
Abstract
Aim:
Harzianoic acids A and B (Hz-A/B) are two rare cyclobutene-containing sesquiterpenes isolated from a marine strain of the sponge-associated fungus Trichoderma harzianum. They display anticancer and antiviral effects, reducing the entry of hepatitis C virus (HCV) into hepatocarcinoma cells. The large extracellular loop (LEL) of the tetraspanin protein CD81 represents a molecular target for both Hz-A and Hz-B.
Methods:
The interaction of Hz-A/B with CD81 has been modeled, using structures of the cholesterol-bound full-length protein and a truncated protein corresponding to the LEL portion. The models mimicked the closed and open conformations of the LEL.
Results:
The best ligand Hz-B can form stable complexes with the open LEL structure, whereas binding to the closed form is drastically reduced. Key H-bonds between the acid groups of Hz-B and the CD81-LEL domain stabilize the ligand-protein complex. A comparison of the interaction with the homologous tetraspanin CD9, which also presents a dynamic open/closed equilibrium, underlined the marked selectivity of Hz-A/B for CD81 over CD9. The cyclobutane-containing monoterpene grandisol, an insect pheromone, has been identified as a fragment that could be modulated to improve its modest interaction with CD81-LEL.
Conclusions:
The modeling docking analysis suggests that Hz-B is a robust CD81 binder, better interacting with the LEL portion of CD81 compared to CD9-LEL. The docking study paves the way to the design of small molecules targeting CD81. The study has implications for a better understanding of CD81 binding properties and the regulation of its activities.
Keywords
Tetraspanin, CD81, CD9, harzianoic acid, molecular modeling, grandisolIntroduction
Trichoderma species have been widely recognized as biofertilizer fungi for their ability to produce phytohormones and enhance plant growth [1]. In particular, the fungus Trichoderma harzianum is considered an efficient biofertilizer to promote the growth of diverse plants such as Citrus aurantifolia [2], Bupleurum chinense, and many others [3, 4]. It affects the composition of the soil microbial flora and thus improves the yield and quality of Radix Bupleuri [5]. Different strains can be used as rhizospheric biocontrol agents, such as the fungal isolates T. harzianum MC2 and T. harzianum NBG, both able to suppress bacterial wilt disease in tomatoes [6]. The commercial T. harzianum strain T22 is often used as a biostimulant and to protect against plant pathogens, notably on tomato fruits [7]. There are also species of interest for applications in the field of pest control [8], animal feed, and biofuels industries, such as the T. harzianum isolate MS5 which produces xylanase hydrolytic enzymes for the degradation of xylan [9]. The potential applications of T. harzianum are numerous, including the development of eco-friendly silver nanoparticles for the biodegradation of complex dissolved organics in polluted soils [10].
T. harzianum fungi are recognized as being relatively safe, although there are a few reports of T. harzianum as a rare opportunistic pathogen found in biological samples (blood serum, skin lesions, sputum) of immune-compromised patients, notably patients with acute leukemia or with a renal transplant [11, 12]. The infection can be fatal because the fungus is essentially resistant to antifungal therapy.
Diverse categories of bioactive molecules have been identified from T. harzianum species. For example, the α-pyrone derivative trichoharzianone and the decalin derivative trichoharzianin were isolated recently from the marine-derived fungus T. harzianum PSU-MF79 together with other decalins and δ-lactones derivatives [13]. Many other compounds have been isolated from T. harzianum strains, such as the cytotoxic cyclodepsipeptides trichodestruxins A–D [14], the antitumor and anti-inflammatory compound trichomicin [15, 16], the antifungal agent trichoharzianol [17], and a variety of di- and tri-terpenes, such as the trichodermanins [18, 19], to cite a few examples. T. harzianum species from marine and terrestrial environments provide a rich source of bioactive compounds, as reviewed recently [20, 21]. Guo and collaborators [21] inventoried 180 natural products isolated from T. harzianum species, including peptides (31%), polyketides (27%), terpenoids (26%), alkaloids (8%), and lactones (8%), of which 91 exhibited bioactivities as antifungal, antibacterial, cytotoxic agents or other activities. Among these many natural products, a specific pair of bioactive compounds has caught attention for both structural and mechanistic reasons: harzianoic acid A (Hz-A) and Hz-B.
