 Review
Review

Affiliation:
Department of Internal Medicine, Azienda Ospedaliero-Universitaria, 41100 Modena, Italy
Email: a.lonardo@libero.it; lonardoamedeo2@gmail.com
ORCID: https://orcid.org/0000-0001-9886-0698
Explor Drug Sci. 2023;1:239–252 DOI: https://doi.org/10.37349/eds.2023.00016
Received: March 05, 2023 Accepted: May 03, 2023 Published: August 28, 2023
Academic Editor: Juergen Reichardt, James Cook University, Australia
Nonalcoholic fatty liver disease (NAFLD), its more rapidly progressive steatohepatitic variant [nonalcoholic steatohepatitis, (NASH)], and the recently defined metabolic dysfunction-associated fatty liver disease (MAFLD) may be collectively alluded to as “metabolic fatty liver syndromes” (MFLS). MFLS is a common clinical complaint for which no licensed drug treatment is available and a public health issue posing a heaven burden on healthcare systems. Iron plays a key role in many of the key pathogenic steps concurring in the development and progression of MFLS, notably including genetics, intestinal dysbiosis, adipositis, insulin resistance, metaflammation, oxidative stress and ferroptosis, endoplasmic reticulum stress, and hepatic fibrosis. This notion raises the logical expectation that iron depletion, which can easily be implemented with venesection, might improve several aspects of MFLS. However, few published studies have globally failed to support these expectations. In conclusion, venesection in MFLS exhibits a strong biological rationale and possible metabolic benefits. However, confronted with failures in hepato-histological outcomes, data call for additional studies aimed to reconcile these inconsistencies.
“Metabolic fatty liver syndromes” (MFLS) was coined by Hansen and Walzem [1] in 1993 to describe steatosis occurring in “chickens, humans, cows, and cats”. Thirty years later, MFLS has been re-proposed as a definition embracing nonalcoholic fatty liver disease (NAFLD)/nonalcoholic steatohepatitis (NASH), and metabolic dysfunction-associated fatty liver disease (MAFLD) [2, 3].
NAFLD is principally a diagnosis of exclusion indicating those frequent cases of hepatic steatosis (HS), steatohepatitis, cirrhosis, and hepatocellular carcinoma (HCC) that occur in the absence of any significant alcohol consumption and other secondary causes of (steatogenic) liver diseases such as viral hepatitis, common endocrinopathies, drugs, and rare genetic conditions [4]. Conversely, MAFLD is a positive diagnosis that highlights the presence of dysmetabolic traits, irrespective of concurrent hepatotoxic noxae [5]. Given the strong (bi-directional) association of NAFLD with the full-blown metabolic syndrome (MetS) and its individual constituents: visceral obesity, impaired glucose disposal, arterial hypertension, and dyslipidemia [6], it is logical to conclude that all MFLS put dysmetabolism to a common factor.
Taking the NAFLD paradigm as a model, MFLS is postulated to have a complex multifactorial pathogenesis in which genetics, intestinal dysbiosis, adipositis, insulin resistance (IR), metaflammation, oxidative stress and ferroptosis, endoplasmic reticulum (ER) stress, and increased fibrogenesis all play key roles [7–10]. Globally, the MFLS poses a heavy clinical and financial burden on healthcare systems [11] and presently no licensed drug treatment is available for their cure [12].
Iron released by collapsing stars accounts for the finding that this metal is the fourth most abundant in earth’s crust and oceans, where all living beings count on the interchangeability of the Fe2+/Fe3+ redox pair to catalyze those electron-transfer and acid-base reactions which are necessary to plants, bacteria, animals, and humans to survive [13]. While being essential for many biological activities such as transport and storage of oxygen; energy production; detoxification; and host defense, iron has an ominous distinctive feature, namely it is highly redox-active and this underlies potentially toxicity when this metal is accumulated in excess [13]. Importantly, iron is a key modifier of many of the above-mentioned steps involved in the pathogenic cascade eventually leading to development and progression of MFLS [14, 15]. Moreover, dietary iron supplementation enhances experimental steatohepatitis induced by long-term high-fat diet feeding in rats [16] and, consistently, iron restriction in rats fed a choline-deficient diet led to a significant reduction of hepatic iron levels, oxidative stress, inflammation, and fibrosis (FIB) [17]. In humans, both NAFLD and MAFLD are pathogenically associated with body iron depots, and ferroptosis, a recently described form of programmed cell death caused by iron-dependent lipid peroxidation, playing a key role in disease progression; in agreement inhibition of ferroptosis is a promising novel target in the treatment of NAFLD [18–22]. Taken collectively, those findings summarized above raise the expectation that depletion of iron stores with venesection may potentially represent a novel therapeutic strategy for the treatment of MFLS.
