Original Article
Original Article

Affiliation:
1Department of Chemistry, Pontifical Catholic University of Rio de Janeiro (PUC-Rio), Rio de Janeiro 22451-900, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0008-7859-3981
Affiliation:
1Department of Chemistry, Pontifical Catholic University of Rio de Janeiro (PUC-Rio), Rio de Janeiro 22451-900, Brazil
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-1036-2825
Affiliation:
1Department of Chemistry, Pontifical Catholic University of Rio de Janeiro (PUC-Rio), Rio de Janeiro 22451-900, Brazil
Affiliation:
2Núcleo de Pesquisa e Desenvolvimento de Medicamentos (NPDM), Federal University of Ceará (UFC), Fortaleza 60430-275, Brazil
ORCID: https://orcid.org/0000-0003-3621-8733
Affiliation:
2Núcleo de Pesquisa e Desenvolvimento de Medicamentos (NPDM), Federal University of Ceará (UFC), Fortaleza 60430-275, Brazil
ORCID: https://orcid.org/0000-0001-9379-2817
Affiliation:
2Núcleo de Pesquisa e Desenvolvimento de Medicamentos (NPDM), Federal University of Ceará (UFC), Fortaleza 60430-275, Brazil
3Department of Physiology and Pharmacology, Universidade Federal do Ceará, Fortaleza 60430-275, Brazil
ORCID: https://orcid.org/0000-0002-4344-4336
Affiliation:
1Department of Chemistry, Pontifical Catholic University of Rio de Janeiro (PUC-Rio), Rio de Janeiro 22451-900, Brazil
Email: camilla-buarque@puc-rio.br
ORCID: https://orcid.org/0000-0002-2639-2220
Explor Drug Sci. 2025;3:1008107 DOI: https://doi.org/10.37349/eds.2025.1008107
Received: December 31, 2024 Accepted: March 26, 2025 Published: April 21, 2025
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
Aim: The development of selective and potent antitumor agents remains a significant challenge. This study aimed to synthesize and evaluate biaryl hydroxy-1,2,3-triazoles and 9H-fluorene-1,2,3-triazole hybrids, inspired by previously identified bioactive 1,2,3-triazoles, for their cytotoxic potential against human cancer cell lines.
Methods: A library of 13 biaryl hydroxy-1,2,3-triazoles and 11 fluorene-1,2,3-triazoles was synthesized using optimized Suzuki and telescopic one-pot reactions, with yields ranging from 16% to 97%. The cytotoxicity of these compounds was tested against HCT-116 (colorectal cancer), SNB-19 (astrocytoma), MDA-MB-231 (triple-negative breast cancer), and MOLM-13 (acute myeloid leukemia, FLT3-ITD mutant) cell lines.
Results: Two fluorene-triazoles, 1-(2-bromophenyl)-4-(9H-fluoren-9-yl)-1H-1,2,3-triazole (
Conclusions: The results highlight the critical role of bromine substitution on the aryl azide-derived ring in modulating cytotoxic activity. The study reinforces the potential of rigid fluorene-based scaffolds as promising leads for the development of targeted therapies against FLT3-mutant leukemia, aligning with previous reports on 1,2,3-triazole hybrids antiproliferative activity in leukemia models.
Cancer has become the leading cause of death worldwide among people under 70 years old [1]. In addition, premature deaths due to various types of cancer are also rising across countries [2]. In 2020, the estimated number of cancer cases was almost 20 million, and 10 million deaths related to cancer. Among the most common and fatal cancers include breast, prostate, lung, colon, and leukemia [3]. Cancer cells are constantly evolving which results in a high tumor heterogeneity with a subpopulation of cells displaying a different molecular landscape [4]. This characteristic leads to drug resistance due to genetic, epigenetic, and metabolic changes that hinder anticancer activity [5, 6]. Another important factor that limits cancer treatment is chemotherapy-related toxicities that affect healthy cells leading to acute toxicities, such as hematological and gastrointestinal [7, 8]. Therefore, there is an urgent need for the development of new anticancer compounds to overcome these limitations.
Among the most promising scaffolds for addressing challenges in cancer therapy are 1,2,3-triazoles, a class of heterocycles known for their exceptional stability and diverse pharmacological properties. Their mechanisms of action include the inhibition of cancer cell proliferation, cell cycle arrest, and induction of apoptosis, making them effective against both drug-sensitive and multidrug-resistant cancers [9].
In recent years, triazole-based hybrid compounds have emerged as promising candidates for cancer treatment, demonstrating remarkable therapeutic potential [10]. These hybrids often exhibit dual or multiple mechanisms of action, addressing the limitations of conventional chemotherapeutics by improving selectivity and reducing side effects. Furthermore, their ability to overcome drug resistance and their promising in vivo activities against various cancer types have solidified their role as valuable frameworks for next-generation anticancer agents. By integrating triazoles into hybrid molecules, researchers aim to unlock new possibilities for more effective and safer cancer therapies [11].
Aryl(1-aryl-1H-1,2,3-triazole-4-yl)methanols, commonly known as hydroxy-1,2,3-triazoles, are a promising and versatile scaffold with significant potential for novel drug development. Our research group has extensive expertise in their synthesis, utilizing both copper-catalyzed alkyne-azide cycloaddition (CuAAC) between propargylic alcohols and aryl azides [12], as well as the metal-free and solvent-free enaminone-azide cycloaddition (EACA) employed in this study to synthesize 4-acyl-1,2,3-triazoles [13]. The EACA method provided a simpler and more efficient route to generate bioactive hydroxy-1,2,3-triazoles through acyl reduction. These compounds presented antileishmanial [14], antiviral [15], and cystic fibrosis activity [13].
9H-Fluorene-based compounds have emerged in past years due to their unique chemical and physical properties, which allow their application in a wide range of scientific disciplines, such as optoelectronics [16, 17], solar cells [18], nanostructures [19, 20], and others. Its great prominence is seen in the field of medicinal chemistry where this scaffold is being used in various bioactive compounds [21–24].
