
Abstract
Peptides constitute an important component of Nature’s pharmacy and they play a significant role in several signaling pathways acting as natural biological messengers. While nature has mastered the cycle of creation, application, and destruction of large and short peptides to the benefit of the host organism, organic and medicinal chemists have in their capacity and small steps, made big developments in the field of peptide synthesis as well as in developing them as therapeutics. In comparison to their big counterparts, i.e. proteins, short peptides encompass several advantages, from the ease of synthesis to their physico-chemical properties. However, the real challenge for in vivo application of therapeutic peptides is to overcome their low plasma availability and their fast enzymatic degradation. This review briefly covers the relevant areas of medicinally important short peptides and the recent developments made to turn these peptides into therapeutics. Also presented in this article are important efforts and strategies used to overcome some of the inherent limitations of peptidic molecules and thereby facilitate their progression in the clinical phases towards approved drugs.
Keywords
Short peptides, peptide drugs, peptide stability, pharmaceutical propertiesIntroduction
Nature is the most relevant source of biologically and pharmacologically relevant small molecules [1]. No wonder, natural products (NPs) have always inspired and challenged organic and medicinal chemists to develop a wide range of NP-inspired new molecules and intriguing synthetic strategies including total and semi-synthesis of complex NPs and their analogues [2, 3]. Drug discovery strategies had remained biased towards non-peptidic small molecules not only because of the ease of synthesis but also for their physchem properties favourably influencing cell permeability and oral bioavailability. Furthermore, the emergence of screening technologies, like high-throughput screening (HTS), fragment-based screening (FBS), and screening of DNA-encoded libraries (DELs) have facilitated the discovery of small molecule ligands [4–9]. Despite the success of small molecule chemotherapies to inhibit oncoproteins, it is notable that only 3,000 out of ~25,000 proteins encoded by the human genome possess binding sites suitable to interact with small molecules. The viability of small molecules against tougher and undruggable targets is intriguing and often questionable too. For instance, protein-protein interactions (PPIs), which are critical to tumorigenesis and metastatic pathways due to the high cellular compartmentalization are highly challenging even as binding targets for small molecules because of the featureless nature of protein interfaces [10–12]. The conventional small-sized drug molecules may suffer from reduced target selectivity and thereby manifesting more side effects. On the other hand, protein-based therapeutics though possess exquisite specificity for their target, they often show unfavorable bioavailability and stability that render them highly challenging pre-clinical, as well as clinical candidates. Small or short peptides are a very interesting class of pharmaceutical compounds, molecularly poised between small molecules and proteins, yet biochemically and therapeutically distinct from both. Peptides mediate several signaling pathways and thereby control and affect many physiological functions. Being NPs and mediating natural biochemical and physiological functions, they present an immense opportunity for therapeutic intervention that closely mimics natural pathways [13].
Since the medical discovery of insulin, peptides have displayed interesting possibilities, thanks to their high biocompatibility, safety, selectivity, and efficacy. In Table 1, a comparison of short peptides versus small and large molecules depicts their advantages and drawbacks as therapeutic molecules [14]. Short peptides possess some similarities to biologics, such as high target specificity and affinity, but with a competitive production price that is yet inaccessible for larger molecules. The cell permeability of small molecules, on the other hand, brings a big advantage as oral drug candidates and peptides in general, are not able to penetrate cell membranes and therefore bind to intracellular targets. However, peptides can engage with a larger surface area of their targets and thereby are far more suitable as inhibitors of PPIs [15].
Peptides compared to small molecules and large molecules (enzymes, antibodies, microbiome-based biologics)
| Molecular size | Advantages | Disadvantages |
|---|---|---|
| Small molecules (0.1–1 kDa) |
|
|
| Peptides |
|
|
| Large molecules |
|
|
Quite similar to proteins, the therapeutic peptides are also prone to proteolytic inactivation (i.e. amide bond is easily hydrolyzed) and thus have poor in vivo stability, low oral bioavailability, and low plasma stability [16]. The enzymatic inactivation by proteases has prompted researchers to devise numerous strategies to overcome this limitation. Several chemical modifications of peptide backbone and development of peptidomimetics (substitution with D-amino acids, peptoids, N-methyl amino acids, and β-amino acids) with both core and side-chain modifications (non-natural amino acids), as well as a number of cyclization strategies have emerged in recent years to improve pharmacokinetic (PK) and pharmacodynamic (PD) properties of the peptides [17–19]. Thus, the major factors driving the development of peptide therapeutics are their high specificity and low toxicity which result from their extremely tight binding to their targets. Identification of peptides with such advantages is plausible due to the large chemical space offered by the side-chain variations in the native amino acids of the peptides.
The current pipeline of therapeutic peptides highlights the success rate in clinical trials in terms of selectivity, high specificity, and low toxicity [20]. Over the last two decades, peptides have performed incredibly well, and in some years even outperformed small molecules at the phase II–III transition, with the rate of clinical approval, contining its ascendance [20]. THPdb (http://crdd.osdd.net/raghava/thpdb/), a well-curated repository of Food and Drug Administration (FDA) approved therapeutic peptides and proteins, provides information on their chemical structure, PK/PD profile, and mode of activity [21]. In particular, short peptides (2–45 amino acids) are intriguing bio-molecules with several advantages compared to small molecules or their larger peptidic analogues [22, 23].
Of the 37 new drugs approved in 2022, two are short peptides [24, 25]: tirzepatide (Eli Lilly, 39 amino acids) for the treatment of type II diabetes mellitus (T2DM; Figure 1A), and PluvictoTM (Novartis, 2 amino acids and 2 pseudo-amino acids), the radiolabeled Lutetium Lu-177 pseudopeptide vipivotide tetraxetan for prostate-specific membrane antigen (PSMA) cancer (Figure 1B) [7, 8].
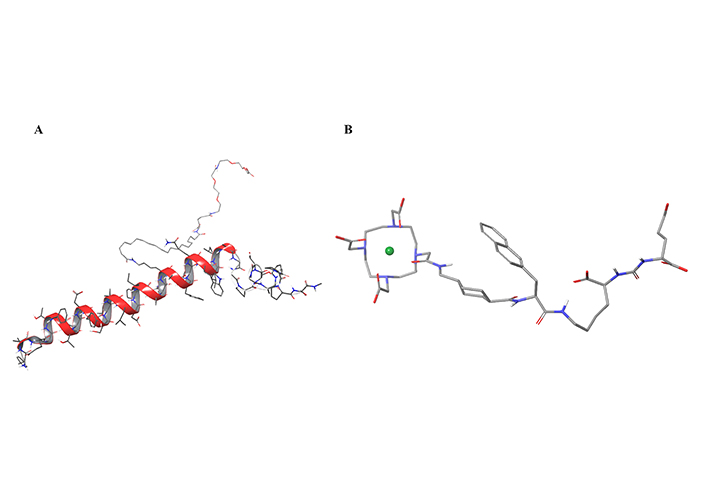
In silico three-dimensional (3D)-models of the two short peptides approved in 2022. A) Tirzepatide; B) Lu-177 vipivotide tetraxetan
There are several excellent review articles covering different aspects of the chemistry and biology of peptides in general as well as proteins [26–33], in this review interrogates the chemical space of short medicinally relevant peptides and the state of the art to improve their physchem properties and thereby their potential therapeutic development into clinical candidates. The field of peptides covering its chemistry and biology is too vast to be covered in a single review. In fact, each of the subtopics touched in this review itself deserves a full review, and therefore, interested readers are suggested to see the cited references.
Medicinally relevant short peptides
Therapeutic peptides are found in all forms of life which show their central role in mediating and managing biological functions [5, 6]. Plants, animals (including humans), bacteria, and fungi create and use biologically active short peptides. Many of these natural peptides have been isolated and well characterized. In particular, arthropods and cephalopods have been recognized as natural sources of peptide-based venoms. Thus, the study of biologically active peptides from insects became a favourite strategy in the last century to develop novel nature-inspired venom- and toxin-based drugs [23].
Cyclosporines, isolated from Fungi imperfecti, are short cyclic peptides that are used as immunosuppressor agents in organ transplantations [34]. They often contain a D-amino acid in their amino acid sequence, that imparts proteolytic stability to these therapeutic peptides. Marine bioactive peptides (i.e. sea snake hydrostatin-SN1) are featured in several reports and endowed with anticancer, anti-inflammatory, and anti-oxidant activities among many others [35].
Although from a pharmaceutical point of view, peptide hormones remain the most clinically used molecules, e.g., the luteinizing hormone-releasing hormone (LHRH) agonists and antagonists for the treatment of prostate cancer [36, 37], over the last decade the peptide market is covering a wide range of therapeutic applications [38], including oncology [39], metabolism [40], endocrinology [41], cardiovascular diseases [42], and as antimicrobial agents [43]. Among the many medicinally relevant short peptides, from both natural and synthetic sources, we focus the first part of this review on endogenous peptides, host defence peptides (HDPs), and non-ribosomal peptides (NRPs), along with their clinical applications.