Venesection (also known as bloodletting or phlebotomy) is a therapeutic option that mankind has retained across millennia; it is cheap, safe, and can easily be implemented in various settings, including a doctor’s surgery [23]. However, while being consolidated in other indications, venesection remains a controversial practice in the MFLS arena [18]. With this backset, the present review article addresses a) definitions, b) biological rationale, and c) metabolic and histological outcomes of published studies regarding venesection in the setting of MFLS.
In 1997, Moirand et al. [24] reported on a cohort of 65 individuals exhibiting the seemingly novel, non-HLA linked, syndromic triad of increased liver iron concentration, hyperferritinemia, and normal transferrin saturation. Given that 95% of these individuals had features belonging to the domain of the MetS, these authors suggested a nexus associated between iron excess and various common dysmetabolic conditions; more recently, Marmur et al. [25] reported that irrespective of its being associated or unassociated with dysmetabolic iron overload syndrome (DIOS), in NAFLD serum hepcidin and hepcidin antimicrobial peptide messenger ribonucleic acid (mRNA) in liver do correlate with body iron content rather than with the severity of NASH or lipid status indicating that DIOS associated with NAFLD is not caused by altered synthesis of hepcidin.
In 2001, Turlin et al. [26] characterized 139 patients with IR-associated hepatic iron overload (IR-HIO) defined as the association of metabolic disorders and hepatic iron overload unrelated to common causes and, being characterized by a mixed pattern with iron deposits in hepatocytes and sinusoidal cells, was histologically different from that observed in genetic hemochromatosis. Periportal FIB was found in the majority (67.4%) of patients, featuring older age, higher sinusoidal iron scores, and a higher prevalence of steatosis and inflammation.
In 2008, Barisani et al. [27] used the definition of dysmetabolic hepatic iron overload (DHIO) to designate their patients in whom hepatic iron overload occurs in the absence of any identifiable mutations and is often associated with liver steatosis and concurrent metabolic disorders. Dysregulated production of hepcidin is the key pathogenic step occurring among individuals with DHIO who exhibit relatively elevated urinary hepcidin levels accounting for the iron phenotype in DHIO: accumulation of iron in macrophages associated with normal-to-low transferrin saturation [27]. Interestingly, hepcidin upregulation negatively affects the process of iron absorption and this decrease is even more pronounced owing to the extra action exerted by iron excess on circulating hepcidin levels [28].
In 2011, Chen et al. [29] used the term “dysmetabolic hyperferritinemia” to describe individuals who had both MetS and hyperferritinemia. In a limited series of 10 patients, these authors were able to conclude that the hallmarks of this condition were elevated serum ferritin levels (median, 672 μg/L) with normal transferrin saturation (median, 38%) and elevated urinary hepcidin (median, 1,584 ng/mg of creatinine) [29].
Experts believe that dysmetabolic hyperferritinemia and DIOS are not truly different conditions but rather mutually related disorders occurring across a pathogenic and clinical spectrum linking IR and disordered iron metabolism [30]. It is logical to assume that the same notion holds true for IR-HIO and DHIO. On these grounds, all the above definitions will be considered interchangeably in the present review.
Articles cited in the present review were retrieved by searching PubMed database for “iron depletion”[Title/Abstract] AND “nafld”[Title/Abstract]; (venesection [Title/Abstract]) AND (nafld[Title/Abstract]). “iron” [Title/Abstract] and “MAFLD” [Title/Abstract]. Additionally, cross-references from the above retrieved articles and the author’s personal archive were also evaluated. Only those publications deemed to be relevant to the ends of this review were eventually retained.
A detailed description of the physiology of iron homeostasis in health states has been covered by excellent reviews over the last few years [31–36] and is out of the scope of the present review. The role of iron metabolism in some of the multiple key steps occurring across the complex, multifactorial process of development and progression of MFLS [9, 37] is discussed hereafter (Figure 1). For clarity’s sake, these pathogenic steps are addressed separately; however, they are strictly interconnected biologically as highlighted below.