Given their structural characteristics (conformational rigidity with significant aromaticity structure), which intensify their pharmacophoric characteristics [25, 26], it is possible to combine them with other structures, such as 1,2,3-triazoles. These synthetic heterocycles have great potential and applicability, mainly due to their trans-amides bioisosteric proprieties that possess broad-spectrum biological activity, low cytotoxicity, and significant structural versatility. These properties allow triazoles to act both as pharmacophores, directly engaging biological targets, and as linkers in hybrid molecules, tethering different pharmacophores to enhance efficacy.
The development of fluorene-triazole hybrid compounds is a little-explored field with great potential. In literature, some examples are present so far, mainly in functional materials chemistry [27–31]. With a view to biological applications, examples can be found, however, most of them bonded together by fluorene’s aromatic rings [32–35].
Fewer examples are found when it comes to 9H-fluorenes-triazole hybrids. Yang et al. [36] proposed the synthesis of fluorene-triazoles containing the pentafluorosulfanyl group, with emphasis on the CuAAC between 3,5-bis(pentafluorosulfanyl)phenyl azide (1a) and 9-ethynyl-9H-fluoren-9-ol (2) as presented in Figure 1a. The authors verified the ability of compound 3 to trigger cell death in human leukemic monocyte lymphoma U937 cells obtaining a half-maximal inhibitory concentration (IC50) value of 6.29. Compound 3 was also compared to Caspase-3, an important enzyme responsible for apoptosis, which has been proven to be 3.4 times more efficient than this enzyme. These results demonstrate the antitumor potential of fluorene-triazole hybrids.
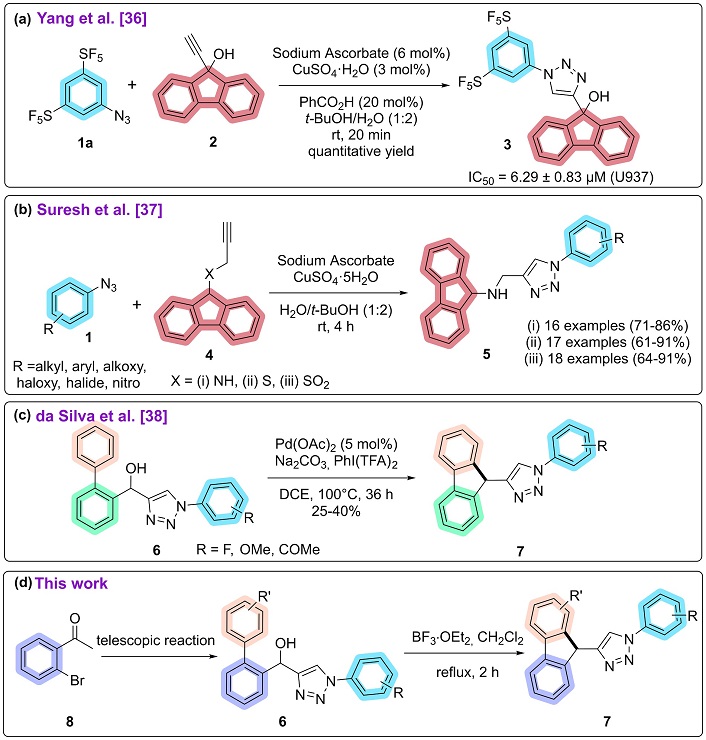
Summary of 9H-fluorene-1,2,3-triazoles hybrids. (a) Synthesis of fluorene triazoles according to Yang et al. [36]. (b) Synthesis of fluorene triazoles according to Suresh et al. [37]. (c) Synthesis of fluorene triazoles according to da Silva et al. [38]. (d) This work. DCE: 1,2-dichloroethane. rt: room temperature
Looking for analogs of structures reported in the literature as Mycobacterium tuberculosis (M. tb) InhA (NADH-dependent 2-trans-enoyl-acyl carrier protein reductase) inhibitors, Suresh et al. [37] rationalized such molecular skeletons as essential elements for InhA inhibition. In this way, the authors presented three 9H-fluorene structures derived from fluorene-triazoles (5) with a good variety of substituents by reacting fluorene-based alkynes (4) and azidobenzenes (1) [37] (Figure 1b).
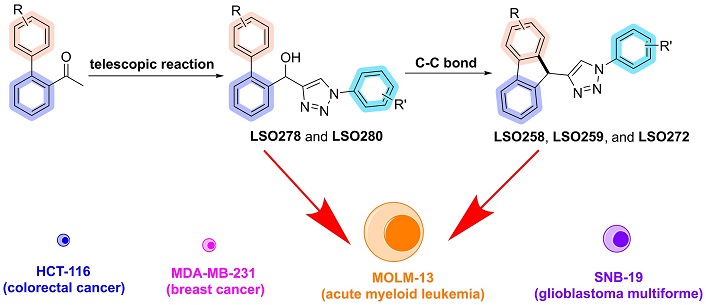
More recently in 2020, our research group reported 3 examples of 9H-fluorenes-triazole hybrids employing a Friedel-Crafts alkylation. Starting from substituted biaryl hydroxy-1,2,3-triazoles (6) (previously synthesized by CuAAC between biaryl-propargyl alcohols and azidobenzenes), in the presence of Pd(OAc)2, Na2CO3, PhI(TFA)2 in dichloroethane, 100°C for 36 hours, with yields of 25 to 40% [38] (Figure 1c). Since the metal-free and solvent-free EACA to obtain 4-acyl-1,2,3-triazoles is a simpler and more effective alternative than CuAAC, our research group decided to employ this methodology for obtaining biaryl hydroxy-1,2,3-triazoles (6) as precursors of 9H-fluorene-triazole (7) (Figure 1d). Given the biological potential of these compounds, another goal was to evaluate the antitumor potential by maximum growth inhibition (MGI) of the new biaryl hydroxy-1,2,3-triazoles and fluorene-triazole hybrids against different cell lines.