Endogenous peptides
Before the invention of solid-phase peptide synthesis (SPPS) in 1963 [44], the solely chemically synthesized peptides were the endogenous human hormones vasopressin and oxytocin [45]. This leap forward, together with the advent of recombinant technology, yielded the production of a large amount of natural and nature-inspired peptides. Insulin, vasopressin, oxytocin, somatostatin, glucagon-like peptide or glucagone-like peptide-1 (GLP-1), and gonadotropin-releasing hormone (GnRH) are some of the endogenous human short peptide hormones. These molecules represent the large family of peptide hormones with a short number of amino acids [46]. GLP-1 (37 amino acids) is a physiologically active short peptide that controls insulin levels. The awfully short half-life of insulin in plasma (1.5–5 min) limits its clinical use and prompted its sequence modifications to improve its stability. Semaglutide, liraglutide, and dulaglutide are commercially available GLP-1 receptor agonists for the treatment of T2DM [47]. As further discussed in Orally available peptides, while their alanine residue substitution enabled them to avoid degradation by dipeptidyl peptidase-IV (DPP-IV); the incorporation of an alkyl chain allowed improved binding to plasma proteins.
Human neurohormone vasopressin is a naturally occurring nonapeptide, which is exclusively found in mammals and synthesized in the hypothalamus. It is a vasoconstrictor and a water homeostasis regulator [48, 49]. As its use was marred with limitations, such as short half-life, target specificity, and side effects, some efforts emerged to discover novel and improved vasopressin analogues [50]. These include the natural lypressin (half-life 5–7 min)—in clinical use as a nasal spray for the treatment of diabetes insipidus [51], and the synthetic terlipressin (half-life 4–5 h), and ornipressine (half-life 1–2 h)—useful in liver cirrhosis hepatorenal syndrome (HRS) [52]. Lypressin differs from vasopressin for having the arginine in position 8 replaced by lysine, and for showing a conformational flexibility in the disulfide portion essential for biological activity [50]. Terlipressin is considered a pro-drug due to a three glycine residue at the N-terminal that are cleaved to release the active metabolite lypressin [53]. Ornipressin, the arginine in position 2 is replaced by ornithine, and is successfully used in combination with tranexamic acid acting on microcirculation.
The use of long-term terlipressin and ornipressin, explored for the coronavirus disease-19 (COVID-19) as well. Indeed, it was demonstrated that arginine vasopressin (AVP) is produced in COVID-19 patients with high concentrations of cytokines to counterbalance the high blood viscosity. High AVP levels in COVID-19 patients, induce hyponatremia, inflammatory disorders, and other complications by activation of nuclear factor-kappaB (NF-κB) and NOD-, LRR- and pyrin domain- containing protein 3 (NLRP3) inflammasome with release of pro-inflammatory cytokines. AVP antagonists might mitigate AVP-mediated inflammatory disorders and hyponatremia and could prove a potential therapeutic modality in treating COVID-19. To this end, extensive structure-activity relationship studies of vasopressin and its analogues have been reported in some recent investigations in 2020–2021 [54].
Another class of short endogenous peptides is opioids, produced mainly in the central nervous system (CNS). They are delivered to the target tissues where they act as neuromodulators [55]. For example, endorphins and enkephalins are involved in several physiological pathways, including pain regulation. Endorphins (~20 different derivatives), ubiquitous within the body, are neurotransmitters derived from the prohormone pro-opiomelanocortin (POMC), and their opioid activity is due to the C-terminal binding to opioid receptors [56]. Enkephalins are encoded from the gene pre-proenkephalin A and encompass short peptides such as 5-methionine (Met5)-enkephalin and 5-leucine (Leu5)-enkephalin. Their main functions as opioid receptor agonists are neurotransmission and pain modulation and are involved in analgesia treatment [57]. The activity of enkephalin analogues was shown to be dependent on the C-terminal amino acid, which can furthermore influence the selectivity of opioid receptors [58].
The great therapeutic potential of endogenous peptides, attributed to their interaction and activation of G-protein-coupled receptor (GPCR) targets, has drawn immense interest from researchers, and the major challenges in their development [i.e. metabolic stability, blood-brain barrier (BBB) permeability, and bioavailability] are in high demand [59].
HDPs
HDPs are crucial components of innate and adaptive immunity with antimicrobial properties. Natural HDPs are synthesized both ribosomally as well as non-ribosomally, by all complex animals, insects, and plants, and generally have adequate direct activity against a broad range of microorganisms [60]. They kill bacteria in response to external stimuli and involve interconnected signaling pathways [61]. The antimicrobial activity of some HDPs could be linked to their immunomodulatory functions. However, the role of HDPs in immunity is very complex, involving various receptors, signaling pathways, and transcription factors, and remains a field of active research. The promising therapeutic potential of HDPs as antimicrobial agents against diverse pathogens are drawing attention [62]. The antimicrobial peptide (AMP) database (https://aps.unmc.edu) encompasses a large collection of natural AMPs (~3,000) [30]. Notably, only seven AMPs are clinically used: daptomycin (natural and synthetic cyclic lipopeptide, 13 amino acids, inhibition of bacterial growth), gramicidin S (natural cyclic peptide, 12 amino acids, membrane disruption, and depolarization) [63], colistin (natural cyclic peptide, 10 amino acids, membrane lysis) [64], oritavancin (nature-inspired lipoglycopeptide, 7 amino acids, membrane lysis and inhibition of cell wall synthesis) [65], dalbavancin (naturally-inspired lipoglycopeptide, 7 amino acids, inhibition of cell wall synthesis) [66], telavancin (nature-inspired, lipoglycopeptide, 7 amino acids, membrane lysis and inhibition of cell wall synthesis) [67], and vancomycin (natural, 7 amino acids, inhibition of cell wall synthesis) [68]. Colistin was first approved for acute and chronic Gram-negative bacterial infections, and despite its significant renal and neurologic toxicity, it remains the last resort treatment for some multi-drug resistant (MDR) species such as Pseudomonas aeruginosa and Acinetobacter spp. [69].
Recently, two reviews [43, 70] have summarized their classification based on their chemical structure (linear, cyclic, and lipopeptides), and depicted their potential applications, including as immunomodulator agents.
The immense scaffold complexity and diverse mechanisms of action make AMPs the potential solution to fight the huge issue and challenge of antibiotic resistance [43]. The mesmerizing ability of certain HDPs in enhancing innate immune responses to control infections as well as control inflammation makes them attractive therapeutic candidates for both anti-infective and anti-inflammatory indications. There are, however, the challenges—the ‘usual suspects’ in short peptide drug development, i.e. limited bioavailability, associated toxicity, and high manufacturing costs of cationic peptides that the scientific community needs to address urgently.
NRPs
NRPs represent an important source of short peptides with therapeutic relevance produced by a different pathway to the traditional ribosomal biosynthesis [71]. They usually consist of less than 10 amino acid residues and are characterized by a complex structural diversity, such as the incorporation of non-proteinogenic amino acids, alternative linkages, and post-translational modifications [72]. Indeed, their structural complexity and diversity present a challenge for organic chemists. Interestingly, NRPs are more resistant to proteases and therefore display higher plasma stability in vivo.
The antibiotic cyclosporin A (Figure 2) and its analogues [73, 74] present a fine example of how the developments in synthetic chemistry pave routes to complex molecules endowed with a broad range of biological activities. Some natural NRPs with therapeutic applications include penicillin precursor ACV-tripeptide, daptomycin, cyclosporin, vancomycin, and bacitracin.
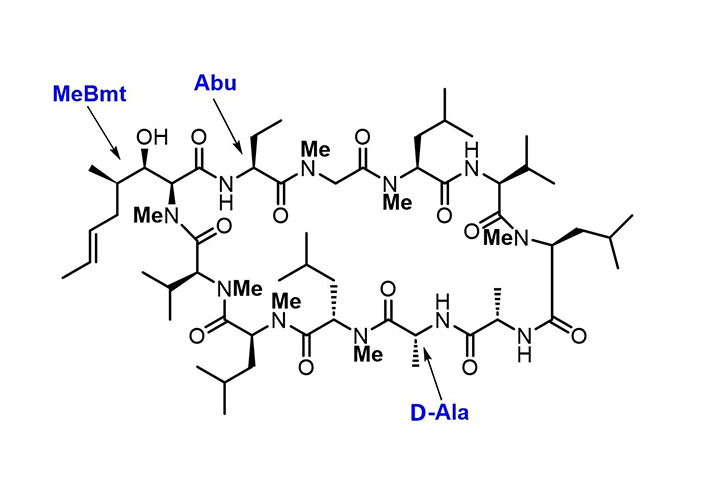
Chemical structure of the NRP cyclosporin A. In NRPs, particular features differ from “typical” peptides, such as the extensive N-methylation, the presence of D-amino acid, and non-coded amino acid residues. Cyclosporin A contains the non-encoded residues (4R)-4-[(E)-2-butenyl]-4,N-dimethyl-L-threonine (MeBmt), α-aminobutyric acid (Abu), and D-Ala
The NRP sequences often are decorated with non-proteinogenic amino acids. Some of the latter are shown in Figure 3. The discovery of more than 800 non-proteinogenic amino acids, unique building blocks typical of NRPs that confer them a great scaffold diversity, has revolutionized the development of therapeutic peptides, especially AMPs against MDR pathogens [75].