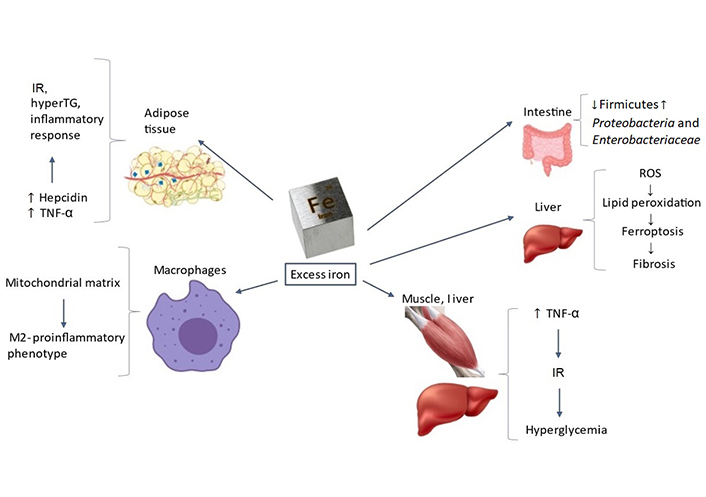
Targets and mechanisms through which excess iron may promote development and progression of MFLS. Further to being involved in many physiological processes such as energetic homeostasis, oxygen transport and storage, and detoxification, iron targets subcellular organelles, and various tissues. Therefore, excess iron deposits may promote MFLS development and progression through intestinal dysbiosis, adipositis, IR, inflammatory response, lipid peroxidation, ferroptosis, and hepatic FIB. TNF-α: tumor necrosis factor α; ROS: reactive oxygen species; hyperTG: hyper-triglyceridemia
Although deranged iron metabolism has repeatedly been reported in association with NAFLD/MAFLD (reviewed in [38]), a cause-and-effect association remains unproven in observational studies. Wang et al. [38] conducted Mendelian randomization analyses to address this research question regarding causality. By utilizing data from the UK Biobank, the Estonian Biobank, the eMERGE network, and FinnGen consortium, these authors were able to conclude that, at sex-stratified analysis iron status predicted genetically was specifically associated with increased odds of NAFLD and FIB/cirrhosis in men [38].
In humans, iron fortification carries dysbiosis via an unfavorable ratio of fecal Enterobacteria to Bifidobacteria and Lactobacilli [39, 40]. Conversely, phlebotomy conducted among individuals with haemochromatosis is associated with a reduction of fecal iron and increased concentrations of bacterium Faecalibacterium prausnitzii which, in turn, is associated with improved colonic health [41].
Ten years ago, Dongiovanni and colleagues [42] shaped an experimental model of DIOS. These authors showed that, in C57Bl/6 male mice, a diet enriched in iron induces IR and hyperTG and affects the metabolism of visceral adipose tissue through up-regulation of hepcidin [42]. Another study conducted in vitro showed that free iron-induced an inflammatory response selectively in pre-adipocytes, documented by increased interleukin-6 secretion, which was blocked by co-incubation with either iron chelators or radical scavengers [43].
Macrophages exhibit a functional, phenotypic, and metabolic polarization with M1-like cells exhibiting pro-inflammatory action and being deemed to have predominantly glycolytic metabolism. M2-like macrophages are anti-inflammatory and have an oxidative metabolism. Obesity is typically associated with the preferential accumulation of inflammatory (M1) adipose tissue macrophages (ATMs), carrying an increased risk of developing local inflammatory changes in the adipose tissue and systemic IR [44]. Interestingly, mitochondria of ATMs and adipocytes themselves exhibit a state of iron overload. Abundance of iron in ATM mitochondria is strictly connected with dysregulation of systemic metabolism as proven by the finding that, in mice, depletion of ATM mitochondrial iron reverses the metabolic effects of a high-fat diet [45]. These elegant studies imply that it is the variability of iron abundance in the mitochondrial matrix of macrophages that affects their polarization, leading to parallel variations in adipocyte iron abundance that lastly result in effects on systemic metabolic health [46].