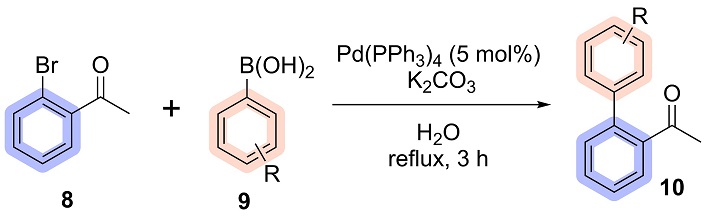
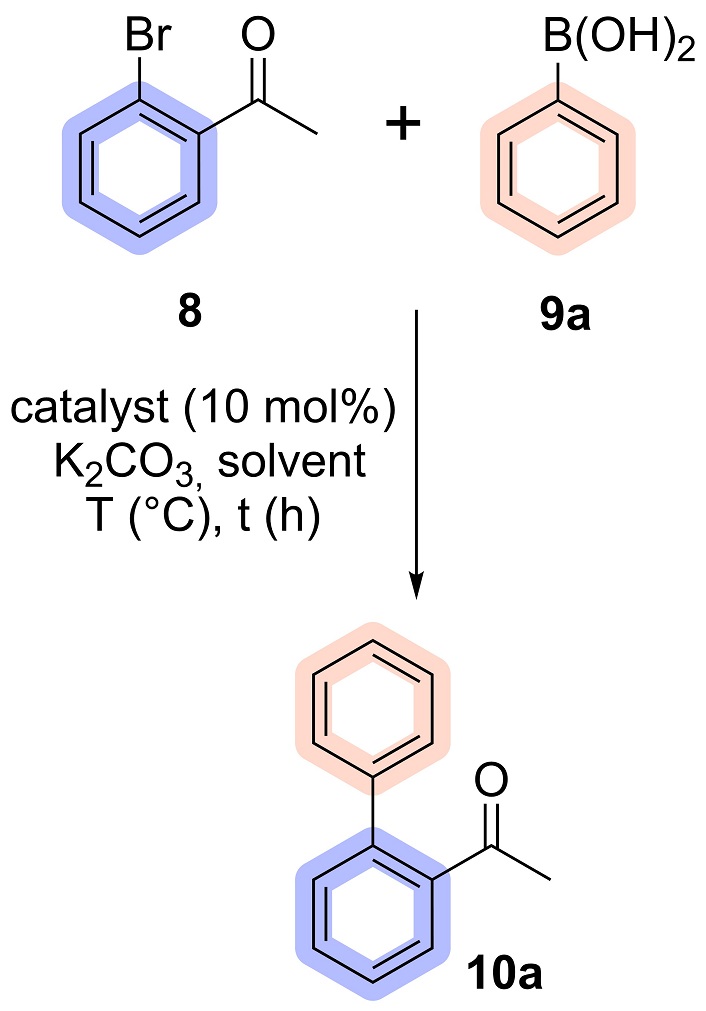
In a 25 mL screw-top test tube, 2-bromoacetophenone (8) (0.199 g, 1.00 mmol), phenylboronic acid (9) (2.00 mmol), K2CO3 (0.276 g, 2.00 mmol), Pd(PPh3)4 (0.05 mmol) and 2.0 mL of water were added. The reaction was stirred constantly for 3 hours at reflux temperature under an argon atmosphere (Figure 2). At the end of the reaction, the organic phase was extracted with ethyl acetate (EtOAc), dried with anhydrous sodium sulphate (Na2SO4) and filtered through celite. The solvent was evaporated under reduced pressure and the product (10) was purified by flash column chromatography using EtOAc/hexane (10:90) as the eluent.
In a 25 mL screw cap test tube, biaryl acetophenone (10) (1.00 mmol), aryl azide (1) (1.50 mmol), and N,N-dimethylformamide dimethyl acetal (DMF-DMA) (0.238 g, 2.00 mmol) were added. The reaction was stirred constantly for 2 hours at 150°C under an argon atmosphere (Figure 3). At the end of the reaction, the product (11) precipitated pure in ice-cold ethanol and vacuum filtered.
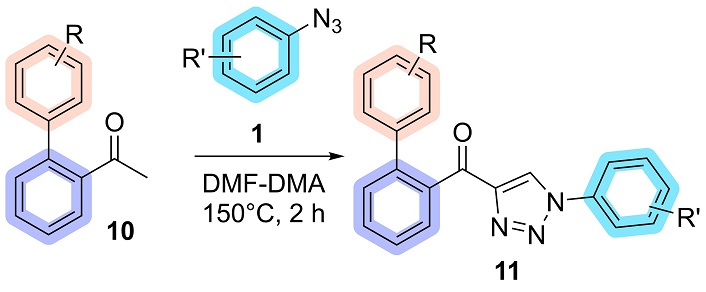
General procedure for the synthesis of biaryl-acyl-1,2,3-triazoles. DMF-DMA: N,N-dimethylformamide dimethyl acetal
Adapting the methodology from Azevedo et al. [13], starting with the addition of biaryl acetophenone (10) (1.00 mmol), aryl azide (1) (1.50 mmol), and DMF-DMA (0.238 g, 2.00 mmol) in a 25 mL screw-capped test tube the reaction was stirred constantly for 2 hours at 150°C under an argon atmosphere. Followed by the evaporation of the DMF-DMA, the tube was then cooled to room temperature, and NaBH4 (0.151 g, 4.00 mmol) and 2.5 mL of methanol were added. The reaction was stirred constantly for 2 hours at reflux temperature (Figure 4). At the end of the reaction, the product (6) was filtered into ice-cold ethanol or extracted with EtOAc followed by purification via flash column chromatography using EtOAc/hexane (50:50) as the eluent.