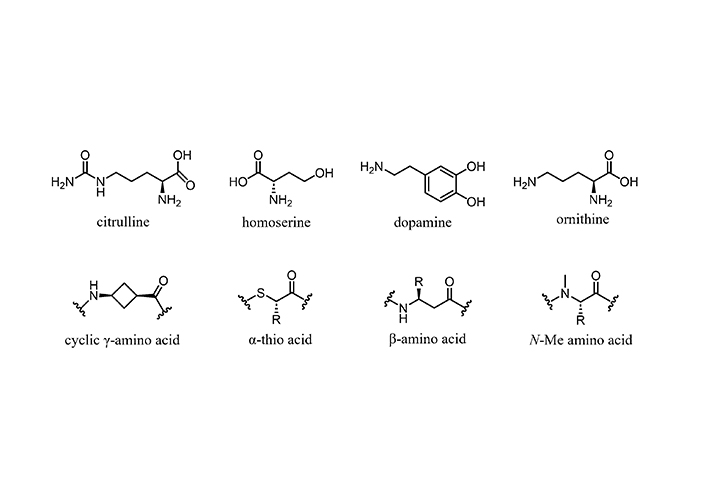
Short peptides in medicinal chemistry
The strengths, weaknesses, opportunities, and threats (SWOT) of therapeutic short peptides describes their crucial features from a PK/PD point of view [22, 23], and their metabolism produces predictable non-toxic degradation products. Only ultra-short peptides (≤ 7 amino acids), a subclass of short peptides, are able to penetrate cell membranes [78] and, therefore, are now used in a wide range of applications [79], such as drug delivery [80], 3D-bioprinting [81], imaging [82], and cell culture [83].
The evolution has selected short peptides mainly to act in the endocrine signaling pathway as natural secondary messengers. Once completed their function, peptides are quickly metabolized (i.e. the signal is turned off by irreversible post-translational modifications) [84]. Therefore, it is not surprising that our body is programmed to deactivate peptide circulation: indeed, they are first proteolytically cleaved, and then rapidly processed via renal filtration. Peptidases (also called proteases, proteolytic enzymes, or proteinases) are the most populated classes of enzymes, and they hydrolyze peptidic bonds [85]. The peptidases database (MEROPS, www.ebi.ac.uk/merops/) encompasses more than 4,000 proteolytic enzymes of which 609 come from human beings (Homo sapiens) [86]. The high sequence similarity in the catalytic sites of a number of existing homologues of peptidases makes the development of protease-resistant therapeutic peptides an extremely challenging task. In 2018, Klein et al. [87] classified known proteases based on their proteolytic mechanism and proteolytic cleavage site (Table 2), The knowledge of the proteolytic mechanisms offers a common strategy to overcome peptide instability by replacing suspect residues of the cleavage site with other amino acids [85, 87].
Main proteases with their cleavage site and proteolytic mechanism
| Proteases | EC | Proteolytic mechanism | Cleavage site |
|---|---|---|---|
| Asn lyases* | 4.3.2 | Elimination reaction | Asn-Ala bond |
| Aspartic peptidases | 3.4.23 | Acid-base | Cys |
| Cys peptidases | 3.4.22 | Catalytic | C-terminal Asn |
| Glutamate peptidases* | 3.4.23 | Hydrolysis | NAA-Glu |
| Metallopeptidades | 3.4.24 | Catalytic | Metal-binding residues (i.e. His, Glu, Asp) |
| Serine peptidases | 3.4.21 | Acyl-enzyme intermediate | Aromatic amino acids (i.e. Phe-Thr, Tyr-Ser) |
| Threonine peptidases | 3.4.25 | N-terminus hydrolysis | N-terminal Met |
* At present not found in mammals (isolated from fungal and bacterial sources). EC: enzymatic class; NAA: N-acetyl aspartate; Asn: asparagine; Ala: alanine; Cys: cysteine; Glu: glutamic acid; His: histidine; Asp: aspartic acid; Phe: phenylalanine; Thr: threonine; Tyr: tyrosine
The short in vivo half-life of peptides is a key challenge for the medchem community to address in the development of peptidic drug candidates and many research groups had put their efforts to address the issue. Due to their rapid clearance, peptide half-lives span from mere seconds up to a few hours. The in vivo life of peptides is largely determined by the amino acid sequence, terminal modifications, dose, and the route of administration. PEPlife (http://crdd.osdd.net/raghava/peplife) is a manually curated database showing the experimentally calculated half-life of peptides [88]. This data repertoire contains the parameters used to determine the half-life of peptide: name, primary sequence, chain length, chemical modifications at N- and C-termini, biological properties, and the assay used for the experimental calculations. Gentilucci et al. [89] overviewed the major chemical modifications in peptides that improved or extend their half-life (e.g., stereochemical inversion, the use of non-natural amino acids as pseudo-proline and ornithine, N- and/or C-terminal capping, PEGylation, glycosylation, and lipidation). Peptide chain cyclization and the insertion of a lipid fragment also increase the half-life by hampering proteolytic degradation [90, 91]. One example of successful lipidation is the once-daily drug liraglutide which is identical to GLP-1 except for Lys34Arg substitution [92]. Lipopeptides will be discussed properly in Lipopeptides.
Cell-penetrating peptides
In general, cells internalize hydrophilic macromolecules via an energy-dependent mechanism, called endocytosis. Alternatively, macromolecules can pass across the membrane in an energy-free pathway (passive diffusion) [93]. Several energy independent mechanisms for cell-penetrating peptide (CPP) internalization are known, including the pore-formation or the barrel-stave model [94], the carpet model [95], and the toroidal-pore mode [96]. Although some larger proteins possess the ability to serve as transporter after conjugation (i.e. cargoes) with peptides, peptide nucleic acids (PNA), and oligonucleotides, the discussion here is focused on short CPP with sequences up to 45 amino acids.
The possibility to target biological molecules intracellularly with short peptides is a very significant task that may confer a number of applications in disease diagnosis and therapy [97]. However, the low membrane permeability of peptides remains an obstacle in biomedical research, and therefore strategies to deliver them to intracellular targets are highly desired. To this aim, different delivery approaches, such as the use of liposomes [98] and nanoparticles [99], were developed to translocate drugs to the targets, but their potential immunogenicity and toxicity remain of concern [100]. CPP are short peptides with particular physicochemical properties that enable them to enter cells and serve as a cargo delivery system [101]. The online repository of CPPs (https://webs.iiitd.edu.in/raghava/cppsite/) contains 1,855 sequences (number of CPP updated in February 2023), of which 1,753 are linear, and 102 are cyclic peptides. This database provides a lot of information, including chemical modifications, in vitro/in vivo model systems, and a list of several cargos delivered by CPPs. In fact, CPPs have been extensively employed for efficient intracellular delivery of nucleic acids [102], proteins [103], imaging [104], and anti-cancer agents, as well as small molecules [105]. CPPs are usually classified based on their source, i.e. if they are protein-derived (41.73%), synthetic (54.82%), or chimeric (3.45%). The composition of short peptides including their sequence and nature of amino acid residues defines their physical/chemical properties and their ability to cross the membranes. In general, they can be broadly distinguished as hydrophobic, cationic, and amphipathic CPPs. Penetratin, a 16 amino acids peptide derived from Anthennapedia homeodomain, was the first short peptide that displayed an ability to penetrate the plasma membrane [106].