It is widely accepted that both dietary iron and iron overload are strong risk factors for diabetes and that this association is mechanistically mediated by pancreatic beta cell failure, IR, and regulation of energy metabolism in iron-sensing adipocytes [47]. While the molecular mechanisms mediating these effects are not fully understood, oxidant stress, modulation of adipokines, and intracellular signal transduction pathways are all deemed to play a role [47].
As regards the specific NAFLD arena, in 2002, Lonardo et al. [48] reported in their case-control study that—at univariate analysis the relative risk of NAFLD (defined as ultrasonographic evidence of steatosis in individuals without any competing causes of steatogenic liver disease) significantly decreased with increasing transferrin. However, at multiple logistic regression analysis, only fasting insulin and uric acid were found to be independent predictors of NAFLD [48]. The primacy of hyperinsulinemia in NAFLD has more recently been confirmed by Bril et al. [49] who have shown that fasting intact insulin concentrations (measured with mass spectrometry) accurately predict NAFLD among non-diabetic individuals.
In 2014, Wlazlo et al. [50] prospectively investigated the associations of biomarkers of iron metabolism with IR at various anatomic sites (e.g., muscle, liver, and adipocytes) and, importantly, with impaired glucose tolerance. To this end, a cohort of 509 individuals was followed up for a 7-year period. The study concluded that biomarkers of iron metabolism are associated with IR in muscle, liver, and adipocytes, eventually leading to hyperglycemia [50].
In 2020, Mayneris-Perxachs et al. [51] investigated the plasma lipidome, IR, biomarkers of iron metabolism, together with a profile of cytokines and adipokines in 65 overweight/obese individuals. Data have shown that several lipid classes belonging to the phosphatidylcholines, lisophosphatidylinositol, dihydroceramides, phosphatidylserine, diacyliglerols, and triacylglycerols lipid classes, were positively associated with serum ferritin concentrations, an established biomarker indicating iron storage. Given that many of these lipid classes are reportedly involved in IR and type 2 diabetes in both experimental studies and in humans, it appears logical to conclude that the entity of iron depots may modulate glucose metabolism through lipidomic changes. In this regard, the key issue is whether lipids or iron are major determinants of IR. To address this research question, Martin-Rodriguez et al. [52] conducted a cross-sectional study of 104 healthy non-diabetic Caucasians submitted to quantitative magnetic resonance imaging (MRI) T2 gradient-echo technique to gauge hepatic iron and 1H-magnetic resonance (1H-MR) spectroscopy to measure intra-hepatic triglyceride (TG; IHT) content. Data have shown that IR was more closely associated with IHT than intra-hepatic iron (although some interplay occurred between them). Interestingly, the association of NAFLD with IR was mediated by high serum TNF-α concentrations, while increased IHT appear to affect the development of hepatic iron overload in individuals who were free of both diabetes and hemochromatosis [52].
Gut microbiota plays a key role in metaflammation. In rats, a high-fat high-fructose (HFHF) diet induces hepatic metaflammation and metabolic disorders via disturbed homeostasis of gut microbiota and altered innate immune system, activation of lipopolisaccaride-Toll-like receptor 4 (LPS-TLR4) signaling pathway, and messengers such as α-hydroxyisobutyric acid and other organic acids [53]. Given its critical role in the development of metaflammation and metabolic disorders, it is important to investigate the relation of gut microbiota with iron metabolism. In this connection, whenever more iron is available in the gastrointestinal tract, the composition of intestinal microbiota is altered, with a reduced number of the beneficial Firmicutes species to the advantage of noxious species such as Proteobacteria, particularly Enterobacteriaceae [54].
Iron participates in vital biological pathways such as the red blood cell-mediated transport of oxygen and oxygen reduction to water during respiration. While this metal generally has a limited availability, pathological accumulation of iron within tissues elicits toxic effects via increased generation of ROS and enhanced oxidative stress [55]. Recent studies suggest that ferroptosis, which may be triggered by various stimuli notably including intracellular accumulation of iron and lipid peroxidation, underlies ROS-dependent regulated cell death [56]. Iron present in the cytosol promotes ROS formation through the Fenton reaction (i.e., H2O2 is transformed in hydroxyl radicals) which caused lipid peroxidation to play a key role in ferroptosis [56]. Additionally, lipid peroxidation occurring within the liver results in damage to hepatocyte mitochondria and lysosomes, which are deemed to participate in the processes of cell necrosis and apoptosis, ultimately leading to the fibrogenic process in the liver [57], which is also discussed below under point 4.8.