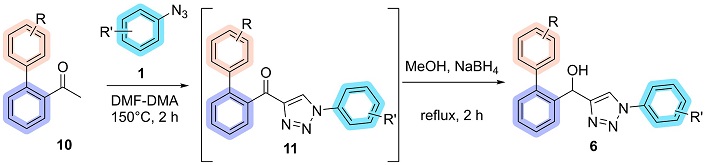
General procedure for the synthesis of biaryl hydroxy-1,2,3-triazoles. DMF-DMA: N,N-dimethylformamide dimethyl acetal
In a 4 mL vial, biaryl hydroxy-1,2,3-triazole (6) (0.125 mmol), BF3∙OEt2 (0.050 mL, 0.40 mmol) and 2.0 mL of dichloromethane were added. The BF3∙OEt2 was added slowly in an ice bath under constant stirring, then the reaction was brought to reflux temperature and continued for 2 hours (Figure 5). At the end of the reaction, the solvent was evaporated under reduced pressure and the product (7) was purified by column flash chromatography using EtOAc/hexane (10:90) as eluent.
Cell lines were obtained from the National Cancer Institute (Bethesda, MD). All cell culture media (Gibco™, Thermo Fisher Scientific, Cat. No. 31800022) contained 10% fetal bovine serum (Gibco™, Thermo Fisher Scientific, Cat. No. 12657029) and 1% penicillin/streptomycin (Gibco™, Thermo Fisher Scientific, Cat. No. 15140122). For the experiments, cells were seeded in 96-well plates (Nest™, Wuxi NEST Biotechnology, Cat. No. 701001) in the following concentrations 7 × 104 cells/mL (HCT-116), 2 × 105 cells/mL (MDA-MB-231), 1 × 105 cells/mL (MOLM-13 and SNB-19), and 1.5 × 105 cells/mL (HEK-293) and cells were maintained at 37°C with 5% CO2 in a humidified incubator.
The effects of biaryl hydroxy-triazoles and fluorene-triazoles compounds on human cancer cell viability, expressed as the percentage of cell growth inhibition, were determined by the MTT (3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide) (PerkinElmer, VICTOR Nivo Multimode Plate Reader, Waltham, MA). Colorimetric assay, as previously described [39, 40]. Cells were grown in 96-well plates (Vetec™, Sigma-Aldrich, Cat. No. V900888) at defined concentrations and exposed to a single concentration of 50 μM for 72 h. The negative control was treated with 0.5% DMSO (ACS™, ACS científica, São Paulo, BR). Afterward, the plates were centrifuged, and the medium was replaced by fresh medium (150 μL) containing 0.5 mg/mL MTT. Three hours later, the MTT formazan product was dissolved in 150 µL DMSO, and absorbance was measured using a multiple reader (PerkinElmer, VICTOR Nivo Multimode Plate Reader, Waltham, MA). Compound effect was quantified as the percentage of control (DMSO 0.5%) absorbance of the reduced dye at 595 nm. The compounds with MGI ≥ 75% in at least one human cancer cell line were selected for IC50 evaluation.
The IC50 evaluation of the selected fluorene-triazole hybrids was determined by the MTT colorimetric assay, as previously described [39, 40]. The research aimed to assess the IC50 of 5 compounds on four different human cancer cell lines: MOLM-13 [acute myeloid leukemia (AML)], SNB-19 (astrocytoma), MDA-MB-235 (breast cancer), HCT-116 (colon carcinoma cell line) and one non-tumor cell line, HEK-293 (human embryonic kidney). The tested compounds were dissolved in DMSO, and then added to each well, followed by incubation of 72 h at concentrations ranging from 0.4 to 50 μM. The negative control was treated with 0.5% DMSO. Afterward, the plates were processed as described above. Experiments were carried out in triplicate and repeated at least three times. The experiments were analyzed by linear regression using the GraphPad Prism program, version 6.01, to determine each IC50 value.
The selectivity index (SI) is based on the IC50 obtained for each cell line by calculating the ratio of IC50 on the non-cancer cell line over the IC50 for the cancer cell line. A compound is considered selective towards a tumor cell line with a SI ≥ 2 [41, 42].
SI = IC50non-cancer cells/IC50cancer cells
Given that the most bioactive compounds for leishmaniosis and cystic fibrosis identified by our research group, such as (4-bromophenyl)(1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methanol and (2-bromophenyl)(1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methanol, feature bromine in the aryl ring derived from aryl azide, we designed biaryl hydroxy-1,2,3-triazoles and fluorene derivatives to incorporate the bromine group. So, we started exploring the Suzuki reaction using 2-bromoacetophenone as a substrate to obtain biaryl acetophenone 10a.
The reaction, conducted under the conditions detailed in Table 1, entry 1, and adapted from the methodology reported by da Silva et al. [38], yielded the desired product in 65% yield after purification via column chromatography. To further optimize the reaction, different catalysts were evaluated in entries 2 and 3. Among the tested catalysts, Pd(PPh3)4 delivered the highest yield and was selected for subsequent studies. The next phase of optimization targeted the solvent and reaction time. With the reaction time fixed at 3 hours, various solvents were tested, including toluene, EtOH:H2O mixtures (20% and 50% ethanol), pure H2O, and solvent-free conditions. Both EtOH:H2O mixtures and pure water exhibited comparable performance. Pure water was ultimately chosen due to its significant advantages in environmental sustainability, safety, and cost efficiency [43, 44].