According to their therapeutic use, CPPs find important applications in anticancer therapies [107], in drug resistant infections [108], and for diabetes treatment [109]. Several CPP-based therapies are currently under preclinical development [107]. In particular, poly-arginines [110], human immunodeficiency virus (HIV)-trans-activator of transcription (TAT)-derived CPPs, and penetratin are well investigated and considered as the models for the further design of novel CPPs. The naturally occurring tat sequence i.e. RKKRRQRRR is derived from TAT and is a cationic peptide that has been extensively used in imparting cell permeability characteristics to the peptides and also is employed as a delivery system. The TAT-modified tobacco mosaic virus (TMV-TAT) was developed by decorating the TAT peptide on the exterior surface and the method was used to explore novel small interfering RNA (siRNA) carriers [111]. Introduction of CPP enhanced cell internalization and thereby TMV-TAT acquired endo/lysosomal escape capacity without inducing lysosomal damage, resulting in both high efficiency and low cytotoxicity. Several other examples are being studied for their use in cancer treatment [112], including the intracellular delivery of chemotherapeutic agents and thus improving their efficiency.
In vivo, the application of CPPs is hampered by low tissue selectivity and in vivo stability. Ideally, for CPP target delivery, the short half-life could be an advantage as they are fast degraded and cleared easily after delivery. However, various research groups have explored structural modifications of peptides to confer them a better cell entrance and more in vivo stability, for example by tuning their net charge (i.e. cationic peptides) or by the conjugation with hydrophobic moiety able to interact with membranes. In 2003, Elmquist and Langel [113] studied the cellular internalization and stability of peptide vascular endothelial-cadherin (pVEC), a CPP derived from murine vascular endothelial cadherin, containing 18 amino acids that act as a vector carrier crossing the membrane with a non-endocytotic mechanism and without any toxicity. Its half-life was determined to be 10.5 min in PBS with trypsin, and 44 min in PBS with carboxypeptidase A. The designed all-D analogue was however not degraded by these proteases, and could even maintain a comparable in vitro uptake.
Further improvement of the cell-penetration ability may be achieved either by masking the backbone amide groups, by incorporating a guanidinium pattern, or by stapling the side-chains [114]. Enhancing the lipophilicity of peptides (by conjugation with hydrophobic moieties) may help the passive membrane permeability [113–115]. Cyclosporin A (Figure 2), a fungal metabolite with immunosuppressive activity used in organ-transplant patients, is a highly N-methylated cyclic short peptide that is orally available [116], and that has inspired the design of passively penetrating peptides [117].
Somatostatin analogues are short peptides that can be used in peptide receptor radionuclide therapy (PRRT). Somatostatin receptors (1–5 types) are over-expressed in tumoral tissues [118–120]. Radiolabelled somatostatin analogues [labelled with radioactive substance like Yttrium-90 or Lutetium-177] have been employed in targeted radiopeptide therapy and proven to significantly improve survival and quality of life in patients suffering from metastatic or unresectable neuroendocrine tumors (NETs) [121]. In a recent work, in vivo, experiments were performed wherein tumors were targeted by the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10- tetraacetic acid (DOTA) coupled with highly potent somatostatin receptors agonists or antagonists [122]. The somatostatin receptor subtype-3 (sst3) antagonist DOTA-sst3-ODN-8 (Figure 4) showed no internalization but high binding affinity and selectivity, preventing agonist-stimulated internalization. Although the agonist natIn-DOTA-NOC can induce sst3 internalization, all antagonists labelled 76 times more sst3 sites in cultured HEK-sst3 cells than the agonist, plausibly owing to their higher hydrophobic content in antagonists than agonists, and thus, a stabilization in the lipidic bilayer of receptors, and a longer time of action.
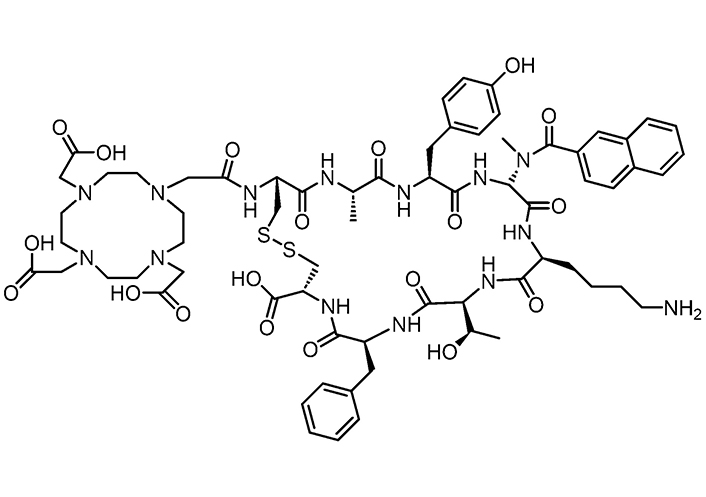
Structure of the sst3 antagonist DOTA-sst3-ODN-8, useful as radioligand to target tumors in vivo
Many AMPs respond to the offensive environment by adopting mechanisms that render them permeable to bacteria [94], and they are found both in vertebrates and invertebrates. In these peptides, the secondary structure and conformational changes lead them to lyse the microbial membrane. It was demonstrated that the α-helix content is not necessary for selective membrane lysis [123]. A non-cell selective lytic natural short AMP from bee venom, melittin (26 residues), possesses a broad-spectrum bactericidal activity against several clinical strains [124].
Recently, the use of CPP-conjugated drugs is emerging as a novel strategy to combat antibiotic resistant infections, drug-resistant tumors, and other pathologies. For instance, conjugation of AMPs with CPPs enhances the antibacterial activity [108]. Lee et al. [125] described the generation of arginine rich magainin (R9-megainin, RRRRRRRRR-GGGGIGKWLHSAKKFGKAFVGEIMNS) that showed higher antimicrobial activity against gram-negative bacteria than magainin. The same effect was observed in R9-M15 (RRRRRRRRR-GGGKWKKLLKKPLKLLKK). Notably, R9 alone does not possess antimicrobial activity.
Conjugation of CPP with glycopeptide antibiotics is also interesting example reported in the literature [126]. Indeed, in 2018, Antonopolis and co-workers reported the conjugation of vancomycin with octa arginine, conjugation of vancomycin-D-octaarginine (V-r8) (Figure 5), that exhibited stronger activity and superior efficacy than vancomycin alone against gram-positive bacteria, including methicillin-resistant Staphylococcus aureus (MRSA) biofilms and persister cells [126]. Persisters-active peptide based drugs are generally polycations that unselectively disrupt the bacterial cell membrane curvature. The eptide-nucleoside Relacin [127, 128] and its analogues have been the first example of a tailored approach against a specific bacterial target (Rel proteins) [129] that unfortunately showed limited activities and cell penetrating properties. In this regard, we have been active in the targeting of Rel proteins with a structure-based approach [130–132] that led to interesting amino acid-derived compounds.
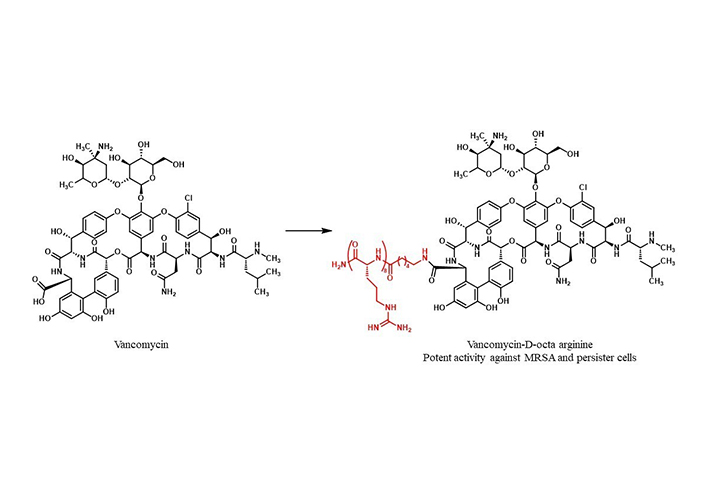
In 2019, the synthesis of vancomycin analogues with the addition of single amino acid residues was reported demonstrating that only one arginine, V-r, led to significantly improved efficacy against gram-positive bacteria, including MRSA [133].
CPP-conjugation with polymers or with supramolecular nanostructures is an alternative shielding strategy to protect CPPs from proteases attack [134]. Remarkably, D-JNKI-1, a conjugated CPP, is found in vivo application in Alzheimer’s disease since it is able to cross to the BBB and penetrate the brain parenchyma till its target in brain mitochondria [135].
Linares et al. [136] in an interesting recent research described the synthesis of cell-penetrating peptide conjugate (C-POC), a novel peptide-platinum (IV) CPP with a high anticancer efficacy and a reduced impact on non-tumoral cells compared to the standard platinum-based therapy.
