which have cancer-related functions [74]. The ALDH1 subfamily is associated with stemness, poor prognosis, tumor formation, and chemotherapy resistance [67, 75]. ALDH1 proteins (ALDH1A1, ALDH1A2, ALDH1A3), primarily localized in the cytoplasm, oxidize retinal and aliphatic aldehydes. ALDH1A is particularly significant in retinoid signaling, catalyzing the oxidation of retinol to retinoic acid (RA). RA then translocates to the nucleus to regulate downstream gene transcription [76]. Ginestier et al. [77] demonstrated that breast CSC differentiation depends on retinoid signaling, with ALDH1 serving as a key modulator.
ALDH contributes to therapy resistance by actively oxidizing aldophosphamide and detoxifying various anticancer drugs. Hilton [78] showed that high ALDH1 expression enhanced leukemia cell line resistance to cyclophosphamide, findings later corroborated by Yoshida et al. [79] and Kastan et al. [80]. In lung cancers, ALDH1A-positive cells correlate with poor prognosis and demonstrate tumor-forming capacity in vivo [71]. However, Dimou and colleagues [81] reported contradictory findings, where ALDH1A1-negative lung cancer patients showed shorter survival times compared to ALDH1A1-positive cases. These findings have prompted interest in ALDH as a therapeutic target for CSCs through inhibitors or antibody-based approaches [67].
CD133
CD133, a transmembrane protein, functions as a CSC marker capable of initiating tumor growth and development [82]. While expressed in hematopoietic stem cells, glial stem cells, and endothelial progenitor cells, CD133 rarely appears in normal tissue cells [83, 84]. Its expression has been documented in various cancers, including colorectal [85], pancreatic [86], ovarian [87], lung [88], and hepatocellular cancers [89], and glioblastomas [90]. Meta-analysis has shown that high CD133 expression correlates with metastasis, poor prognosis, multiple drug resistance (MDR), and decreased five-year survival [91], highlighting its potential as a therapeutic target.
EpCAM
EpCAM, a transmembrane glycoprotein, mediates cell-cell adhesion, signaling, and epithelial tissue integrity. In normal epithelial tissues, it maintains tissue structure and cell polarity while contributing to cellular signaling and growth. EpCAM overexpression in epithelial cancers, including colon [51], breast [52], and pancreatic cancers [53], promotes cancer cell migration, invasion, and proliferation. This CSC marker serves as a valuable biomarker for cancer diagnosis and prognosis.
Techniques for studying CSCs
Various techniques are employed to study CSCs, focusing on identifying, isolating, and characterizing these cells within tumors. However, it is challenging since CSCs and normal stem cells are similar with respect to not only biomarkers but also transcription factors that regulate gene expression and signaling pathways that control their function. Some of the commonly used techniques include the following.
Tumor sphere assay
The tumor sphere assay represents a significant advancement in cancer research as a three-dimensional (3D) cell culture technique that permits studies on cellular behavior in a physiologically relevant environment. The history of this method dates to the 1940s when researchers first observed that tumor cells grown in suspension could form spheroids that mimicked the in vivo tumors. While the technique was formally established in the 1970s, the technique was only adopted extensively in the 1990s for applications in drug screening and cancer research and is now considered a gold standard for CSC research [92, 93].
The methodology involves culturing cells under non-adherent conditions in specialized media containing growth factors that promote the formation of 3D structures. This approach leverages the unique properties of CSCs, particularly their ability to display anchorage-independent growth. Scientists use this method extensively to study CSC biology, investigate mechanisms of drug resistance, and analyze tumor heterogeneity. However, it is important to note that while spheroid formation suggests the presence of CSCs, these structures may not fully replicate the complexity of the tumor microenvironment, necessitating validation through in vivo models. The reason for this is that the tumor microenvironment essentially consists of non-tumor cells, such as immune cells, ECM, blood vessels, and other somatic cells, such as fibroblasts. These are missing in the tumor sphere assays. More recently, organoid-based assays have been developed wherein some of these cells have been included to better represent an in vivo tumor [94, 95]. In addition, patient-derived organoid models are being developed to not only study the biology of cancer but also to utilize the model as a personalized medicine strategy [96].
Aldefluor assay
The Aldefluor assay has evolved significantly since its initial development in the 1990s for identifying hematopoietic stem cells [80]. Today, it serves as a valuable tool for detecting and isolating CSCs from various solid tumors, including breast, colorectal, and prostate cancers [97–99]. This method operates by measuring the activity of the ALDH enzyme, which acts as an intracellular marker for CSCs. The assay’s mechanism relies on the cleavage of a fluorescent substrate called BODIPY-aminoacetaldehyde (BAAA) to its product BODIPY-aminoacetate (BAA). The negatively charged BAA accumulates within cells, making those with high ALDH activity readily identifiable through flow cytometry [100, 101]. While this method offers a quick and effective way to isolate viable CSC populations, it requires expensive flow cytometry equipment and is specific to ALDH1A without distinguishing between various ALDH isoforms.
Side population assay
The side population assay exploits one of the defining characteristics of CSCs: their overexpression of ABC transporter proteins. This method was first discovered by Goodell and colleagues [102] in 1996 during their studies of bone marrow cells. The technique utilizes Hoechst 33342 dye, which permeates the membrane and binds to nuclear DNA. The researchers observed that certain cells, termed “side population” cells, could actively pump out this dye through ABC transporters, particularly the ABCG2 transporter [103].
ABCG2 has become recognized as both a phenotypic marker for CSCs and a key player in drug resistance [104]. The assay works by identifying unstained cells through flow cytometry and examining their self-renewal and differentiation capabilities. As Hoechst 33342 binds to AT-rich regions in DNA’s minor groove, the fluorescence intensity varies based on factors such as DNA content, chromatin structure, and cell cycle stage. This method proves particularly valuable for identifying CSCs that lack known cell surface markers.
Flow cytometry-based method
Flow cytometry stands as a cornerstone technique in CSC research, offering sophisticated analysis of single cells or particles as they pass through laser beams. This robust method enables researchers to identify and isolate CSC populations by examining both cell surface markers and intracellular molecules through fluorescence-activated cell sorting (FACS) with specific antibodies.
The technique’s strength lies in its ability to analyze dispersed light and fluorescent signals emitted by cells under laser illumination. It has earned its status as the gold standard for cell sorting due to its capability to isolate rare cell populations, study heterogeneity using multiple surface markers, and achieve high-purity cell isolation through both positive and negative selection [105]. However, researchers must contend with its time-consuming nature and potential impacts on cell viability, which can affect experimental outcomes.
Magnetic bead separation method
Magnetic bead separation offers an alternative approach to flow cytometry for isolating CSC populations [106]. This method employs magnetic beads conjugated with antibodies or ligands that specifically target CSC surface markers. Available through various systems such as Dynabeads, EasySep, magnetic activated cell sorting (MACS), and Mojosort, this technique combines high specificity with efficient processing of large samples. While magnetic bead separation provides a quicker alternative to flow cytometry, it comes with certain limitations. The method cannot isolate mixed populations using multiple markers simultaneously, instead requiring serial processing that can become time-consuming when handling numerous samples. Additionally, the labeling and separation steps may alter CSC phenotypes and affect their viability. Nevertheless, its accessibility and efficiency in processing large samples make it a valuable tool in CSC research.
CSC signaling pathways
CSCs rely on intricate signaling networks that govern their critical functions in tumor development, growth, spread, and therapeutic resistance. Multiple interconnected pathways maintain the CSC state, including Wnt/β-catenin, Notch, Hippo, Hedgehog (Hh), PI3K/Akt/mTOR, JAK/STAT (signal transducer and activator of transcription), and nuclear factor kappa B (NF-κB) [107–113]. These pathways work in concert to regulate CSC self-renewal and differentiation capabilities.