Reaction conditions optimization for the Suzuki reaction on 2-bromoacetophenone
| Reaction for the Suzuki reaction on 2-bromoacetophenone | Entry | Catalyst | Solvent | Temperature | Time (h) | Yield (%) |
|---|---|---|---|---|---|---|
 | 1 | Pd(OAc)2 + PPh3a | Toluene | Reflux | 16 | 65 |
| 2 | PdCl2b | Toluene | Reflux | 16 | 65 | |
| 3 | Pd(PPh3)4b | Toluene | Reflux | 16 | 80 | |
| 4 | Pd(PPh3)4b | 20% EtOH:H2O | Reflux | 3 | 90 | |
| 5 | Pd(PPh3)4b | 50% EtOH:H2O | Reflux | 3 | 93 | |
| 6 | Pd(PPh3)4b | H2O | Reflux | 3 | 92 | |
| 7 | Pd(PPh3)4b | - | 200°C | 3 | 70 | |
| 8 | Pd(PPh3)4c | H2O | Reflux | 3 | 95 | |
| 9 | Pd(PPh3)4d | H2O | Reflux | 3 | 60 | |
| 10 | Pd(PPh3)4c, e | H2O | Reflux | 3 | 87 |
a Catalyst: 10 mol% and 20 mol% respectively. b Catalyst: 10 mol%. c Catalyst: 5 mol%. d Catalyst: 1 mol%. e Scale up to 10 mmol from 0.2 mmol. -: no solvent
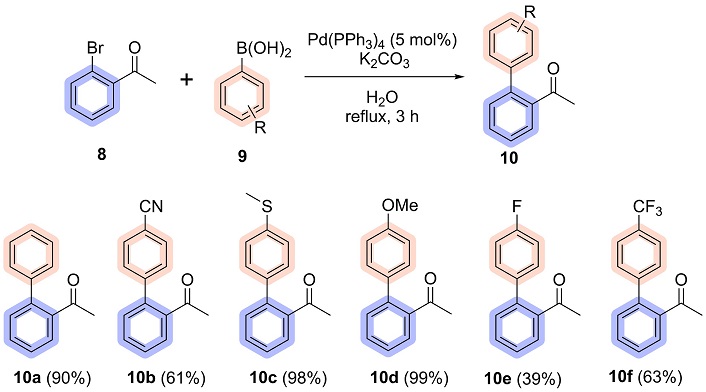
The optimized conditions identified in Table 1, entry 8, emerged as the most efficient and reliable for the synthesis, using 5 mol% Pd(PPh3)4 in water as the solvent under reflux for 3 hours, achieving yields of 95%. To further validate their robustness and scalability, the reaction was performed on a 50-fold scale, maintaining excellent efficiency with a slightly reduced yield of 87% after purification. From this optimized condition, it was possible to synthesize 6 biaryl acetophenones, with yields ranging from 39 to 99%, as shown in Figure 6.
According to the literature, in general, electron-rich organoboranes and electron-deficient halides are the most reactive substrates for the Suzuki reaction, thus resulting in a positive effect on the reaction yield [45, 46]. Given that the halide used is 2-bromoacetophenone, characterized by the presence of an electron-withdrawing group, and based on the scope presented above, the good yields obtained are in line with the trends reported in the literature. Additionally, boronic acids bearing electron-donating substituents consistently led to higher yields compared to those with electron-withdrawing groups, further corroborating established reactivity patterns.
The subsequent step involved applying the substituents previously established by the methodology for synthesizing 4-acyl-1,2,3-triazoles (11) from acetophenones and aryl azides [13]. The biaryl acetophenone 10a reacted with 4-bromophenyl azide 1b and DMF-DMA at 150°C for 2 hours. The mechanism for this reaction is a combination of previously described mechanisms for enaminone formation by Thomas et al. [47], as well as a mechanism described by the research group in 2022 [48]. This reaction successfully yielded the biaryl-acyl-1,2,3-triazole 11ab, which was isolated in 40% yield following precipitation in ice-cold ethanol, with the NMR spectra corresponding to these molecules available in the supplementary material (Figures S1 and S2). The reduction of compound 11ab was carried out using NaBH4 in methanol under reflux for 2 hours. This reaction yielded the biaryl hydroxy-1,2,3-triazole 6ab with complete conversion of the starting material (Thin Layer Chromatography-determined). The hydroxy-triazole precipitated directly from the reaction mixture upon the addition of water and was isolated by filtration, affording a yield of 93% (Figure 7).
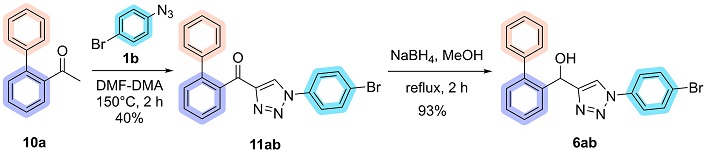
Synthesis of biaryl-acyl-1,2,3-triazole 11ab and biaryl hydroxy-1,2,3-triazole 6ab. DMF-DMA: N,N-dimethylformamide dimethyl acetal
Attempts to synthesize hydroxy-triazoles with other substituents revealed a significant limitation during the isolation of the acyl-triazoles. Unlike the previous case, none of the type 6 triazole precipitated in pure form, necessitating purification via column chromatography. The isolation process was further complicated by the similar retention factors of the 11 and the corresponding 10, making their separation particularly challenging.
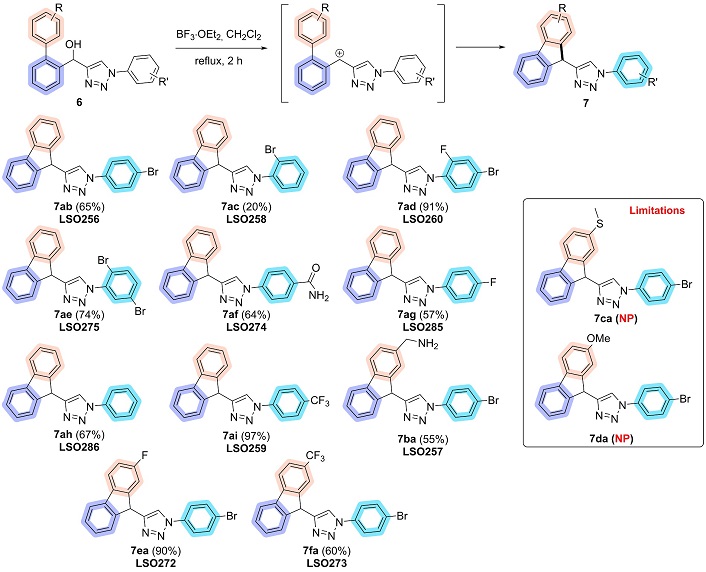
To address this challenge, a telescopic approach was proposed, applying the known properties of hydroxy-triazoles, which readily precipitate in pure form upon the addition of water. This one-pot methodology involved the in situ generation and consumption of intermediates, eliminating the need to isolate the acyl-triazole [49]. After evaporating DMF-DMA, the acyl group was directly reduced, enabling the synthesis of hydroxy-triazoles without requiring purification of the intermediate. Using the new telescopic approach, a diverse set of 13 novel biaryl hydroxy-1,2,3-triazoles (6) was successfully synthesized by varying the biaryl acetophenones and aryl azides, obtaining yields ranging from 16% to 89% (Figure 8).