Orally available peptides
As the oral bioavailability of a molecule is determined by its PK properties, several factors need to be considered in designing orally available peptides. After oral administration, only the fraction that passes through epithelial cells, blood circulation, and reaches the target, is the amount that correlates with the efficacy of the drug [137]. In general, natural linear peptides are not orally available since they are rapidly attacked by proteases, leading to their degradation and clearance. Among the first strategies towards improved oral availability involved using constrained conformations of peptides which make them less prone to proteolytic degradation [138]. Indeed, the constrained conformation induced by cyclization can prevent endopeptidase cleavage of peptides, while other modifications can avoid exopeptidase deactivation [139].
Subsequently, several strategies including prodrug approaches, PEGylation [140], cyclization [138], lipidation [141], and other chemical modifications (i.e. the N-methylation and the use of D-amino acids mentioned above), have been extensively used to improve peptide stability and bioavailability.
Interestingly, the “Lipinski rule of 5” prescribed for orally available small molecule drugs [142, 143], cannot be applied to peptides. An exhaustive review collecting 125 cyclic peptides reported that only a few peptides are marketed drugs, and even fewer are orally absorbed [144]. The commercially available orally administered peptides are all short and cyclic peptides (e.g., cyclosporine—the immunosuppressant, desmopressin—the antidiabetic, linaclotide—for chronic idiopathic constipation, vancomycin hydrochloride and colistin sulfate—antibiotics, taltirelin hydrate—for spinocerebellar degeneration, glutathione—for AIDS-related cachexia and cystic fibrosis and tyrothricin—for pharyngitis) [145]. In 2019 FDA approved the Novo-Nordisk insulin, semaglutide, as the first orally available GLP-1 receptor agonist. Other oral insulins are in phase I–III trial studies [30].
The scientific data of orally available molecules suggest to reducing hydrogen bond donors (HBDs) by the replacement of amide backbone with N-alkyl groups or other surrogates in peptides and the incorporation of nitrogen into heterocycles appended around the backbone. In 2008, Biron et al. [117] reported the synthesis of 31 orally available somatostatin analogues by N-methylation on the cyclic hexapeptide named Veber-Hirschmann peptide c(PFwKTF) [146] (Figure 6A), selective for somatostatin receptors 2 and 5. Indeed, the short half-life of somatostatin (< 2 min) prevents its use as a drug. Its fast degradation is attributed to the secondary structure (conformation) and to the presence of natural unmodified amino acids in the sequence. Further modifications on somatostatin structure (Figure 6B) based on conformational rigidity, led to the development of the improved drug octreotide.
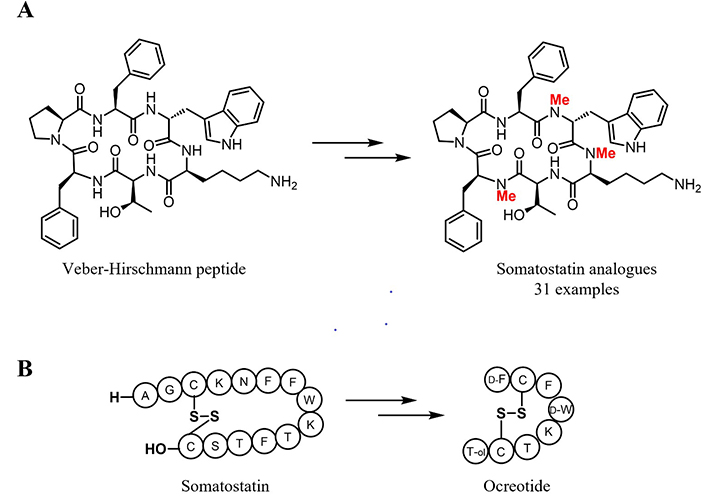
Development of somatostatin analogues. A) An example of successful N-methylation for the synthesis of somatostatin analogues. Positions, where the N-methylation was inserted are highlighted in red; B) somatostatin and octreotide structures
Octreotide (Sandostatin®) is used in acromegalia treatment and in intestinal endocrine tumors [147]. Its higher metabolic stability (half-life 110 min) is mostly owed to the β-turn stabilization induced by the incorporation of substitution with a D-amino acid [148]. Although this synthetic somatostatin analogue possesses a longer half-life, it is still not orally bioavailable. This demonstrates how demanding the task of taming peptides into oral drugs and calls for further research.
Lipopeptides
Lipopeptides, i.e. oligopeptides linked to a fatty acid portion, are a promising family of drugs as they encompass key advantages, including an extended half-life, the ability to cross the BBB easier than non-lipidized counterparts, and to bind plasma proteins [149, 150]. In particular, their binding to plasma proteins (such as albumin) [151] confers protease stability. Daptomycin (Cubicin®, Figure 7), a branched cyclic lipopeptide synthesized by non-ribosomal bacterial synthetases, is the only antibiotic lipopeptide approved for clinical use [152]. Its calcium-dependent mechanism of action results in the depolarization of the bacterial membrane in anaerobic and aerobic Gram-positive strains [153]. Daptomycin is intravenously administered once daily (half-life 8–9 h) to treat MDR infections.
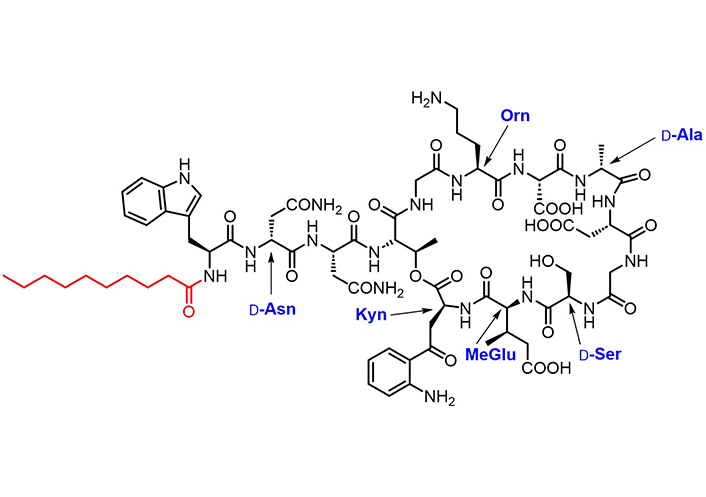
Chemical structure of daptomycin. The decanoyl moiety is marked in red. Orn, D-Asn, D-Ser, D-Ala, (2S,3R)-methylglutamate (MeGlu), and Kyn are non-proteinogenic amino acid residues
In 2022, Wan et al. [154] described the advances in the Iturins family, the cyclic lipopeptides extracted from Bacillus subtilis with a broad-spectrum activity against fungi and bacteria, and they are mostly used in plant infections and pesticides production [155]. The iturin family is characterized by the peculiar presence of β-amino acids and fatty acid chains, but their clinical use is limited as they can cause hemolysis in human erythrocytes [156].
Liraglutide, a palmitoylated GLP-1 possesses a very long half-life after subcutaneous administration (11–15 h) [157], attributed to albumin binding [158], allowed its marketing for the treatment of T2DM and obesity.
Lipidated hormones are a distinguished class of lipopeptides. Ghrelin (28 amino acids and an octanoyl acid moiety)—the hunger hormone, is the most representative example of a natural lipopeptide, produced in the human stomach and is among the very few peptides able to penetrate the BBB [159]. Ghrelin is active only if modified with octanoic acid and it finds application in eating disorders, obesity, and in growth-hormone deficiencies.
Synthesis of short peptides and optimization
Peptide synthesis has a long journey of method development and reagent discovery and has matured as an art of creating peptide based small and large molecules. Several techniques can be used for peptide synthesis [160, 161], using either the classical solution phase synthesis (SPS) [162] or the SPPS [44]. The latter, in particular, has smoothened the peptide synthesis, so much that automation is largely used for SPPS. Recently, convergent approaches were developed to overcome the limitation of SPPS, including native chemical ligation (NCL) [163, 164], the α-ketoacid-hydroxylamine (KAHA) ligation [165], the Diels-Alder ligation [166], the copper(I)-catalyzed azide−alkyne cycloaddition (CuAAC) [167], and the Staudinger ligation [168] (Figure 8).