The Wnt/β-catenin signaling pathway
The Wnt/β-catenin pathway represents a highly conserved signaling network that orchestrates fundamental cellular processes, including proliferation, survival, differentiation, migration, and stem cell maintenance [114, 115]. This pathway exists in two distinct forms: the canonical (β-catenin-dependent) and non-canonical (β-catenin-independent) pathways. In humans and mice, approximately 19 different Wnt genes produce secreted proteins that can activate either pathway. In the canonical pathway, Wnt proteins bind to frizzled and low-density lipoprotein receptor-related protein 5/6 (LRP5/6), triggering a cascade that involves the protein Dishevelled [116]. This interaction disrupts the β-catenin destruction complex, which normally comprises adenomatous polyposis coli (APC), Axin, casein kinase 1 alpha (CK1α), and glycogen synthase kinase-3 beta (GSK-3β). As a result, the destruction of β-catenin is lost, leading to its accumulation in the cytoplasm, which then can subsequently translocate to the nucleus to activate target gene transcription. The non-canonical pathway operates independently of β-catenin, instead utilizing small GTPases (Rho and Rac) and calcium signaling to influence cell behavior, polarity, and adhesion [117] (Figure 1).
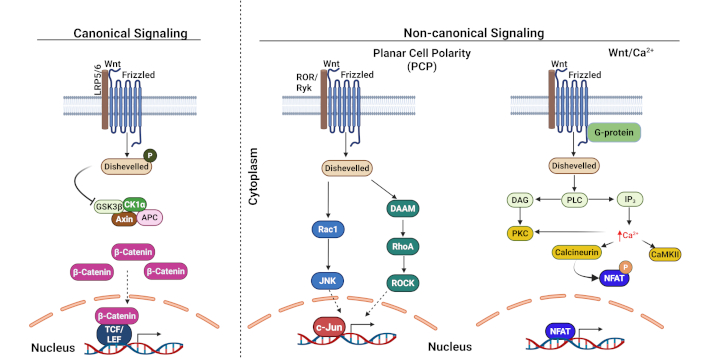
Canonical and non-canonical Wnt signaling pathways. LRP5/6: low-density lipoprotein receptor-related protein 5/6; GSK3β: glycogen synthase kinase 3 beta; CK1α: casein kinase I alpha; APC: adenomatous polyposis coli; TCF/LEF: T-cell factor/Lymphoid enhancer-binding factor; DAAM: dishevelled-associated activator of morphogenesis; Rac1: ras-related C3 botulinum toxin substrate 1; JNK: c-Jun N-terminal kinase; RhoA: ras homolog gene family, member A; ROCK: Rho-associated coiled-coil containing protein kinase; DAG: diacylglycerol; PLC: phospholipase C; IP3: inositol triphosphate; PKC: protein kinase C; CaMKII: calcium/calmodulin-dependent protein kinase II; Ca2+: calcium ion; NFAT: nuclear factor of activated T-cells. Adapted with permission from Chae and Bothwell [118], © 2018 Elsevier Ltd. Created in BioRender. Opara, J. (2025) https://BioRender.com/a91k218
Dysregulation in Wnt/β-catenin signaling in CSCs: aberrant activation of Wnt/β-catenin signaling in cancers typically stems from loss-of-function mutations in APC or AXIN1 or activating mutations in β-catenin itself. CSCs consistently show heightened Wnt pathway activity compared to non-CSCs across various malignancies, including colorectal, breast, gastric, and pancreatic cancers [119–121]. This hyperactivation enhances CSC properties such as self-renewal, tumorigenic potential, DNA repair capacity, immune evasion, and therapy resistance [122]. Recent research has revealed specific mechanisms linking Wnt signaling to cancer therapy resistance. For instance, studies in colonospheres demonstrated that Wnt3a-mediated pathway activation promotes survival through the upregulation of anti-apoptotic proteins Bcl-2 and survivin, conferring resistance to 5-fluorouracil (5FU) [123]. In colorectal cancer, APC mutations increase β-catenin accumulation in leucine-rich repeat-containing G protein-coupled receptor 5 (LGR5)-positive stem cells, promoting their transformation into CSCs and subsequent tumor formation [124, 125]. Additionally, enhanced Wnt signaling has been shown to improve DNA damage repair, contributing to resistance against poly (ADP-ribose) polymerase (PARP) inhibitors in ovarian cancer [126]. Furthermore, studies have reported the increased expression of Wnt5a (which stimulates β-catenin independent signaling) to correlate with poor prognosis in gastric cancer and melanoma [127, 128].
The pathway’s role extends to hematological malignancies, where it normally regulates hematopoietic stem cell function and homeostasis. However, dysregulated Wnt signaling can disrupt normal hematopoiesis, as demonstrated in mouse models where constitutive β-catenin expression led to severe hematopoietic abnormalities and mortality [129]. These findings collectively suggest that targeting the Wnt pathway could potentially sensitize CSCs to conventional therapies, offering a promising therapeutic strategy.
Notch signaling pathway
The Notch pathway represents a fundamental evolutionary conserved signaling system that regulates cell development, differentiation, and tissue homeostasis. Recent research has revealed that dysregulated Notch signaling plays a pivotal role in CSC maintenance and metastasis [130].
The pathway consists of transmembrane receptors (Notch 1–4) that interact with ligands (Delta-like 1, 3, 4, and Jagged 1-2), which are expressed in neighboring cells. Once the ligand engages with the Notch receptor, a two-step proteolytic process is triggered: first, the tumor necrosis factor-α-converting enzyme (TACE) engages the receptor in the Notch extracellular domain (NECD) and cleaves this domain. NECD is then internalized by the ligand-expressing cell, where it is believed to be degraded. Upon cleavage of the NECD, the receptor changes conformation, enabling the γ-secretase complex to access the receptor in the intracellular region of the receptor, wherein it cleaves and releases the Notch intracellular domain (NICD). Within the nucleus, the three-domain transcription factor CSL, along with other proteins, forms a nuclear repressor complex that blocks transcription. However, when NICD migrates to the nucleus, it interacts with CSL and forms a transcriptional activation complex along with other co-activators such as mastermind (MAML), SKIP, and histone acetyltransferases (HATs). This complex modifies the chromatin and activates gene expression [130, 131]. Beyond this canonical pathway, Notch can also signal through non-canonical mechanisms that operate independently of ligands, γ-secretase, or CSL. This alternative pathway involves complex interactions with other signaling networks, including Wnt, mTORC2, PI3K, AKT, hypoxia-inducible factor-1α (HIF-1α), and NF-κB, both in the cytoplasm and nucleus [132] (Figure 2).
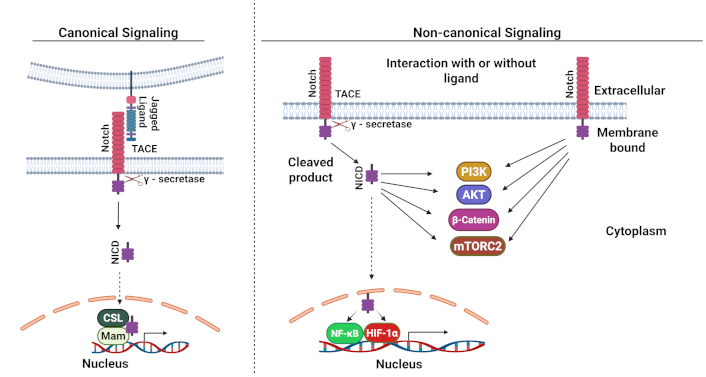
Canonical and non-canonical Notch signaling pathways. TACE: tumor necrosis factor-α-converting enzyme; NICD: Notch intracellular domain; NF-κB: nuclear factor kappa B; HIF-1α: hypoxia-inducible factor-1α. Adapted from Ayaz and Osborne [133], CC BY. Created in BioRender. Opara, J. (2025) https://BioRender.com/s65s201
Dysregulation of Notch signaling in CSCs: Notch signaling is a key pathway in CSCs, and its abnormal activation affects various stem cell properties and functions, including self-renewal, differentiation, therapy resistance, invasion, and migration [130]. Elevated Notch1/2 mRNA levels have been observed in renal cell carcinoma (RCC) CSCs, which correlates with enhanced chemoresistance due to the upregulation of stemness-associated markers such as MDR1, OCT4, and KLF4 genes [134]. Research in non-small cell lung cancer (NSCLC) has demonstrated that Notch1 activation drives EMT, leading to chemotherapy resistance that facilitates tumor growth, invasion, and metastasis [135]. In colorectal cancer, elevated Notch1 expression correlates with advanced disease characteristics, including tumor stage, lymph node involvement, invasion depth, and differentiation status [136]. Studies in pancreatic cancer have revealed that hyperactive Notch signaling disrupts normal neurovascular development and tumor angiogenesis, compromising drug delivery efficiency [137]. This finding has important implications for treatment strategies. Notably, experiments in NSCLC mouse models carrying KRAS mutations with wild-type p53 showed that Notch inhibition, whether through pharmacological or genetic approaches, effectively suppressed tumor growth [138]. Similarly, blocking Notch signaling in pancreatic cancer enhanced sensitivity to gemcitabine by activating intrinsic apoptotic pathways [139]. These findings collectively suggest that targeting the Notch pathway could represent a promising therapeutic approach for eliminating CSCs and overcoming treatment resistance.