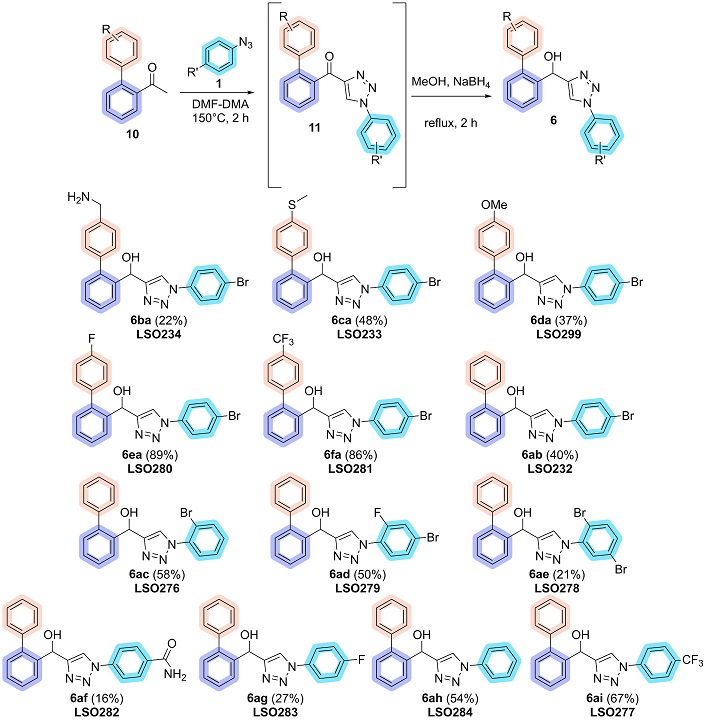
Scope of biaryl hydroxy-1,2,3-triazoles. DMF-DMA: N,N-dimethylformamide dimethyl acetal
The yields obtained showed significant variability, likely influenced by challenges in the synthesis of compound 11, particularly the in situ formation of the enaminone, which is essential for its subsequent reaction with the azide to produce the triazole. Additionally, variations in the solubility of the reduced product during precipitation may contribute to inconsistencies in the final yield.
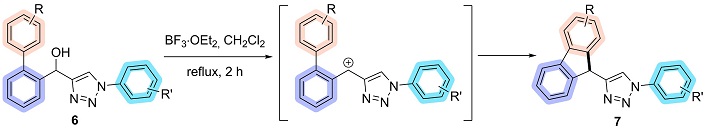
Once the bi-arylated precursors were synthesized, a methodology was required to access the desired fluorene-triazoles (7) boron trifluoride (BF3) was selected due to its strong Lewis acid character, which effectively coordinates with and activates the secondary alcohol, facilitating the formation of the carbocation intermediate. This method not only allows for the use of mild reaction conditions but is also commercially available, easy to handle under standard laboratory conditions, and can be conveniently removed after the reaction, thereby streamlining the overall process [50–52].
The formation of fluorene was achieved via an intramolecular Friedel-Crafts alkylation approach with a carbocation as an intermediate [53]. This methodology was applied to biaryl hydroxy-1,2,3-triazoles, which were reacted with BF3∙OEt2 in dichloromethane under reflux for 2 hours. Upon completion of the reaction, the solvent was evaporated, yielding 11 new 9H-fluorene-1,2,3-triazole in 20 to 97% yield (Figure 9).
The observed variation in yields can be attributed to the position of substituents relative to the cyclization attack site. Optimal yields are achieved when this site has high electronic density, which is enhanced by electron-withdrawing substituents in the meta position and electron-donating substituents in the ortho and para positions. The failure to obtain compounds 7ca and 7da is likely due to the presence of donor groups in the meta position, which reduces the electronic density at the target site, rendering it less reactive and thus deactivating the cyclization process. All products were thoroughly characterized by NMR spectroscopy, while all fluorene-triazoles were analyzed using both NMR and FTIR-ATR (Figures S1–S73).
As previously described, our research group identified the (4-bromophenyl)(1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methanol and (2-bromophenyl)(1-(4-bromophenyl)-1H-1,2,3-triazol-4-yl)methanol, featuring bromine in the aryl ring as important targets for leishmaniasis and cystic fibrosis. However, testing (4-bromophenyl)(1-(4-bromophenyl)-1H-1,2,3-triazole-4-yl)methanol (LSO24), in these tumor cell lines yielded no so positive results (Table S1).
To identify LSO24 analogs with enhanced potency against tumor cell lines and assess the influence of flexible ligands biaryl hydroxy-triazoles or scaffolds with conformational rigidity fluorene-1,2,3-triazoles, the cytotoxic effect of the newly synthesized biaryl hydroxy-triazoles and fluorene-triazoles hybrids were evaluated in a single concentration for MGI and in a range of concentration for IC50 determination against human cancer cell lines HCT-116 (colorectal cancer), SNB-19 (astrocytoma, glioblastoma multiforme), MDA-MB-231 (triple-negative breast cancer, metastatic) and MOLM-13 (AML). MGI and IC50 from all compounds are summarized in Table S1.
The MGI evaluation showed that the compounds LSO258, LSO272, LSO276, LSO278, and LSO281 achieved satisfactory growth inhibition in at least one cell line (Figure 10). Consequently, they were selected for IC50 determination in four human cancer cell lines and one non-cancerous human cell line.