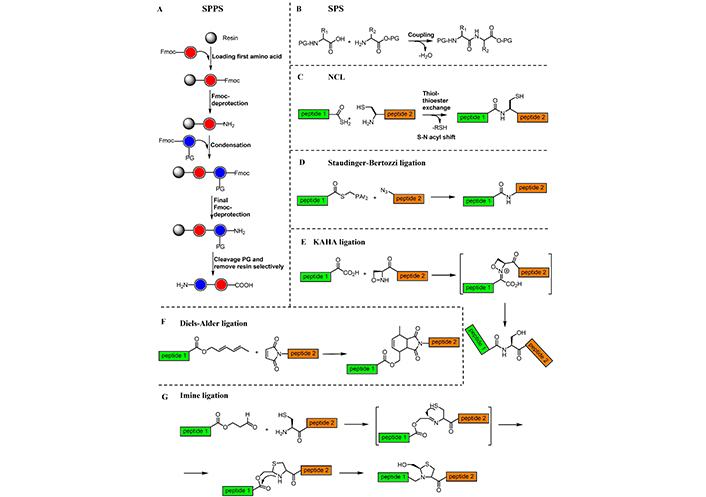
Chemical approaches for peptide synthesis. A) SPPS illustrated used 9-fluorenylmethoxycarbonyl (Fmoc)-chemistry; B) SPS; C) NCL; D) Staudinger-Bertozzi ligation; E) KAHA ligation; F) Diels-Alder ligation; and G) imine ligation
Although the standard application of solution phase peptide synthesis allows to condense convergently a peptide with high purity and yield, the long-reaction time and the laborious preparation of building blocks, and purifications remain big disadvantages. The advent of SPPS allowed synthetic improvements, permitting the preparation of long chain peptides with modifications at any point of the sequence and without the time-consuming purifications after each step. SPPS strategy requires the presence of a resin as solid support which is linked to the C-terminus of the amino acid, and with a typical NH2 protecting group, i.e. the base labile Fmoc or the acid labile tert-butyloxycarbonyl (Boc) [169, 170]. Microwave-assisted SPPS is a high-performing development of SPPS that allows the synthesis of peptides in high yields and with a low rate of racemization [171]. The advent of automated instruments has facilitated the synthesis of long peptides with the insertion of chemical modifications and conjugations [172]. The Fmoc strategy is the mainly used method, which is continuously updated with numerous improvements made in reagents and reaction conditions [173–176]. The most common reagents and conditions in microwave-assisted SPPS are shown in Table 3.
Updated reaction protocols in microwave-assisted SPPS
| Strategy | Reagents and conditions | Ref. |
|---|---|---|
| Boc | Solvent: DMF Deprotection: 100% TFA Coupling: HOBt, DIC, and DIPEA or HBTU and DIPEA (for Boc-Asn) | [177] |
| Fmoc | Solvent: DMF Deprotection: 10% (w/v) piperazine or 20% (v/v) piperidine Coupling: Oxyma pure and DIC | [178, 179] |
| Fmoc | Solvent: DMF Deprotection: 10% (w/v) piperazine or 20% (v/v) piperidine Coupling: HOBt and DIC or HBTU (or HATU) and DIPEA | [171, 180–183] |
| Fmoc | Solvent: NMP Deprotection: 10% (w/v) piperazine or 20% (v/v) piperidine Coupling: HBTU (or HATU) and DIPEA | [184] |
Ref.: reference; DMF: dimethylformammide; TFA: trifuoloacetic acid; HOBt: hydroxybenzotriazole; DIC: N,N-Diisopropylcarbodiimide; DIPEA: N,N-Diisopropynethylamine; HBTU: 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate; NMP: N-Methyl-2-pyrrolidone; HATU: 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate
For decades, piperidine has been preferred as Fmoc deprotecting reagent, despite its higher toxicity as compared to piperazine. One interesting study, in 2016, reported the comparison between 4-methylpiperidine, piperidine, and piperazine as Fmoc-deprotection reagents, confirming their interchangeability in terms of purity and yield of the desired product [185].
A recent and brilliant review described the development over the years for sustainable peptide synthesis, with particular attention on the technical innovation of SPPS and peptide anchored (PA) liquid phase peptide synthesis [186].
A time-lapse on ligation methods
Development of chemical ligation methods towards chemoselectively linking unprotected amino peptide fragments in an aqueous medium was a major leap in the synthetic methodology of peptides [163]. The peptide ligation methods encompass reactions that involve the reactivity of the α-nitrogen, or the participation of the side chain (Figure 8A–C) [187].
Saxon et al. [168] and Saxon and Bertozzi [188] reported modifications of the Staudinger ligation, the chemoselective formation of an amide group from a functionalized phosphine and an azide (Figure 8D). They introduced a new reaction (called “traceless” Staudinger ligation), that uses a phenol ester and N-acylimidazole phosphines, to rapidly form amide bonds. Staudinger-Bertozzi ligation has found many applications in peptide synthesis as well as in enabling the study and manipulation of biological processes. For instance, in 2013, the Staudinger-Bertozzi ligation was found to be ideal for the incorporation of molecular photoswitches such as azobenzenes into biomolecules [189].
In general, the Staudinger ligation is a widely used approach to conjugate biomolecules for in vitro therapeutic targeting, but some drawbacks regarding the biocompatibility, such as the incomplete removal of unreacted phosphane-biotin that interferes with background fluorescence [190], had been reported [191, 192]. More biocompatible techniques were recently explored, introducing the so-called click chemistry that employs copper(I) [3 + 2] N3 cycloaddition reactions [193–195].
In 2006, de Araújo et al. [166] reported a novel strategy for a site-specific chemoselective ligation of peptides and proteins by applying Diels-Alder cycloaddition reaction executed between a dienyl ester and a maleimide dienophile under mild conditions (Figure 8F). Other selective ligations have explored reactions of hydroxylamines and hydrazines with aldehyde-ketones to form oximes and hydrazones [196, 197], and by reactions between aldehyde with cysteine (or serine), via imine ligation, that rearrange to form a stable pseudoproline (Figure 8G) [198, 199]. An important evolution of NCL is presented by the approach involving N-terminal serine and threonine residues (Ser-Thr peptide ligation) [200, 201]. Recently, this method was expanded in an elegant one-pot ligation-desulfurization reaction by the exploration of C-terminal proline salicylaldehyde esters [202]. The resulting reaction between N-terminal penicillamine and C-terminal proline salicylaldehyde esters was proposed as an example of cysteine/penicillamine ligation, useful for chemical protein synthesis [202].
Post-synthesis covalent modifications with an orthogonal double crosslinking of genipin and sulfosuccinimidyl 4-(N-maleimidomethyl)cyclohexane-1-carboxylate (Sulfo-SMCC; through the formation of a maleimide) were described by Ciulla et al. [179] (Figure 9) to increase the stiffness in linear short peptides and in order to induce a higher β-sheet content capable of mimicking physiological microenvironments, such as the extra-cellular matrix (ECM), and with constructs closer to stiffnesses of cardiac tissue, osteoids, and skin [179].
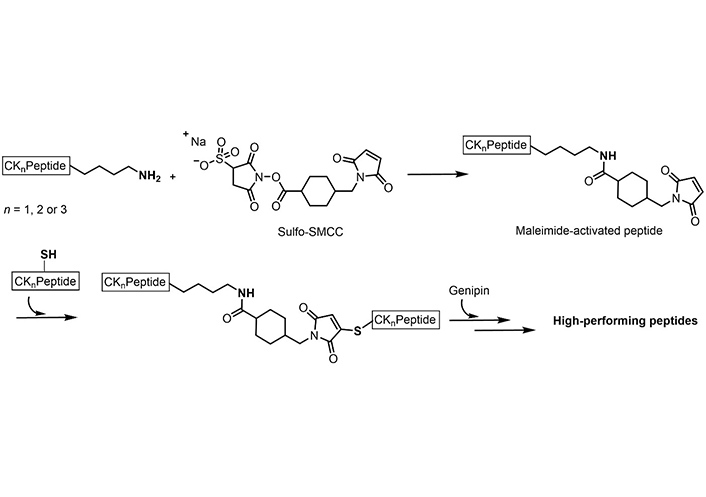
Orthogonal crosslinking of Sulfo-SMCC and genipin on short peptides containing both cysteine and lysine
Note. Adapted from “Boosted cross-linking and characterization of high-performing self-assembling peptides,” by Ciulla MG, Pugliese R, Gelain F. Nanomaterials (Basel). 2022;12:320 (https://doi.org/10.3390/nano12030320). CC BY.
Synthetic strategies to improve pharmaceutical properties
Structural modifications can improve the physicochemical properties of peptides. Modifications that confer rigid conformations, as well as proteolytic resistance are the desired ones. To this end, backbone modification and cyclization are two classical strategies applied to stabilize peptides in their bioactive conformation and improve resistance to biodegradation. Cyclization aims to introduce a conformational constraint into the recognition sequence and the use of non-standard amino acids and/or small molecule fragments etc. enhance their stability into the bioactive conformation, decreasing the entropy loss due to receptor binding and, therefore, increasing their affinity for the target [203].
Cyclization
Many chemical methods were developed over the years to improve PKs profiles of peptides. Constrained peptides have attracted greater attention because of their better pharmaceutical properties as compared to the linear and non-strained counterparts [204]. The literature is rich in both synthetic and enzymatic methods for peptide cyclization, including direct amide bond formation (between carboxylic acid and amine of two groups), through NCL, using bioorthogonal reactions (i.e. KAHA ligation, Figure 8E), by the use of disulfide bond formation [204], and employing enzymes to catalyze highly efficient and selective cyclization (e.g., transglutaminase or subtiligase) [205]. Here the authors would like to highlight some cyclization examples applied in therapeutic short peptides which led to a successful lead optimization with improved PK properties and peptide stability (Figure 10).