The ER defines the sub-cellular organelle that, through ER-resident enzymatic activities and chaperones, governs proper folding of linear polypeptides and proteins [58]. This is a vital process given that failure to shape the proper three-dimensional architecture of proteins will eventually lead to storage of misfolded or unfolded proteins. Perturbed ER homeostasis, eventually culminating in the so-called ER stress, is triggered either by accumulated misfolded/unfolded proteins or lipotoxicity. In response to ER stress, the unfolded protein response (UPR) is activated to reestablish ER homeostasis (hence considered to be an “adaptive UPR”), or, conversely, to culminate in cell death (in this case ER stress is “maladaptive”) [58]. UPR participates in a variety of pathophysiological states, spanning from inflammatory response, nutrient disorders, and viral infection as well as development and progression of various liver conditions, notably including NAFLD and others [58, 59].
Recent studies have highlighted the connections linking the liver hormone hepcidin with ER stress. High hepcidin concentrations (which may mirror iron body status, inflammatory signals, or ER stress) result in iron being stored intracellularly at various sites (e.g., duodenal enterocytes, hepatocytes, and macrophages) [59]. In other words, accumulation of liver fat induces ER stress which, in turn, promotes hepatic lipogenesis, thus creating a positive-feedback loop, which may not only contribute to the development of HS but also to the maintenance of hepatocellular injury and FIB progression within the pathogenic NASH spectrum [59].
Liver FIB describes the accumulation of extracellular matrix components that result from persistent liver injury. While the fibrotic hepatic response was selected by evolution to keep tissue integrity, whenever exceeding a physiological amount, it paradoxically impairs liver regeneration thus threatening hepatic health and function [60]. Irrespective of the type of inciting insult, repeated bouts of hepatocyte damage, followed by the wound-healing fibrosing response, may potentially result in progressive hepatic FIB disease as well as cirrhosis, namely a distorted liver histology exhibiting abundant amounts of scar tissue; development of nodular (as opposed to lobular) hepatic architecture and eventually resulting in the ominous triad of portal hypertension, liver failure, and primary liver cancer [60].
The clinical and mechanistic aspects of iron metabolism relevant to the development of hepatic FIB have been reviewed elsewhere [61]. Data indicate that excess iron deposition, which is frequently observed in chronic liver diseases owing to variable etiology, feeds the Fenton reaction to generate large amounts of free radicals that severely harm cells and tissues, ultimately perpetuating fibrosing signals both in the parenchymal and non-parenchymal liver cells [61]. Collectively, these phenomena exacerbate disease progression and aggravate liver histology.
Few studies, published from 2002 to 2018, have evaluated the effects of iron depletion obtained with phlebotomy on metabolic outcomes and histological features in NAFLD. These are summarized in Table 1 [62–71].
Studies evaluating the effect of iron depletion with phlebotomy in NAFLD
| Author, year [Ref] | Method | Findings | Conclusion |
|---|---|---|---|
| Facchini et al., 2002 [62] | Iron depletion to a level of NID was induced by phlebotomy in 17 NAFLD patients. | Although, at the baseline, they did not have any supranormal levels of body iron, at NID, NAFLD individuals exhibited a significant improvement of both fasting and glucose-stimulated plasma insulin concentrations and a significant decrease to almost-normal serum ALT. | Iron depletion exerts an insulin-sparing activity. |
| Sumida et al., 2006 [63] | Eleven individuals with biopsy-proven NASH were phlebotomized biweekly until NID (i.e., serum ferritin concentration < 30 ng/mL). | Paralleling a significant drop in ferritin levels, ALT activity significantly decreased while body weight remained unaltered. Even though 2 patients withdrew, no significant side effects of repeated phlebotomy were observed. | NID is associated with improved ALT activity irrespective of body weight changes. |
| Valenti et al., 2007 [64] | Sixty-four individuals submitted to phlebotomy were compared to matched controls (based on age, sex, ferritin, obesity, and ALT levels) undergoing lifestyle changes alone. | Compared to lifestyle changes alone and independent of confounding factors (changes in BMI, baseline HOMA-IR, and presence of the MetS), iron depletion was associated with a significantly higher reduction in IR (assessed with RIA and the HOMA-IR index, at baseline and after 8 months). The effect of iron depletion in reducing HOMA-IR was higher among those with higher ferritin concentrations and in carriers of the mutations in the HFE gene. | Independent of confounding factors, phlebotomy reduces IR. |