Hh signaling pathway
The Hh pathway plays a fundamental role in embryonic development by regulating cell proliferation, fate determination, and stem cell maintenance [140]. This complex signaling network operates through three distinct ligands: Sonic Hh (Shh), Indian Hh (Ihh), and Desert Hh (Dhh). These ligands interact with the transmembrane receptors Patched (Ptch) and Smoothened (SMO) to activate downstream Gli transcription factors.
Canonical signaling mechanism: in the canonical pathway, Hh ligands bind to Ptch1, a twelve-pass transmembrane receptor. This interaction triggers Ptch1 endocytosis and degradation, releasing its inhibition of SMO, a G-protein coupled receptor (GPCR)-like protein. β-arrestins and the motor protein KIF3A then facilitate SMO translocation to the plasma membrane, where it undergoes phosphorylation by multiple kinases (PKA, CKIα, and Gprk2). Activated SMO releases Gli proteins from their suppressor SuFu, enabling their nuclear translocation and subsequent target gene activation. Without Hh ligand binding, Ptch1 maintains SMO suppression, preventing Gli nuclear translocation and transcriptional activity [141] (Figure 3).
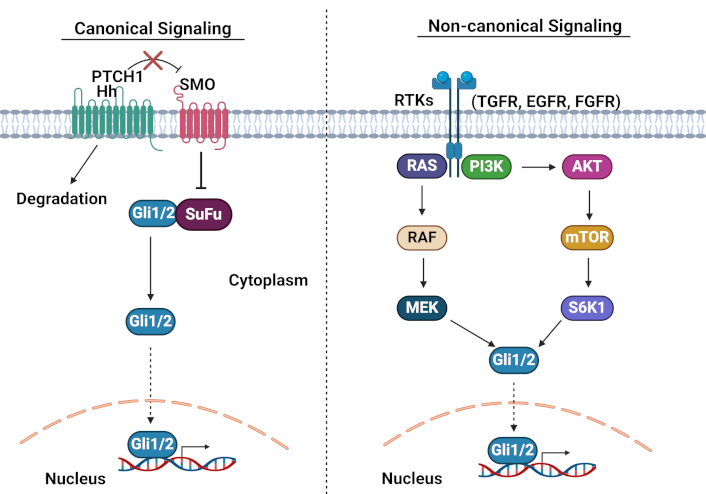
Canonical and non-canonical Hedgehog signaling pathways. PTCH1: patched1; SMO: Smoothened; Hh: hedgehog; RTKs: receptor tyrosine kinases; TGFR: transforming growth factor receptor; EGFR: epidermal growth factor receptor; FGFR: fibroblast growth factor receptor. Adapted from Pietrobono et al. [142], CC BY. Created in BioRender. Opara, J. (2025) https://BioRender.com/p92l297
Non-canonical signaling: the pathway can also signal through non-canonical mechanisms, either SMO-dependent but Gli-independent (involving small GTPases like Rac1, RhoA, or Src) [143], or through Gli activation independent of SMO. The latter involves crosstalk with other pathways, including Wnt/β-catenin, MAPK, epidermal growth factor receptor (EGFR), and transforming growth factor-beta (TGF-β) [144–147] (Figure 3).
Dysregulation of Hh signaling in CSCs: several studies have reported that aberrant Hh signaling contributes significantly to chemotherapy resistance and cancer progression. The pathway promotes EMT, β-catenin nuclear localization, and Snail expression across multiple cancers, including ovarian, breast, and basal cell carcinoma [148]. Clinical studies of ovarian epithelial tumors have revealed elevated SMO, Gli, and Ptch protein levels in both primary tumors and cisplatin-resistant cases compared to chemotherapy-sensitive patients [149]. Hh signaling maintains the CSC phenotype by regulating key stemness genes, including Sox2, Nanog, and Oct4 [150]. This has been particularly well-documented in several contexts. In glioblastoma, neurosphere-conditioned media dramatically increases Gli1 activity, while pathway inhibition with cyclopamine reduces Gli1 levels and suppresses stemness gene expression [151]. In pancreatic cancer, SMO inhibition reduces CSC-mediated tumor formation and impairs self-renewal, EMT, invasion, and lung metastasis [152]. In colon cancer, CD133+ CSCs show significantly elevated levels of Gli1, PTCH1, Gli2, and Shh compared to CD133– cells [153]. These findings collectively suggest that targeting the Hh pathway, particularly through Gli1 inhibition, could provide an effective strategy for eliminating CSCs and overcoming therapeutic resistance.
PI3K/AKT/mTOR signaling pathway
The PI3K/AKT/mTOR pathway represents a fundamental signaling cascade that orchestrates multiple essential cellular processes, including proliferation, growth, survival, inflammation, metabolism, and immune responses [154]. The cascade is initiated when extracellular signaling molecules, such as cytokines and growth factors, bind to their corresponding receptors—either receptor tyrosine kinases (RTKs) or GPCRs. This binding event triggers the activation of PI3K, which then catalyzes a critical molecular transformation; the conversion of phosphatidylinositol (4,5)-bisphosphate (PIP2) to phosphatidylinositol (3,4,5)-trisphosphate (PIP3). The resulting PIP3 molecule creates a specific docking site for AKT, enabling its subsequent activation through phosphorylation by phosphoinositide-dependent kinase-1 (PDK1). Once activated, AKT proceeds to phosphorylate several downstream target proteins, notably NF-κB and the mTOR, which ultimately promote cellular survival, enhance metabolism, and stimulate growth [155, 156] (Figure 4).