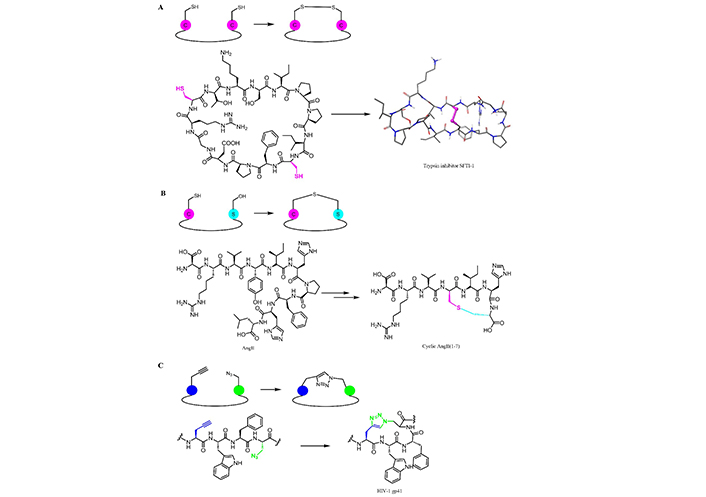
Synthetic strategies for peptides cyclization to improve pharmaceutical properties. AngII(1-7): angiotensin II(1-7)
Backbone modifications and cyclization are, indeed, two classical strategies applied to stabilize peptides in their bioactive conformation and improve resistance to biodegradation. One of the most employed cyclization approaches includes head-to-tail, side chain-to-side chain, tail-to-side chain, and head-to-side chain closures [206]. The resulting cyclic peptidic structures are decorated with stable bonds (an amide, ether, thioether, or disulfide bonds, etc.), either inherently or protected in the resulting conformation of the cyclic structure thereby making the peptides resistant to hydrolysis by peptidases.
Recently, Li et al. [207] synthesized a cyclic analogue of GLP-1 with a prolonged half-life, showing a long-acting anti-diabetic activity (half-life 120 h). Moreover, advances in peptide-based therapy for type II diabetes and obesity revealed the success of long-acting GLP-1R agonists exendin-4 in reducing blood glucose concentration [47]. These molecules are resistant to DPP-IV attack due to a substitution in Ala2 residue, and due to the improved binding with albumin induced by an additional octadecanoic acid in Lys20 residue.
The formation of disulfide bonds greatly enhances peptide stability (Figure 10A) by introducing conformational restrains. Sunflower trypsin inhibitor-1 (SFTI-1) is an interesting example of a naturally occurring cyclic peptide, isolated from sunflower seeds. This peptide contains an intrachain disulfide bond that reduces the flexibility of the peptide thus improving its stability [208, 209]. The SFTI-1 structure (14 amino acid residues) contains similarities to the trypsin-reactive loop region of the Bowman-Birk family of serine protease inhibitors [210], a group of molecules from soybean and termed Bowman-Birk inhibitors [211]. They primarily inhibit serine peptidases of the S1 family but also inhibit S3 peptidases. SFTI-1 is essentially a head-to-tail bicyclic peptide bridged by a disulfide bond between Cys3 and Cys11. In 2014, its total synthesis was reported using an intramolecular hydrazide-based ligation as the key step [212].
Kluskens et al. [213] reported the introduction of a thioether bridge in AngII(1-7) (Figure 10B), resulting in a molecule completely resistant to ACE degradation. More recently, CuAAC was employed to build stable and hydrolysis resistant cyclic peptides. A side chain-to-side chain CuAAC was performed to synthesize HIV-1 gp41 (Figure 10C), and stabilize its secondary structure [167]. The 1,2,3-triazole linkage found utility in peptide chemistry as it is considered an isostere of an amide bond: the CuAAC reaction is, therefore, a particularly powerful tool to generate cyclic peptides or for the ligation of small molecules onto the peptides [214].
In 2016 Astrazeneca signed a collaboration with the UK company Bicycle Therapeutics (https://bicycletherapeutics.com) with the aim to advance the development of highly constrained bicycle drug candidates (BDC) for cancer treatment (Figure 11). Bicycle Therapeutics developed BT1718, that in 2018 completed its phase I dose escalation and is a potential first-in-class bicycle toxin conjugates (BTC) against tumor antigen membrane type 1 matrix metalloproteinase (MT1-MMP) [215].
The cyclic pentapeptide cilengitide (Figure 12) c(RGDf(NMe)Val) is an excellent example of head-to-tail cyclization. The molecule was discovered by Haubner et al. [216] in the early 1990s, then developed by the German company Merck-Serono, reaching late-stage clinical trials for cancer treatment [217].
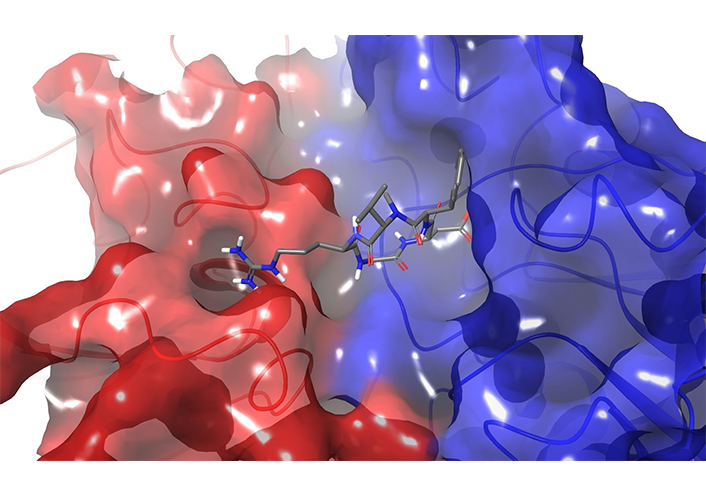
Crystal structure of cilengitide in the integrin αVβ3 binding pocket (PBD ID: 1L5G)
Unfortunately, phase III cilengitide trials faced some dose regimen and efficacy issues. Nonetheless, this drug candidate was the first anti-angiogenic short peptide targeting the αv integrins, and it can be considered as a prototypic example of the design strategy of a highly selective peptide inhibitor [218, 219].
The crystal structure of the αVβ3-Mn complexed with cilengitide (Figure 12) was obtained by Xiong et al. [220] in 2002. Integrins are transmembrane receptors that recognize the tripeptide sequence Arg-Gly-Asp (RGD) contained in the ligands of the extracellular matrix. RGD, indeed, is a crucial binding motif for several receptors, but as such the tripeptide is a bad drug candidate due to its short half-life and poor selectivity. Since integrins are involved in several pathological processes, they are considered very promising therapeutic targets [221].
The unraveling of the bioactive conformation of cilengitide bound to integrin αVβ3 [222] and the rationalization of the biological affinity provided by Kessler’s studies inspired and guided rational designs of several peptidomimetic ligands [222–225], including some integrin inhibitors which entered to clinical phases [221]. In the pioneering work of Mas-Moruno et al. [226], a systematic stepwise strategy was developed to design cyclic peptides under “conformational control”. In the absence of a receptor structure, Aumailley et al. [227] embedded the RGD sequence into hexa- or pentapeptides, using turn-inducing D-amino acid to stabilize the bioactive conformation. They synthetized libraries of cyclic peptides varying the position and the nature of the RGD flanking residues. By a combination of NMR and molecular dynamics studies [227] it was revealed that the position of the D- rather than their natural L-residues, mainly determined the conformational preferences of the peptide. The biological evaluation identified the sequence c(RGDfV) as a lead peptide sequence that was further optimized by applying a N-methyl scan and resulting in the final structure of cilengitide (Figure 12). It was demonstrated that an extra N-methylation in cilengitide resulted in analogues with augmented selectivity [218].
Recent structure-activity relationships (SAR) studies on an RGD-containing cyclic octapeptide (called LXV, where X is an aromatic residue) and αvβ3 integrin revealed that the hydrophobic and aromatic residues, such as X7, are essential for the binding with integrin [228].
Eptifibatide (Integrilin), a cyclic heptapeptide based on a peptide recognition sequence found in snake venom, and tirofiban, na RGD-mimic, are the only approved small molecules drugs that bind selectively to the platelet integrin αIIbβ3 (Figure 13A and B). Eptifibatide is a cyclic peptide with a bond between the cysteine and mercaptopropionyl residue, and it is used to treat angina and certain types of heart attack by intravenous injection. Tirofiban is a small molecule that binds within the ligand-binding pocket of αIIbβ3 receptors to compete with fibrinogen and von Willebrand factor and it is indicated in patients with acute coronary syndrome [229].