| Valenti et al., 2011 [65] | Italian multicenter (6-month to 8-month) observational study of 198 non-hemochromatosis NAFLD patients, (79 submitted to phlebotomy and 119 to counselling only). IR was measured with HOMA. | Venesection was significantly associated with normal HOMA and normal ALT. These findings were confirmed in 57 pairs matched for propensity and in the cohort of those with normal baseline ferritin (< 350 ng/mL). | Among NAFLD patients, venesection is associated with normal IR and normal ALT. |
| Beaton et al., 2014 [66] | Fifty-six NAFLD patients were submitted to liver biopsy with assessment of liver iron concentration at the entry and 6 months after phlebotomy conducted until the patient had either low serum ferritin or manifested anemia. | Iron removed with phlebotomy was correlated with the decrease in serum ferritin. However, no significant correlations were found between serum ferritin and ESR, CRP, or grade of liver inflammation. | Serum ferritin mirrors liver iron storage in NAFLD. Phlebotomy, by decreasing body iron stores, induces a parallel decrease in serum ferritin. However, inflammation is not the cause of hyperferritinemia observed in NAFLD. |
| Adams et al., 2015 [67] | Prospective 6-month RCT evaluating the impact of phlebotomy in 74 NAFLD subjects (33 assigned to phlebotomy compared to 41 controls). Primary endpoints of the study: HS (assessed with MRI) and liver injury (assessed with ALT and CK-18). Secondary endpoints: IR (assessed with ISI and HOMA), and systemic lipid peroxidation (assessed with plasma F2-isoprostane levels). | Those assigned to phlebotomy exhibited a significantly greater reduction than controls in ferritin levels over 6 months. At 6 months, there was no difference between phlebotomy and control groups in HS or CK-18 levels. At the end of the study, there was no difference in ISI, HOMA, or F2-isoprostane levels between cases and controls. There was no difference in any of the endpoints in patients with baseline hyperferritinemia nor in relation to the number of phlebotomy sessions. | In NAFLD patients, reduction in ferritin obtained with phlebotomy is not associated with improved liver enzymes, intra-hepatic fat content, or IR. |
| Jaruvongvanich et al., 2016 [68] | Meta-analytic review including 4 interventional studies totaling 438 participants. | Phlebotomy was significantly associated with lower HOMA-IR, reduced ALT and TG levels, and increased HDL-C levels. | Phlebotomy in NAFLD individuals decreases IR and transaminase and improves lipid profile. |
| Lainé et al., 2017 [69] | Prospective, RCT in 146 nondiabetic DIOS patients (randomly assigned to receive venesections with LFDA and 128 to LFDA only) with hepatic iron > 50 μmoL/g at MRI. Study outcomes: metabolic and hepatic outcomes of one-year maintenance of serum ferritin levels < 50 μg/L by phlebotomy. | Iron depletion was associated with significantly reduced values of ferritin and HOMA; body weight increased; no significant changes were observed as regards glycemia ALT, AST, GGT, FLI, and FIB-4. As a side effect, fatigue occurred significantly more often among individuals submitted to phlebotomy than in controls. The cohort of those patients who lost weight,exhibited improved values of glycemia, HOMA, serum ferritin, lipid profile, and liver enzymes irrespective of phlebotomy. | Phlebotomy fails to improve dysmetabolic traits and liver enzymes. Moreover, it is associated with weight gain and is not invariably well tolerated. |
| Britton et al., 2018 [70] | This study compared paired serum samples (at baseline and after 6 months) in 28 controls and 23 patients submitted to phlebotomy. HIC was assessed with MR FerriScan. | Unexpectedly, this study found that baseline serum adiponectin concentration was positively associated with HIC (this finding was strengthened after correction for modifiers). Moreover, significant inverse correlations between HIC and measures of IR [adipose tissue IR (Adipo-IR)], serum insulin, serum glucose, and homeostasis model assessment of IR, HbA1c, and HS were found. Finally, a positive correlation was noted with the insulin sensitivity index. Serum adipokines did not differ between the controls and phlebotomy groups. | In NAFLD patients, there is a positive correlation between serum adiponectin and insulin sensitivity. |