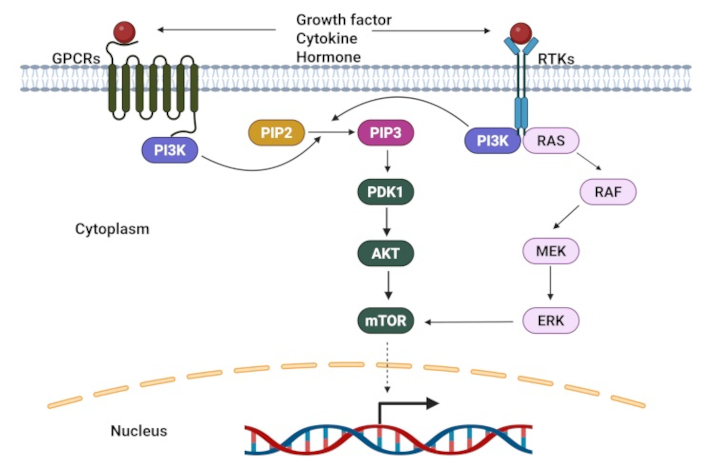
A schematic of PI3K/AKT signaling pathways. GPCRs: G protein-coupled receptors; RTKs: receptor tyrosine kinases; PDK1: phosphoinositide-dependent kinase-1; PIP2: phosphatidylinositol (4,5)-bisphosphat. Adapted from Yang et al. [157], CC BY. Created in BioRender. Opara, J. (2025) https://BioRender.com/d35o868
Dysregulation of PI3K signaling in CSCs: aberrant activation of the PI3K/AKT/mTOR pathway is found in numerous cancer types, encompassing both hematological malignancies and solid tumors. Current research underscores its pivotal role in CSC biology [158, 159]. Studies demonstrate significant impacts across multiple cancer types. In prostate cancer, heightened PI3K/AKT/mTOR signaling was found to promote radiation resistance through enhanced EMT and CSC characteristics [160]. In the context of brain tumors, AKT was found to specifically, rather than mTOR, drive ABCG2 expression in glioma stem-like cells—a mechanism subsequently verified in both AML and acute lymphoblastic leukemia (ALL) stem cells [161]. The pathway’s influence extends to lung cancer, where its abnormal activation increases chemokine receptor CXCR4 in NSCLC [162]. This CXCR4 upregulation sustains stem-like properties through STAT3 signaling enhancement. The pathway further contributes to chemoresistance through dual mechanisms: activation of nuclear factor erythroid 2-related factor 2 (Nrf2) [163] and HIF-1α [164]. HIF-1α plays a particularly complex role, simultaneously promoting cell survival and migration while triggering the production of reactive oxygen species (ROS) and glutathione (GSH), factors that enhance drug resistance [165]. Given these wide-ranging effects on CSCs and drug resistance mechanisms, the PI3K/AKT pathway emerges as a compelling therapeutic target for CSC elimination.
JAK/STAT signaling pathway
The JAK/STAT signaling pathway serves as a critical intracellular mechanism governing essential cellular processes, including proliferation, differentiation, inflammation, and immune regulation [166]. This pathway is initiated when specific growth factors or cytokines bind to their respective receptors, triggering the activation of tyrosine kinase JAK, which subsequently phosphorylates STAT proteins. Upon phosphorylation, these STAT proteins form dimers and translocate to the nucleus, where they regulate the transcription of their target genes (Figure 5). Research has demonstrated that dysregulation of this pathway contributes significantly to cancer progression, particularly through its roles in uncontrolled cell proliferation, survival, and the maintenance of CSC characteristics [167, 168].
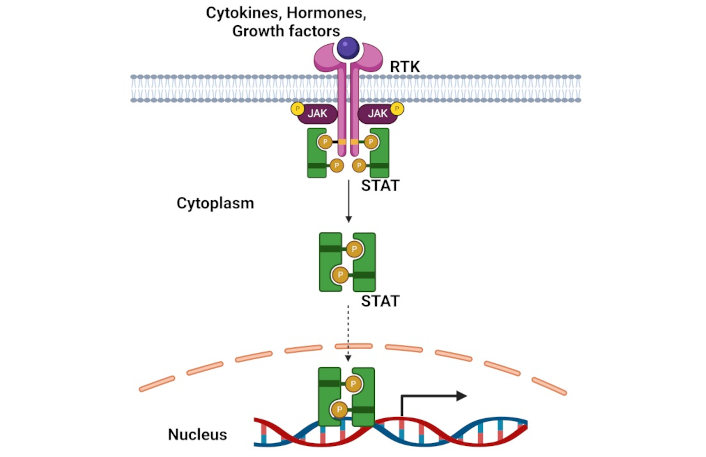
A schematic of JAK/STAT signaling pathway. STAT: signal transducer and activator of transcription; RTK: receptor tyrosine kinase. Created in BioRender. Opara, J. (2025) https://BioRender.com/r81x669
Dysregulation of JAK/STAT signaling in CSCs: The JAK/STAT pathway’s influence extends beyond basic cellular regulation to play a crucial role in CSC-related tumor metastasis through its impact on the EMT process. Choi and colleagues [169] revealed a significant mechanism wherein a positive feedback autocrine loop between osteopontin and the JAK/STAT3 pathway facilitates the EMT process, thereby contributing to sustained enhancement of CSC-associated tumor metastasis. Furthermore, within NSCLC, IFN/JAK/STAT signaling was found to regulate CSC populations [170]. Type I interferon (IFN-I), secreted by the CSCs, is in turn bound to receptors on the CSCs themselves, creating an autocrine loop that further activates the JAK/STAT1 pathway, ultimately enhancing SOX2 expression and maintaining CSC stemness. Additionally, the pathway's complexity is further evidenced by its ability to interact with other significant signaling cascades, including PI3K/AKT and MAPK/ERK pathways, which collectively contribute to chemoresistance in cancer cells.
Natural products as anti-CSC agents
Natural products have played a fundamental role in disease treatment since ancient Egyptian times and continue to gain prominence in Western medicine due to their therapeutic efficacy, minimal toxicity, and cost-effectiveness [171, 172]. Their significance in cancer prevention has been well-documented through epidemiological studies, with research demonstrating their antiproliferative, proapoptotic, and antioxidant properties against various cancers and CSCs [173, 174].
The search for effective cancer treatments has highlighted natural products as promising therapeutic agents, particularly for targeting CSCs. These compounds possess unique characteristics that make them ideal for drug discovery, including structural diversity, steric complexity, and low hydrophobicity [175–177]. Their therapeutic applications extend beyond cancer to include cardiovascular diseases [178], multiple sclerosis, and other conditions. The versatility of natural products has attracted attention across multiple industries, from cosmetics to pharmaceuticals, with substantial evidence supporting their role in reducing cancer incidence and recurrence [179, 180].
Throughout history, naturally occurring compounds and their modified derivatives have served as the foundation for many pharmaceuticals approved for global use [181]. The efficacy of these plant-derived compounds stems from their rich composition of phytochemicals, including flavonoids, alkaloids, saponins, tannins, and phenolic compounds. These components work synergistically to target CSCs by modulating multiple signaling and metabolic pathways that are typically dysregulated in cancer [182, 183]. Their molecular rigidity and diverse bioactive compound profile surpass that of synthetic compounds, making them particularly effective at targeting protein-protein interactions crucial for modern drug development [184].
The significance of natural products in cancer treatment is exemplified by their contribution to over 50% of antitumor drugs. Notable examples include vincristine and vinblastine, isolated from Catharanthus roseus (Madagascar periwinkle) [180], taxanes (taxol) from Taxus brevifolia Nutt (Pacific yew), and podophyllotoxin analogs from the North American mayapple Podophyllum peltatum L [185–187]. Clinical studies have consistently demonstrated that these plant-derived compounds offer multiple advantages: they are safe, minimally toxic, and highly effective against various CSC properties [188]. This combination of safety and efficacy makes natural products particularly valuable in the ongoing development of cancer treatments.
This review summarizes a few natural products that target CSCs in various cancer types and their significant CSCs.
Polyphenols targeting CSCs
Polyphenols are compounds naturally found in fruits, beverages, vegetables, cereals, etc. Foods like cocoa, berries, coffee, tea, olives, beans, and red wine are very high in polyphenols. Most of the natural products used in pharmaceutical industries are polyphenols and they have been demonstrated to modulate angiogenesis, apoptosis, cell growth, inflammation, and invasiveness in vitro due to the abundance of antioxidants present in them [189].
Curcumin
Curcumin, a natural polyphenol derived from the rhizome of turmeric (Curcuma longa) in the ginger family, has emerged as a compound of significant medical interest [190]. Research has established its diverse therapeutic properties, including antimicrobial, anti-inflammatory, anticancer, and antioxidant effects [191–194]. Extensive investigation has revealed curcumin’s significant impact on CSCs across multiple cancer types.
Studies have demonstrated curcumin’s mechanisms of action in various cancers. In pancreatic cancer, curcumin was observed to suppress tumor xenograft growth in vivo in a dose-dependent manner by inhibiting cyclooxygenase 2 (COX2) overexpression, thereby inducing apoptosis [195]. Furthermore, curcumin modulates notch signaling by downregulating notch target genes in prostate cancer cells [196]. In breast cancer, curcumin exhibits multiple therapeutic effects.