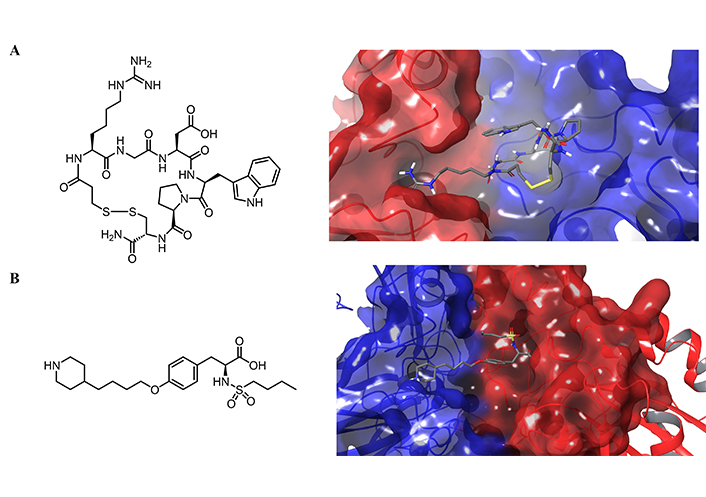
Chemical structure and their corresponding crystal structures of the molecules that bind selectively integrin αIIbβ3. Chemical structure of A) eptifibatide; B) tirofiban
Peptidomimetics
The peptidomimetic concept was introduced in the 1980s and consists of the art of transforming peptides into potential drugs with the aim of maintaining crucial key interactions and affinity for the endogenous receptor or intracellular target while improving the PK properties. There are several successful examples of rational design of peptidomimetic ligands that led to an approved drug [230]. It is beyond the scope of this review to cover all the chemical space explored by this category of compounds, given the broadness of the subject [231–233]. The design of peptidomimetic ligands depends on what is known about the target (i.e. structure and function) and the features of the ligand-protein interaction. Two main drug design strategies have been applied over the years in this field [234]. One approach is a classical medicinal chemistry approach, where, starting from a linear peptide, the molecular groups are successively replaced by non-peptide moieties. The second is a rational design approach that generates cyclic or acyclic peptidomimetic ligands using scaffolds to block the bioactive conformation of the peptide or to properly orient the pharmacophoric groups. Both approaches can be combined in a hierarchical workflow that starts with the generation of a first library of peptidomimetics with local modifications of the peptide recognition sequence (e.g., bioisosteres, N-alkylation, see [235] for an example), followed by a hit-to-lead optimization through a rational design approach that transforms them to candidates as oral drugs. When the bioactive peptide is unknown, a screening of large peptidomimetic libraries is the alternative [236].
According to Pelay-Gimeno et al. [237], four classes (A–D) of peptidomimetics can be identified based on the degree of structural similarity to the parent peptide: peptidomimetics with few local modifications that well-overlap the structure of the native peptide belong to class A (such as stapled peptides, N-alkylation, L- to D-amino acid substitution and macrocyclization), while mimetics that are the least similar to the native peptide, such as those obtained in the lead optimization process, belong to class D (i.e. tirofiban). Classes A and B are modified peptide compounds while C and D are nonpeptide small molecules.
Stapled peptides
The stapling strategy consists in an external side-chain crosslinking that forces the peptide into assuming an α-helical structure (Figure 14) [238]. To the best of our knowledge, the literature reported a maximum of two staples in the same sequence, usually i, i + 3 and i, i + 4 [239, 240]. The enhanced helicity confers enhanced lipophilicity to the structure and allows the peptides to enter into biological membranes [240]. Furthermore, an interesting property of stapled peptides is the proteolytic resistance associated both with helical structures and with the stapling chain.
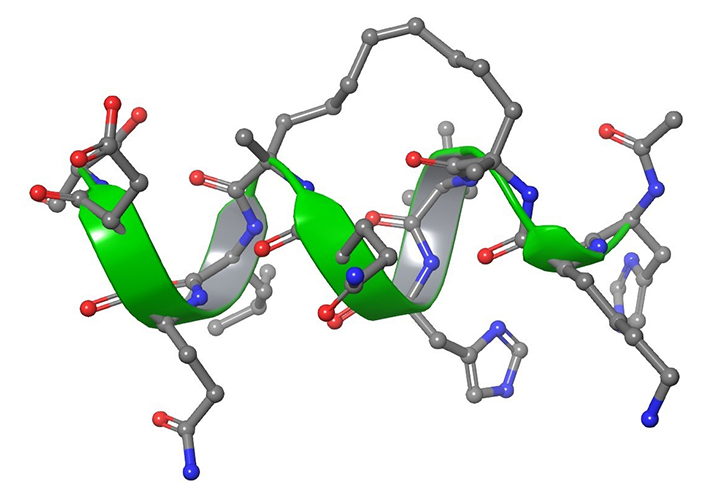
Graphical representation of peptide stapling. The amino acids involved in the stapling are located at the right distance, i, i + 4. The stapling showed is NMR solution experiments derived and it represents a stapled peptide, developed in 2011, targeting estrogen receptors (PBD ID: 2LDA) [241]
The development of stapled peptides has, however, some prerequisites: it is necessary that the presence of functional groups introduced in the sequence does not affect the chirality, and that the stapling bridge formation does not interfere with activity.
Recently, Cromm et al. [242] in an excellent review described the therapeutic use of hydrogen stapled peptides as modulators of biological functions, resembling potential inhibitors of numerous PPIs. In this framework, an interesting example of successful stapling was reported over the last decade by Walensky et al. [243] which reported the in vivo activity of a BCL-2 protein-derived stapled peptide targeting an intracellular PPI.
Stapled peptides can engage with intracellular targets with high selectivity and efficiency and the stapling can also finely tune the physiochemical properties of these molecules, therefore representing a relatively underexplored but potentially highly rewarding field of research [244, 245].
Conclusions
Short peptides are endowed with a remarkable ability to mediate and modulate several biological functions, and therefore, represent an underexplored class of peptidic molecules and behold immense potential to be used as innovative therapies in almost all branches of medicine. This century itself is witness to the explosion of high-quality research crystallized in this area. The emerging technological revolution has only enhanced the potential of the short peptides by solving some key limitations associated with peptides; be it related to their synthesis and purifications or their unraveling of their co-crystal structures with their targets or the ability to deliver them right to the desired intracellular targets. A steady growth observed in research addressing the key inherent disadvantages embodying short peptides, such as cell permeability, oral bioavailability, and their in vivo stability, is highly encouraging and brings hope for a positive future prospect of peptide-based medicines. Further developments in our understanding of, and the ability to modulate suboptimal parameters of peptides such as poor PKs, immunogenicity, high synthesis cost, unfavorable PDs, and lack of delivery options, will open new avenues for several biological and biomedical research areas, including chemical biology, biotechnology, and drug discovery. A continuous improvement in the computational approaches or in silico abilities in chemo- and bioinformatics will further play a key supporting role in the development of peptide-based drugs. Short peptides are easier to handle synthetically and are modifiable with non-natural and synthetic fragments, be in non-natural amino acids, or other small carbo- or heterocyclic fragments and will continue to find applications in diverse areas of medicinal chemistry, including peptidomimetics, and hopefully remain a more productive source of clinical candidates of many different disease indications.
Abbreviations
| AMP: |
antimicrobial peptide |
| AVP: |
arginine vasopressin |
| BBB: |
blood-brain barrier |
| Boc: |
tert-butyloxycarbonyl |
| COVID-19: |
coronavirus disease-19 |
| CPP: |
cell-penetrating peptide |
| CuAAC: |
copper(I)-catalyzed azide alkyne cycloaddition |
| Fmoc: |
9-fluorenylmethoxycarbonyl |
| GLP-1: |
glucagone-like peptide-1 |
| HDPs: |
host defence peptides |
| KAHA: |
α-ketoacid-hydroxylamine |
| MDR: |
multi-drug resistant |
| NCL: |
native chemical ligation |
| NPs: |
natural products |
| PD: |
pharmacodynamic |
| PK: |
pharmacokinetic |
| PPIs: |
protein-protein interactions |
| RGD: |
Arg-Gly-Asp |
| SFTI-1: |
sunflower trypsin inhibitor-1 |
| SPPS: |
solid-phase peptide synthesis |
| sst3: |
somatostatin receptor subtype-3 |
| T2DM: |
type II diabetes mellitus |
| TAT: |
trans-activator of transcription |
Declaration
Author contributions
MGC, MC, SS, and KK: Conceptualization, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2023.