| Murali et al., 2018 [71] | Meta-analytic review of 9 published studies globally enrolling 820 patients with DIOS and/or NAFLD (427 submitted to phlebotomy and 393 to lifestyle changes only). Study quality was evaluated with the Cochrane collaboration tool. | Compared to lifestyle changes alone, iron depletion was not associated with changes of the HOMA index nor AST in DIOS and/or NAFLD patients (five studies, 626 patients). There was a statistically significant, mild improvement in ALT, but the effect size was very small. Also, in the cohort of those exhibiting both NAFLD and hyperferritinemia, phlebotomy did not improve HOMA, insulin level, ALT, or AST. Finally, no study showed that iron reduction was associated with significantly improved the grade of hepatitis or the stage of FIB. | Compared to lifestyle changes alone, among DIOS and/or NAFLD patients, phlebotomy fails to induce any significant improvement in IR, liver enzymes, or liver histology. |
ALT: alanine transaminase; AST: aspartate transaminase; BMI: body mass index; CK-18: cytokeratin-18; CRP: C-reactive protein; ESR: erythrocyte sedimentation rate; FLI: fatty liver index; GGT: gamma glutamyl transferase; HbA1c: hemoglobin A1c; HDL-C: high-density cholesterol; HIC: hepatic iron concentration; HOMA: homeostasis model assessment; ISI: insulin sensitivity index; LFDA: lifestyle and diet advice; NID: near iron deficiency; RCT: randomized, controlled trial; RIA: radio-immuno assay
Taken collectively, the above studies have yielded conflicting findings. On the one hand, they possibly suggest that metabolic outcomes are dissociated from liver histology features. More specifically, a few studies support the notion that iron depletion corrects IR and hypertransaminasemia, and dylipidemia also improves [68]. It is probable that this beneficial effect occurs irrespective of confounding factors, notably including changes in body weight [64]. On the other hand, other studies found no such metabolic improvement [69, 70].
However, there seems to be universal agreement on iron depletion failing to improve liver histology in NAFLD [66, 67]. Of concern, one study reports, fatigue occurred ten times more often among patients submitted to venesection than in controls (25.3% versus 2.3% P < 0.0001) [69].
Fernandez et al. [18], in their recent critical re-evaluation of published studies have emphasized that “the studies are inconclusive due to heterogeneity in eligibility criteria, sample size, randomization, hepatic iron measurement, serial histological endpoints, target ferritin levels, length of venesection, and degree of confounding lifestyle intervention” [18]. Accordingly, these authors have called for a novel, sufficiently powered, trial should be conducted to surmount the above listed drawbacks of published research [18].
A robust body of evidence supports the notion that iron overload negatively affects several steps of the complex pathogenesis of MFLS, such as genetics, intestinal dysbiosis, adipositis, IR, metaflammation, oxidative stress, and ferroptosis, ER stress, and hepatic FIB.
However, the data published so far (which are summarized in Table 1 and discussed under the section “
The recent attempts to bring NAFLD research and practice further through the introduction of the novel disease entity, MAFLD, offer the opportunity to re-assess the role, if any, of phlebotomy-induced iron depletion in MFLS. This line of research has several practical implications spanning from lifestyle advice to management [75].
Additionally, in future trials, it will probably be important to dissociate the individual effects of iron depletion on the risks of de novo lipogenesis, IR, and incident type 2 diabetes from the specific effects on NASH histology. Finally, innovative therapeutic strategies will have to consider the effects of intracellular re-distribution of iron (particularly in the ATMs) rather than the global body/hepatic iron homeostasis.
ATMs: adipose tissue macrophages
DHIO: dysmetabolic hepatic iron overload
DIOS: dysmetabolic iron overload syndrome
ER: endoplasmic reticulum
FIB: fibrosis
HS: hepatic steatosis
IHT: intra-hepatic triglyceride
IR: insulin resistance
MAFLD: metabolic dysfunction-associated fatty liver disease
MetS: metabolic syndrome
MFLS: metabolic fatty liver syndromes
MRI: magnetic resonance imaging
NAFLD: nonalcoholic fatty liver disease
NASH: nonalcoholic steatohepatitis
ROS: reactive oxygen species
UPR: unfolded protein response
AL: Conceptualization, Data curation, Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