Research has shown that breast CSCs typically display high nuclear translocation of β-catenin, which promotes EMT genes while suppressing E-cadherin transcription [174]. Curcumin treatment effectively blocks this β-catenin nuclear translocation, thereby inhibiting breast CSC migration, restoring E-cadherin expression, and suppressing EMT. Chung et al. [197] identified that curcumin also inhibits the STAT3 signaling pathway by preventing STAT3 phosphorylation and its subsequent nuclear translocation, reducing the expression of STAT3-NF-κB target genes and CD44, a key breast CSC marker. Additionally, curcumin suppresses mammosphere formation in a dose-dependent manner, with 50% inhibition at 50 μM and complete inhibition at 100 μM [198]. An interesting discovery is that curcumin prevents breast cancer cell metastasis by inhibiting micro-tentacle formation in suspended cells [199]. More comprehensive evidence of curcumin’s anti-metastatic properties was shown through its ability to inhibit cell proliferation, migration, and invasion in MCF-7 and MDA-MB-231 cell lines while downregulating key CSC-related genes including Nanog, Oct4, and Sox2 [171].
Curcumin’s therapeutic potential extends to other cancer types as well. In glioblastoma, curcumin treatment decreased stem cell proliferation, reduced sphere formation, increased ROS production, enhanced MAPK pathway activation, and reduced STAT3 activation [200]. Furthermore, the treatment of glioblastoma cells with demethoxycurcumin resulted in the inhibition of the JAK/STAT3 pathway [135]. In esophageal cancer, curcumin suppresses CSC formation in a dose-dependent manner, in part through the inhibition of ALDH1A1 and NF-κB [201]. In colon cancer, as well, curcumin and its natural analogs bisdemethoxycurcumin, demethoxycurcumin, and tetrahydrocurcumin were shown to effectively suppress the Wnt/β-catenin pathway by downregulating the transcriptional coactivator p300 [202].
Researchers have discovered that curcumin, a natural compound found in turmeric, can make cancer cells more vulnerable to chemotherapy treatments—a process known as chemosensitization. This finding has generated considerable interest because it addresses two major challenges in cancer treatment: the toxic side effects of chemotherapy drugs and the tendency of cancer cells to develop drug resistance. The evidence for curcumin’s chemosensitizing effects comes from several groundbreaking studies. In breast cancer research, Zhou and colleagues [203] demonstrated that combining curcumin with doxorubicin significantly enhanced the drug’s effectiveness. When they added curcumin, they needed only half the usual amount of doxorubicin to achieve the same cancer-fighting effects in MCF-7 and MDA-MB-231 breast cancer cells. Even more remarkably, curcumin made breast CSCs—which are notorious for their resistance to treatment—5 to 15 times more sensitive to mitomycin. This increased sensitivity was accompanied by a dramatic 75% reduction in CD44+CD24−/low cells, which are markers for CSCs. The mechanism behind this enhanced sensitivity may also involve curcumin’s ability to interfere with drug resistance pathways. For instance, in doxorubicin-resistant breast cancer cells, curcumin blocks the function of a protein called ABCB4, which normally acts like a molecular pump that expels chemotherapy drugs from cancer cells [204]. Curcumin achieves this by inhibiting the protein’s ATPase activity—essentially cutting off its power supply—without changing how much of the protein is produced.
Curcumin’s chemosensitizing effects extend beyond breast cancer. In NSCLC, researchers found that curcumin makes cells more sensitive to cisplatin by triggering stress in the endoplasmic reticulum (ER), the cell’s protein-manufacturing center [205]. This stress response involves several key proteins—DDIT3, GRP78, ATF6, and XBP-1—that make the cancer cells more vulnerable to cisplatin’s effects. In colorectal cancer cells, curcumin was found to reverse cisplatin resistance by reducing levels of a long, non-coding RNA called KCNQ1OT1 [206]. This is fascinating because this RNA molecule typically promotes cancer growth by preventing miR-497 from suppressing Bcl-2, a protein that helps cancer cells survive. By decreasing KCNQ1OT1, curcumin allows miR-497 to do its cancer-fighting job more effectively. The benefits of combining curcumin with chemotherapy were further demonstrated in studies of colorectal liver metastases. When curcumin was combined with 5FU, there were multiple beneficial effects observed, including prevention of cancer cells from transitioning into a more aggressive form through EMT, reducing the formation of CSC clusters, and enhancing both the growth-inhibiting and cell-death-promoting effects of the chemotherapy [206]. These studies were performed with patient-derived tissue samples, making the findings particularly relevant for clinical applications.
The first clinical trial examining the effects of curcumin on cancer was conducted in 1987 by Kuttan et al. [207]. In this study, curcumin was applied as a topical ointment to cancerous lesions. The treatment alleviated itching, reduced exudates from weeping ulcers, minimized odor, decreased lesion size, and relieved pain. This study suggests that curcumin could be an effective, low-cost, well-tolerated therapeutic for skin disease treatment. In a phase I clinical trial, Sharma et al. [208] investigated the pharmacodynamics and pharmacokinetics of curcumin in patients with advanced colorectal cancer resistant to standard chemotherapy, using doses ranging from 440 to 2,200 mg/day. The study reported a 59% reduction in lymphocytic GSH S-transferase (GST) activity, a key enzyme involved in cancer development and progression. While no dose-related toxicity was observed, curcumin was undetectable in the blood and urine but was present in the feces, highlighting its poor bioavailability. In a phase IIa randomized clinical trial [209] where curcumin was combined with FOLFOX for metastatic colorectal cancer, daily oral dosing of 2 g curcumin to FOLFOX chemotherapy was found to be safe and tolerable. No additional adverse events were identified in patients given the combination over those given FOLFOX alone, and Curcumin glucuronide was detected in the plasma. In another study by Mahammedi et al. [210], a pilot phase II trial evaluated the effectiveness of combining docetaxel and prednisone with curcumin in chemotherapy-naive patients with metastatic castration-resistant prostate cancer (CRPC). The results showed that 65% of patients experienced more than an 80% reduction in prostate-specific antigen (PSA) levels, while approximately 36% of responders achieved PSA normalization without toxic effects. Furthermore, the combination of imatinib and curcumin significantly reduced nitric oxide levels in patients with chronic myeloid leukemia (CML) [211]. These collective findings suggest that combining curcumin with conventional chemotherapy drugs could lead to more effective cancer treatments while potentially reducing the doses of toxic chemotherapy drugs needed. This dual benefit of enhanced efficacy and potentially reduced side effects makes curcumin a promising candidate for further development in cancer therapy.
Resveratrol
Resveratrol represents a fascinating example of how natural compounds can have powerful effects against cancer. This polyphenol, first isolated in 1940 from white hellebore (Veratrum grandiflorum) roots, is found in many common foods, including blueberries, grapes, peanuts, and cranberries, as well as in Polygonum cuspidatum, a plant used in traditional Chinese medicine [212]. Beyond its well-documented antioxidant, neuroprotective, immunomodulatory, and anti-inflammatory properties, resveratrol has emerged as a promising anti-cancer agent, particularly in its ability to target CSCs across multiple types of cancer [213–219].
In breast cancer, resveratrol demonstrates remarkable effects on CSCs. Research by Fu and colleagues [220] revealed that resveratrol treatment significantly reduces both the number and size of mammospheres—spherical clusters of breast CSC. The mechanism behind this effect involves the activation of autophagy, a cellular “recycling” process, through increased expression of key autophagy-related genes, including Beclin-1, Atg7, and LC3II. Supporting these findings, Pandey’s team discovered that resveratrol also disrupts mammosphere formation by interfering with lipogenesis (fat synthesis) through a dual mechanism: downregulating fatty acid synthase genes while upregulating genes that promote programmed cell death [221].