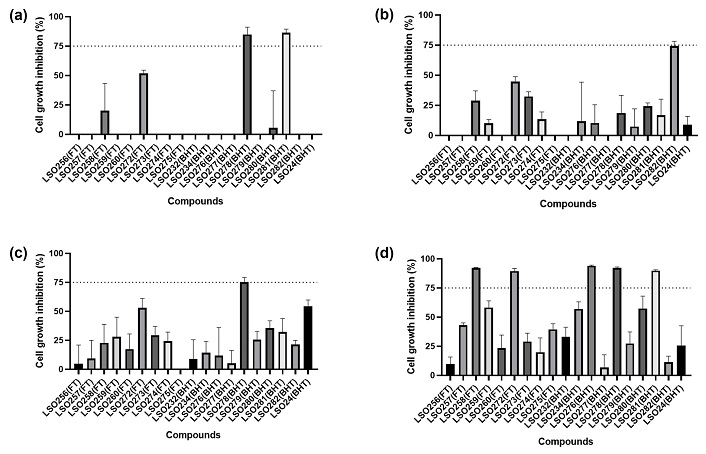
The maximum growth inhibition (MGI) of biaryl hydroxy-triazoles and fluorene-triazoles compounds across a panel of human cancer cell lines after 72 h exposure. (a) HCT-116. (b) SNB-19. (c) MDA-MB-231. (d) MOLM-13
Table 2 presents the IC50 evaluation of biaryl hydroxy-triazole and fluorene-triazole compounds against human cancer cell lines. The results indicate that all compounds exhibited an IC50 below 50 µM for the MOLM-13 cell line, with LSO258, LSO272, and LSO276 demonstrating selectivity for this cell line. The compounds LS0278 and LSO281, both belonging to the biaryl hydroxy-triazoles, were able to achieve an IC50 below 50 µM for more than one cancer cell line. LSO278 presented a cytotoxicity for the cell line HCT-116 and MDA-MB-231 with IC50 values of 23.4 µM and 34.3 µM, respectively. In addition, LSO281 showed an IC50 value of 28.2 µM for the HCT-116 cell line.
IC50 values (µM) of biaryl hydroxy-triazoles and fluorene-triazoles compounds across a panel of human cell lines after 72 h exposure
| Compound | Cell lines | ||||
|---|---|---|---|---|---|
| HCT-116 | SNB-19 | MDA-MB-231 | MOLM-13 | HEK-293 | |
| LSO258 | > 50 | > 50 | > 50 | 25.5 | > 50 |
| LSO272 | > 50 | > 50 | > 50 | 12.5 | 34.5 |
| LSO276 | > 50 | > 50 | > 50 | 24.8 | 42.6 |
| LSO278 | 23.4 | > 50 | 34.3 | 18.7 | 20.8 |
| LSO281 | 28.2 | > 50 | > 50 | 22.7 | 24.7 |
| Doxorubicin | ND | 0.8 | 0.2 | ND | 0.8 |
| 5-Fluorouracil | 19.2 | ND | ND | ND | 6.9 |
| Azacitidine | ND | ND | ND | 0.99 | > 50 |
> 50: not able to determine the half-maximal inhibitory concentration (IC50) value within the range of concentration tested for this cell line. ND: not determined
Additionally, the compounds were evaluated against the human non-cancer cell line HEK-293 to determine their SI. As shown in Table 3, among the tested compounds, only LSO258 and LSO272 exhibited an SI greater than 2 for the MOLM-13 leukemia cell line. In contrast, for the HCT-116, SNB-19, and MDA-MB-231 cell lines, none of the compounds tested showed an SI higher than 1. Notably, LSO272 demonstrated the highest selectivity, with the lowest IC50 value (12.5 µM) for MOLM-13 and an SI of 2.76.
Selectivity index of biaryl hydroxy-triazoles and fluorene-triazoles compounds comparing cytotoxicity on tumor cell lines to non-tumor HEK-293 cell line
| Compound | Cell lines | |||
|---|---|---|---|---|
| HCT-116 | SNB-19 | MDA-MB-231 | MOLM-13 | |
| LSO258 | ND | ND | ND | > 2 |
| LSO272 | ND | ND | ND | 2.76 |
| LSO276 | ND | ND | ND | 1.71 |
| LSO278 | 0.88 | ND | 0.60 | 1.11 |
| LSO281 | 0.87 | ND | ND | 1.08 |
| Doxorubicin | ND | 1 | 4 | ND |
| 5-fluorouracil | 0.35 | ND | ND | ND |
| Azacitidine | ND | ND | ND | > 50 |
ND: not determined
Among the fluorene-triazole compounds evaluated, LSO258 and LSO272, both containing bromine at the blue ring exhibited selective cytotoxicity against the MOLM-13 cell line, with IC50 values of 25.5 µM and 12.5 µM, respectively. In contrast, the flexible biaryl hydroxy-triazole ligands, LSO232 and LSO280, were inactive. In this case, the rigid scaffolds may minimize the entropic loss associated with the ligand adopting a preferred binding conformation, resulting in enhanced potency, improved selectivity, and reduced potential for drug metabolism [25, 26].
Considering the flexible biaryl hydroxy-triazole ligands, compound LSO278, which contains two bromine groups on the blue ring, was the only one to exhibit significant cytotoxicity across three cell lines (HCT-116, MDA-MB-231, and MOLM-13), with IC50 values of 23.4 µM, 34.3 µM, and 18.7 µM, respectively. In contrast, the fluorene-triazole compound LSO275 was inactive even with these two bromine groups, and the conformational rigidity doesn't corroborate the antitumoral activity.
The only instance where both a biaryl hydroxy-triazole (flexible ligand) and a fluorene-triazole (rigid scaffold) demonstrated activity was observed with LSO258 and LSO276, both of which feature bromine in the same position in the blue ring. Compound LSO276 showed cell line-dependent activity, with an IC50 of 24.8 µM in the MOLM-13 acute leukemia cell line and LSO258 with IC50 values of 25.5 µM, both with same potency.