In colorectal cancer, resveratrol’s effects are equally impressive. Reddivari and colleagues’ research demonstrated that resveratrol treatment reduces sphere formation, proliferation, and the number of crypt-containing cells—key indicators of CSC activity [222]. These effects primarily occur through suppression of the Wnt/β-catenin pathway and increased cell death. Buhrmann’s team further showed that resveratrol inhibits EMT factors, reduces NF-κB activation, and helps overcome drug resistance in colorectal cancer cells [217]. Additionally, a pilot study by Howells et al. [223] investigating micronized resveratrol (SRT501) in patients with colorectal cancer and hepatic metastases revealed a significant increase in cleaved caspase-3 immunoreactivity in tumor tissue compared to that of patients in the placebo group, demonstrating its role in the induction of apoptosis. Resveratrol shows promise in treating aggressive brain cancers. In glioblastoma, one of the most challenging cancers to treat, resveratrol reduces the expression of CD133, a key CSC marker, thereby inhibiting the survival of cancer cell colonies [120]. The compound achieves this by blocking the phosphorylation of Akt, a protein crucial for cell survival and proliferation. Additionally, it promotes the expression and activation of p53, which subsequently drives the transcription of its downstream target genes [224]. Additional research by Cilibrasi et al. [225] confirmed resveratrol's ability to decrease both the proliferation and viability of glioblastoma stem cell.
In pancreatic cancer, resveratrol’s mechanisms are especially intriguing. The compound suppresses anti-apoptotic proteins (Bcl-2 and XIAP) while increasing proteins that promote cell death (caspase 3/7) [226]. It also inhibits key stem cell maintenance factors like Nanog and Oct4. Perhaps most significantly, Zhou’s team [227] discovered that resveratrol could enhance the effectiveness of gemcitabine, a common chemotherapy drug, by suppressing SREBP1, a transcription factor that controls the synthesis of cholesterol, fatty acids, and phospholipids. This combination therapy showed promising results both in laboratory cultures and in living organisms, successfully reversing chemotherapy resistance and reducing CSC characteristics.
The compound’s effects on nasopharyngeal carcinoma (NPC) reveal yet another dimension of its anti-cancer properties. In NPC, resveratrol disrupts CSCs’ unique metabolism by increasing ROS and disturbing mitochondrial function [228]. This metabolic disruption leads to reduced stem cell characteristics through the downregulation of key stemness genes (Oct4, Sox2, Klf4, c-Myc, Nanog) and increased expression of tumor-suppressing microRNAs (miR-145 and miR-200c). Importantly, when combined with radiation therapy, resveratrol significantly increases the sensitivity of CSCs to radiation treatment. This is consistent with the combination treatment in pancreatic cancer cells, where combining resveratrol with gemcitabine reversed chemoresistance and enhanced cell killing [227].
These findings collectively paint a picture of resveratrol as a versatile anti-cancer agent that works through multiple mechanisms to target CSCs, enhance traditional treatments, and overcome drug resistance. Its ability to work through diverse pathways while being naturally present in common foods makes it a particularly interesting candidate for further development in cancer therapy.
Epigallocatechin-3-gallate
Epigallocatechin-3-gallate (EGCG) represents the primary bioactive constituent found in green tea, exhibiting significant therapeutic potential through its diverse biological activities. Research has established EGCG’s capacity to function as an antioxidant, reduce inflammation, prevent DNA methylation, and mitigate the risk of various diseases, including diabetes, cardiovascular conditions, and cancer [229–231]. Furthermore, EGCG demonstrates a regulatory role in lipid metabolism through its effects on lipogenic and lipolytic enzymes [232, 233].
Recent scientific investigations have revealed EGCG’s remarkable efficacy against multiple cancer types. In colorectal cancer research, studies demonstrate that EGCG treatment significantly diminishes both the quantity and dimensions of cancer spheroids in established cell lines while concurrently reducing the expression of CSC markers. The compound achieves these effects through modulation of the Wnt/β-catenin signaling pathway [234]. Additionally, EGCG enhances the therapeutic efficacy of conventional chemotherapy agents such as 5FU in resistant colon cancer cell populations through the downregulation of stem cell-related factors such as Notch1, Bmi1, Suz12, and Ezh2 [235].
In the context of lung cancer, research indicates that EGCG administration effectively inhibits spheroid formation across multiple cell lines while promoting apoptosis in CSCs [236]. The compound’s mechanism of action involves the downregulation of β-catenin and its associated target genes. Similarly, investigations in breast cancer have shown that EGCG induces programmed cell death through the upregulation of cellular communication network factor 5, simultaneously reducing mammosphere formation and stem cell marker expression [237]. Studies focusing on HNSCC have revealed that EGCG not only reduces spheroid formation but also enhances the effectiveness of cisplatin treatment [238]. This enhancement occurs through the downregulation of drug-resistance genes, specifically ABCC2 and ABCG2 transporters. Furthermore, research on ovarian cancer demonstrates EGCG’s ability to suppress tumorsphere formation and decrease the expression of critical CSC markers Nanog and PROM1 [239].
These comprehensive findings suggest that EGCG holds considerable promise as an adjunctive therapeutic agent in combination with standard chemotherapy protocols. The compound’s demonstrated ability to target CSCs and enhance chemotherapeutic efficacy across multiple cancer types indicates its potential significance in future cancer treatment strategies. A phase I study of EGCG treatment in combination with chemotherapy (cisplatin and etoposide) and radiation in advanced stage III NSCLC showed significant regression of esophagitis to grade 0/1 [240]. Based on this, they concluded that oral administration of EGCG is safe, feasible, and effective. However, a two-stage single-arm phase II clinical trial in women with advanced-stage ovarian cancer showed that treatment with an EGCG-enriched tea drink was not an effective maintenance intervention after standard treatment [241]. Their treatment adherence was high, and no toxicity was observed, however, they stated stability to be one of the probable causes of ineffectiveness.
Flavonoids targeting CSCs
Flavonoids constitute a significant class of dietary polyphenols that occur naturally across a diverse range of plant-based food sources. These compounds are abundantly present in common dietary items, including fruits, vegetables, tea, red wine, onions, and soybeans [242]. The flavonoid family encompasses several distinct structural subgroups, notably flavones, flavanols, flavonols, and isoflavones, each with unique chemical characteristics and biological properties [243].
Research has established that flavonoids possess a comprehensive array of beneficial properties that make them particularly valuable from a therapeutic perspective. These compounds demonstrate remarkable versatility in their biological activities, exhibiting antioxidant capabilities that help protect cells from oxidative stress [244]. Additionally, flavonoids have shown considerable efficacy as antiviral and antibacterial agents, contributing to their potential role in preventing and managing infections [245]. Further expanding their therapeutic potential, flavonoids demonstrate notable anti-inflammatory properties, suggesting their utility in addressing inflammatory conditions [246]. They have also shown promise in cancer prevention and treatment while additionally offering anti-allergic properties that may be beneficial in managing allergic responses [247, 248]. This broad spectrum of biological activities positions flavonoids as significant compounds in both preventive healthcare and therapeutic applications.
The extensive distribution of flavonoids in common dietary sources, combined with their diverse biological activities, underscores their importance in human health and their potential value in developing new therapeutic strategies. Their natural occurrence in widely consumed foods also makes them particularly accessible for dietary interventions aimed at promoting health and preventing disease.
Quercetin
Quercetin represents a prominent flavonoid compound found abundantly in numerous natural food sources, including cherries, grapes, apples, onion skin, red wine, tomatoes, citrus fruits, and various vegetables [249]. This compound has garnered significant scientific interest due to its diverse therapeutic properties, including antioxidant, anti-inflammatory, anti-allergic, anti-cancer, and anti-toxic effects [250–252]. In cancer biology, quercetin demonstrates remarkable anticancer properties through multiple mechanisms. It regulates various signaling pathways, influences estrogen receptor expression, modulates p53 activity, and inhibits heat shock proteins [253–255]. Research has shown that quercetin’s cell cycle effects vary by cancer type, inducing arrest at different phases: G0/G1 in leukemia, G2/M in breast carcinoma, and S phase in colorectal cancer [256–259].
Studies focusing on prostate CSCs have revealed quercetin’s significant therapeutic potential. When CD44+/CD133+ and CD44+ stem cells were treated with quercetin, researchers observed inhibited proliferation and migration, along with increased apoptosis, occurring in both dose- and time-dependent manner [259]. These effects were attributed to the compound’s ability to inactivate multiple signaling pathways, including PI3K/AKT, MAPK/ERK, and JNK pathways. Additional research using highly invasive prostate cancer cells DU145 subline III demonstrated quercetin’s capacity to inhibit spheroid formation by suppressing the phosphorylation of JNK and JUN, and reduced expression of stem cell marker proteins CD44, ABCG2, Sox2, and Nanog [260].
In breast cancer research, quercetin has shown promise in addressing drug resistance. Studies revealed that quercetin treatment significantly inhibited CSC propagation in MCF-7 cells and enhanced the effectiveness of conventional chemotherapeutic agents such as doxorubicin, paclitaxel, and vincristine [261]. These effects were associated with the reduced expression of P-glycoprotein, a key factor in multidrug resistance.
Quercetin’s effectiveness extends to gastrointestinal cancers, where it has been shown to target gastric CSCs through mitochondria-dependent apoptosis through PI3K/AKT pathway inhibition [262]. In head and neck cancer, quercetin demonstrates chemo-preventive properties by modulating stemness and EMT while significantly reducing ALDH1 activity, inhibiting self-renewal capacity, and suppressing the expression of stemness gene signatures, including Oct4, Nanog, and Nestin [263].
Research has also explored quercetin’s potential in combination therapies. When combined with other natural compounds such as ellagic acid or resveratrol, quercetin exhibits synergistic effects in various cancer types. In leukemia cells, the combination of quercetin and ellagic acid inhibited cell proliferation and induced apoptosis by depolarizing mitochondria, resulting in cytochrome C release and caspase 3 activation in pancreatic cancer [264, 265]. Such combinations have also shown enhanced apoptotic effects through mitochondrial depolarization and subsequent activation of cell death pathways [265]. Furthermore, quercetin’s combination with sulforaphane has demonstrated particular effectiveness in targeting pancreatic CSCs, reducing ALDH1 activity, and inhibiting self-renewal capabilities [266]. These comprehensive findings underscore quercetin’s significant potential as both a standalone therapeutic agent and a complementary treatment in combination with conventional cancer therapies or other natural compounds. Its ability to target CSCs and enhance the effectiveness of standard treatments positions quercetin as a promising candidate for future cancer treatment strategies. It is also noteworthy that quercetin has been reported to have no adverse effects on normal cells [267].
Apigenin
Apigenin is a naturally occurring flavonoid compound that demonstrates significant therapeutic potential, particularly in targeting CSCs across multiple cancer types. This bioactive compound is naturally present in a variety of common dietary sources, including chamomile tea, culinary herbs such as parsley, cilantro, and basil, as well as vegetables like onions, and citrus fruits such as oranges [268].
Research has demonstrated apigenin’s remarkable efficacy in addressing various cancer types through multiple mechanisms. In glioblastoma, apigenin exhibits its anti-cancer properties by inhibiting CSC proliferation through a specific molecular mechanism: blocking c-Met phosphorylation and its associated downstream signaling pathways [269]. This results in a clear reduction in the expression of critical CSC marker proteins, including CD133, Sox2, and Nanog. In the context of triple-negative breast cancer (TNBC), apigenin has shown promise in suppressing the stem-like characteristics of cancer cells. Studies have demonstrated that apigenin treatment effectively reduces the self-renewal capabilities of MDA-MB-231 and MDA-MB-436 cell lines [270]. This effect is achieved through multiple mechanisms, including the reduction of the CD44+/CD24− cell population and the inhibition of mammosphere formation. Furthermore, apigenin’s interference with YAP/TAZ-TEADs protein interactions leads to increased CSC apoptosis. Prostate cancer cells have also shown sensitivity to apigenin treatment. Apigenin demonstrated enhanced therapeutic efficacy when combined with conventional chemotherapy agent cisplatin. This combination therapy works by simultaneously upregulating pro-apoptotic genes, including Apaf-1, p53, and p21, while downregulating anti-apoptotic genes, such as Bcl-2 [271]. Additionally, apigenin independently inhibits prostate cancer CSC survival and migration through its modulatory effects on the PI3K/AKT/NF-κB signaling pathway [272]. Apigenin has also demonstrated significant therapeutic potential in HNSCC, particularly in addressing hypoxia-induced stem cell characteristics. A study utilizing multiple HNSCC cell lines has shown that apigenin effectively counteracts hypoxia-induced expression of various CSC markers [273]. Specifically, the compound shows dose-dependent efficacy in downregulating the expression of critical stem cell markers and effectively neutralizing hypoxia-induced increases in CD44, CD105, and Nanog expression.
The extensive research findings demonstrate that apigenin shows substantial promise as a therapeutic intervention for cancer treatment, with effectiveness in targeting CSCs. Its natural occurrence in common dietary sources, combined with its demonstrated efficacy across multiple cancer types, positions apigenin as a promising candidate for further development in cancer therapy strategies.
Luteolin
Luteolin is a significant flavonoid compound found abundantly in various plant sources, including common vegetables, fruits, and medicinal herbs. Its natural sources encompass carrots, broccoli, apple skin, onion leaves, cabbage, parsley, and peppers [274]. Scientific research has established luteolin’s diverse therapeutic properties, which include anti-inflammatory, antimicrobial, antioxidant, anti-cancer, and anti-allergic effects [275–278]. It demonstrates particularly noteworthy anti-cancer properties through multiple mechanisms. The compound effectively inhibits cancer progression by suppressing angiogenesis, invasion, and metastasis. These effects are achieved through the compound’s ability to suppress survival pathways, regulate cell cycle progression, and induce programmed cell death [279]. Further investigations have established luteolin’s role in counteracting hormone-induced cancer progression. Specifically, the compound has shown effectiveness in addressing medroxyprogesterone acetate (MPA)-induced tumor growth [280, 281]. While MPA typically promotes stem-like properties in breast cancer cells by increasing CD44+ cell populations and ALDH1 activity, luteolin treatment significantly reduces these effects [282]. The compound also demonstrates the ability to inhibit MPA-induced mammosphere formation and VEGF secretion, ultimately reducing tumor growth in multiple breast cancer cell lines.
In the context of chemotherapy resistance, luteolin has shown promise in treating paclitaxel-resistant esophageal cancer. Research indicates that luteolin treatment reduces stem-like characteristics in resistant cells through SOX2 protein downregulation. The compound demonstrates anti-metastatic properties by inhibiting cancer cell migration and invasion through its suppression of the EMT process. These effects are mechanistically linked to luteolin’s inhibition of the PI3K/AKT pathway, specifically through reduced phosphorylation of AKT at the S473 site [283].
These comprehensive findings underscore luteolin’s significant potential as a therapeutic agent in cancer treatment, particularly in addressing CSCs and chemotherapy resistance. Its natural occurrence in common dietary sources, combined with its demonstrated efficacy in multiple cancer contexts, positions luteolin as a promising candidate for further development in cancer therapy strategies, especially in combination with conventional treatments.
Phenethyl isothiocyanate
Phenethyl isothiocyanate (PEITC) represents a significant organosulfur bioactive compound predominantly found in cruciferous vegetables, particularly broccoli. This compound exists naturally in plants as glucosinolates [β-thioglucoside-N-hydroxysulfates (GS)], which are released upon tissue damage [284]. Research has established PEITC’s substantial role in cancer risk reduction through various molecular mechanisms [285, 286].