Overall, most of the compounds evaluated in this study were more potent against MOLM-13, even when IC50 values were not achieved. For instance, LSO259, LSO234, and LSO280 demonstrated an MGI above 55% for MOLM-13, while for cell lines derived from solid tumors, their MGI values were below 35. In addition, most of the compounds evaluated in this study were more potent towards MOLM-13, even those that did not reach an IC50 value. For example, LSO259, LSO234, and LSO280 displayed an MGI above 55% for MOLM-13, while for the cell lines from solid tumors, only an MGI below 35%.
The MOLM-13 cell line represents a subtype of AML, specifically M5a, characterized by wild-type TP53 and a mutated FLT3 gene [52–54]. This FLT3-ITD mutation leads to the constitutive activation of the FLT3 tyrosine kinase receptor, driving uncontrolled cell proliferation and resistance to apoptosis [55, 56]. As a result, AML patients with this mutation often experience poor survival rates. First-generation tyrosine kinase inhibitors targeting the FLT3 pathway have been evaluated in clinical trials; however, their low specificity limited their therapeutic efficacy [57, 58]. To overcome this, second-generation FLT3 inhibitors, such as gilteritinib and quizartinib, were developed and demonstrated significant clinical benefits, leading to their approval for AML treatment in the USA, Japan, and Europe. Nevertheless, treatment resistance remains a challenge, as AML cells can acquire additional mutations in FLT3 or related genes, ultimately reducing the effectiveness of FLT3 inhibitors [56, 59, 60].
Different studies have shown the antiproliferative effect of 1,2,3-triazoles-hybrids against leukemia cell lines. Ashwini et al. [54] synthesized a series of 1,2-benzisoxazole tethered 1,2,3-triazoles with preeminent anticancer activity in vitro. The lead compound PTB presented IC50 ranging from 1–2.5 μM after 96 h exposure in the following cell lines, MOLM-14, MOLM-14 and MV4-11. In another study, novels 1,4-diaryl-1,2,3-triazolo-based ureas compounds were developed as FLT3-ITD inhibitors and tested against cancer cell lines FLT3-ITD dependent (BaF3, MV4-11, and MOM-13) and FLT3-WT cell line. The results showed that different compounds were highly cytotoxic and selective towards FLT3-ITD cell lines compared to the wild-type one. In addition, the molecular docking evaluation indicated that the binding of one of the derivatives was mainly due to the triazole ring [55].
The observed cytotoxicity of bromine-containing fluorene-triazole and biaryl hydroxy-triazole derivatives, particularly LSO258, LSO272, LSO276, and LSO278, aligns with the well-established role of bromine in enhancing drug-target interactions through halogen bonding (X-bonding) [56]. Bromine’s moderate electronegativity, polarizability, and ability to form directional halogen bonds likely contributed to the increased potency of these compounds, particularly against MOLM-13 cells. Halogen bonds with carbonyl oxygens, amides, and π-stacking with aromatic residues may have improved ligand-protein interactions, enhancing selectivity and binding affinity to biological targets [57, 58]. Furthermore, bromine’s lipophilic nature may have facilitated better membrane permeability and cellular uptake, contributing to the observed cytotoxic effects [59].
Additionally, the structural influence of bromine within rigid fluorene-triazole scaffolds suggests that the conformational constraints imposed by this framework may have minimized entropic losses during binding, resulting in more favorable protein-ligand interactions. Interestingly, LSO278, a flexible biaryl hydroxy-triazole bearing two bromine atoms, demonstrated broad activity across multiple cancer cell lines, suggesting that in certain cases, enhanced electronic interactions provided by multiple bromine substituents can compensate for flexibility-related entropic penalties. These findings reinforce the importance of bromine in medicinal chemistry, highlighting its potential to fine-tune molecular recognition, optimize pharmacokinetic properties, and enhance cytotoxicity in drug discovery efforts [60, 61].
In conclusion, we successfully synthesized 13 new biaryl hydroxy-1,2,3-triazoles using a one-pot reaction that eliminated the need for isolating intermediates, achieving yields ranging from 16% to 89%. These triazoles served as precursors for 9H-fluorene-triazole hybrids, resulting in the formation of novel compounds with yields between 20% and 97%. The synthesized structures demonstrated significant potential for inhibiting the growth of several cancer cell lines. Among the compounds evaluated, most exhibited higher potency against MOLM-1, with LSO258, LSO272, LSO276, and LSO278 showing IC50 values of 25.5, 12.5, 24.8, and 18.7 µM, respectively. Notably, LSO278 displayed versatility, exhibiting additional activity against HCT-116 (IC50 = 23.4 µM) and MDA-MB-231 (IC50 = 34.3 µM). While a direct correlation between flexible ligands and rigid scaffolds could not be established, the presence of a triazole moiety and bromine substitution on the blue ring (derived from aryl azide) consistently emerged as key features, as all active compounds shared this structure.
AML: acute myeloid leukemia
CuAAC: copper-catalyzed alkyne-azide cycloaddition
DMF-DMA: N,N-dimethylformamide dimethyl acetal
EACA: enaminone-azide cycloaddition
EtOAc: ethyl acetate
IC50: half-maximal inhibitory concentration
InhA: NADH-dependent 2-trans-enoyl-acyl carrier protein reductase
MGI: maximum growth inhibition
MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide
SI: selectivity index
The supplementary figures for this article are available at: https://www.explorationpub.com/uploads/Article/file/1008107_sup_1.pdf. The supplementary table for this article is available at: https://www.explorationpub.com/uploads/Article/file/1008107_sup_2.pdf.
DCZ: Writing—original draft, Investigation, Data curation. MFMFA: Writing—review & editing, Investigation, Data curation. MSR and RdSB: Investigation. JdBVN: Writing—original draft, Investigation. CdÓP: Validation, Supervision, Funding acquisition. CDB: Conceptualization, Validation, Writing—review & editing, Supervision, Funding acquisition. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
This study was financed in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior-Brazil (CAPES)-Finance Code 001, Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) for the scholarships and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), process number [E-26/202.780/2019], and the National Program for Oncological Care PRONON/NUP [25000.019172/2021-11]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.