In colorectal cancer research, PEITC demonstrates noteworthy therapeutic effects through its ability to decrease stem cell growth via suppression of the Wnt/β-catenin signaling pathway [287]. Studies have revealed that this regulation occurs through interactions with the tumor microenvironment, highlighting PEITC’s complex mechanisms of action [288]. Research focusing on breast cancer has also shown particularly promising results. PEITC effectively reduces tumor progression and cell growth in HER2-positive breast cancer [289]. The compound demonstrates a significant impact on breast CSCs by decreasing mammosphere formation and reducing the expression of stem cell biomarkers through the reactivation of cadherin 1, a known tumor suppressor [290]. In TNBC, PEITC shows remarkable efficacy in suppressing CSC characteristics. Studies demonstrate that PEITC treatment reduces mammosphere formation in MDA-MB-231 chemo and radio-resistant cell lines and decreases CD44+/CD24− populations in a dose-dependent manner [291]. This effect is mediated through ROS modulation, specifically involving the downregulation of metadherin, a protein associated with various cancer signaling pathways and therapy resistance. Further studies have established that PEITC treatment effectively suppresses the levels of key pluripotency factors, Oct4 and Nanog, which are crucial for maintaining stem cell characteristics [292].
Additional studies on chemotherapy resistance have revealed PEITC’s potential as a chemosensitizing agent. Studies using cisplatin-resistant cervical cancer cells demonstrate that PEITC pre-treatment effectively restores cellular sensitivity to chemotherapy [293]. This occurs through multiple mechanisms, including the restoration of intracellular platinum levels, reduction of free GSH levels, increased ROS production, and alterations in mitochondrial membrane potential. Additionally, PEITC treatment leads to the downregulation of several key proteins involved in drug resistance and cell survival.
These findings collectively indicate PEITC’s significant potential as both a standalone therapeutic agent and a chemosensitizer. Its ability to enhance the sensitivity of CSCs to conventional radio- and chemotherapy positions PEITC as a promising candidate for combination therapy approaches in cancer treatment. The compound’s natural occurrence in common dietary sources, combined with its demonstrated therapeutic efficacy, suggests its potential value in developing more effective cancer treatment strategies.
Limitations and translational barriers of natural products in clinical applications
Many signaling pathways, including Wnt/β-catenin, Notch, Hh, Hippo, STAT3, and NF-κB, that play important roles in CSCs also play similar roles in normal stem cells [294, 295]. As such, natural compounds do not affect normal stem or somatic cells, at least at the doses that affect CSCs. However, the possibility exists that some of these compounds may affect normal cells (both normal somatic cells and normal stem cells) at higher doses. In fact, some natural products have been associated with adverse side effects, such as hepatotoxicity and mild gastrointestinal discomfort [296, 297]. This is why before any clinical trial is initiated to test the efficacy of a compound, it would be prudent to perform phase I clinical dose escalation trials to identify the maximum tolerated dose and determine the appropriate phase II testing dose.
There are other barriers with natural compounds, which make it difficult to evaluate their efficacy. The natural compounds appear to have significantly higher activity as complex mixtures than when they are isolated as individual active ingredients. Moreover, it is often difficult to isolate and/or synthesize these active ingredient(s) [298]. Lastly, clinical applications of natural products, especially natural compounds have been limited due to poor aqueous solubility, low bioavailability, poor absorption, instability, unfavorable pharmacokinetics, and rapid metabolism coupled with lack of targeting specificity, making it challenging to maintain therapeutically relevant levels in the bloodstream and tissues [299–301]. To address these limitations, studies have focused on developing innovative formulation strategies, including the use of nanoparticles to enhance absorption, improve bioavailability, and increase selective uptake by cancer cells over normal cells [302, 303]. Furthermore, the generation of analogs of natural compounds has been an approach to circumvent the above issues [304–306]. However, these derivatives no longer fall under the purview of natural compounds and would require extensive drug development strategies before clinical applications. Thus, maintaining a balance between therapeutic efficacy against CSCs and the preservation of normal cell function is essential for developing safe and effective treatments using natural compounds.
Conclusions
Cancer represents one of the most significant global health challenges, characterized by the unregulated proliferation and dissemination of abnormal cells. This complex disease manifests through the formation of tumors, invasion of surrounding tissues, and metastatic spread via circulatory and lymphatic systems. A defining characteristic of cancer is tumor heterogeneity, which manifests both between different tumor types and within individual tumors through genetic and phenotypic diversity. The concept of tumor heterogeneity holds particular significance as it fundamentally influences tumor growth patterns, treatment responses, and disease recurrence. CSCs play a pivotal role in this heterogeneity through their distinctive capabilities of self-renewal and differentiation into multiple types of cancer cells. These cells present a significant challenge to effective cancer treatment by contributing to three major therapeutic obstacles: drug resistance, metastasis, and disease recurrence.
Natural products have emerged as promising therapeutic agents in targeting CSCs, offering several distinct advantages. These compounds demonstrate specific anti-CSC properties while maintaining low toxicity profiles against normal stem cells, attributable to their rich composition of bioactive compounds. Their therapeutic potential is particularly noteworthy in their ability to modulate multiple signaling pathways crucial to CSC maintenance and function. The most significant of these pathways include Notch signaling, Hh signaling, and Wnt/β-catenin pathways, which are fundamental to CSC self-renewal.
Understanding the molecular interactions between phytochemicals and CSCs, particularly their effects on these signaling pathways, represents a crucial avenue for developing novel cancer therapeutics. The evidence presented comprehensively demonstrates the central role of CSCs in tumor progression and treatment resistance while identifying numerous natural products capable of specifically targeting these cells. However, the therapeutic application of natural products in targeting CSCs faces several challenges. These include potential off-target effects due to the presence of CSC biomarkers in normal cells and limited progress in clinical applications. The path forward requires rigorous testing in preclinical animal models and clinical trials, following the successful example of compounds like myristoyl amide, a doxycycline derivative that has demonstrated efficacy in eliminating CSCs and preventing metastasis in animal studies [307].
This comprehensive understanding of CSCs and their interaction with natural products opens new possibilities in cancer treatment. Future research directions should focus on optimizing these natural compounds for clinical application while addressing their current limitations, potentially leading to more effective and targeted cancer therapies.
Abbreviations
| 3D: | three-dimensional |
| 5FU: | 5-fluorouracil |
| ABC: | ATP-binding cassette |
| ALDH: | aldehyde dehydrogenase |
| AML: | acute myeloid leukemia |
| APC: | adenomatous polyposis coli |
| BAA: | BODIPY-aminoacetate |
| CD: | clusters of differentiation |
| CSCs: | cancer stem cells |
| ECM: | extracellular matrix |
| EGCG: | epigallocatechin-3-gallate |
| EMT: | epithelial-mesenchymal transition |
| EpCAM: | epithelial cell adhesion molecule |
| GPCR: | G-protein coupled receptor |
| GSH: | Glutathione |
| HA: | hyaluronic acid |
| Hh: | hedgehog |
| HIF-1α: | hypoxia-inducible factor-1α |
| HNSCC: | head and neck squamous cell carcinoma |
| MAPK: | mitogen-activated protein kinases |
| MDR: | multiple drug resistance |
| MPA: | medroxyprogesterone acetate |
| NECD: | Notch extracellular domain |
| NF-κB: | nuclear factor kappa B |
| NICD: | Notch intracellular domain |
| NPC: | nasopharyngeal carcinoma |
| NSCLC: | non-small cell lung cancer |
| PEITC: | phenethyl isothiocyanate |
| PIP2: | phosphatidylinositol (4,5)-bisphosphate |
| PSA: | prostate-specific antigen |
| Ptch: | Patched |
| RA: | retinoic acid |
| ROS: | reactive oxygen species |
| Siglec-10: | sialic-acid-binding Ig-like lectin 10 |
| SMO: | Smoothened |
| STAT: | signal transducer and activator of transcription |
| TNBC: | triple-negative breast cancer |
Declarations
Author contributions
JKO: Conceptualization, Writing—original draft. SS: Writing—original draft. SMT, SA: Writing—review & editing. All authors read and approved the submitted version.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
SA and SMT are funded through The University of Kansas Cancer Center Support Grant (CCSG) awarded by the National Cancer Institute [P30 CA168524]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.