Hz-A and -B have been isolated from the species T. harzianum LZDX-32-08 (GenBank KY744354) isolated from the sponge Xestospongia testudinaria collected near Leizhou Island, Guangdong Province of China. An in vitro culture of the fungal strain was developed, leading to the isolation of small quantities of Hz-A (5 mg) and Hz-B (2 mg) [22]. The sesquiterpene Hz-A and norsesquiterpene Hz-B possess an unusual cyclobutane nucleus which is relatively rare in nature (Figure 1). For both compounds, the cyclobutane ring is substituted with a carboxylic acid group at position 1, a methyl at position 2, and a 4-carboxypent-3-enyl side chain at position 2 (Figure 1). Hz-A is a diacid small molecule, whereas Hz-B is a slightly bulkier compound with three acid groups. The H-bond donor/acceptor potential of Hz-B is larger than that of Hz-A. Both molecules are soluble in water but their lipophilicity is low (Table 1). The compound scaffold is unique. There are compounds bearing a 2-methylcyclobutane-1-carboxylic acid moiety but no other natural product with similar side chains at positions 1 and 2. To our knowledge, there are no natural analogues of Hz-A/B known at present.
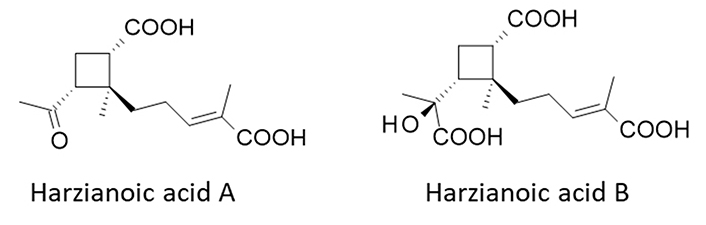
Structures of Hz-A [PubChem compound identity number (CID): 146682324] and Hz-B (CID: 146682325)
Computed physicochemical properties of Hz-A and Hz-B
| Products | Hz-A | Hz-B |
|---|---|---|
| Molecular weight | 268.3 | 314.3 |
| Dipole moment (D) | 2.9 | 2.8 |
| Total solvent accessible surface area (SASA; Å2)* | 525.3 | 552.2 |
| Hydrophobic SASA | 290.3 | 247.5 |
| Hydrophilic SASA | 224.8 | 294.5 |
| Molecular volume (Å3) | 911.0 | 978.2 |
| H-bond donor | 2 | 4 |
| H-bonds acceptor | 5 | 7 |
| log P (octanol/water) | 1.9 | 1.8 |
| log S (aqueous solubility) | –2.9 | –2.7 |
* SASA calculated with a radius probe of 1.4 Å. Drug properties were calculated with the Biochemical and Organic Simulation System (BOSS) 4.9 software. Partition coefficient (log P)
The structural originality of the products is associated with an innovative mechanism of action, even if we cannot establish a link between the cyclobutane scaffold and the protein target. Both Hz-A and -B were found to reduce the entry of hepatitis C virus (HCV) into hepatocarcinoma Huh7.5 cells. Their efficacy is modest, with calculated half maximal inhibitory concentration (IC50) values of 35.5 μmol/L and 42.9 μmol/L, respectively. Interestingly, the two compounds revealed a high affinity for the HCV-coreceptor CD81, with measured Kd values of 0.38 μmol/L and 0.66 μmol/L, respectively [22]. The tetraspanin CD81 (Figure 2A) is a co-receptor for HCV and several other viruses, including human immunodeficiency virus type 1 (HIV-1), herpes simplex virus 1 (HSV-1), influenza A virus (IAV), Chikungunya virus, and a few others [23–26]. CD81 is also considered an anticancer target, implicated in cancer cell proliferation and mobility, and in tumor metastasis. CD81 signaling contributes to the development of colorectal, liver, and gastric tumors, and has been implicated in the aggressivity of B-cell lymphomas [27–29]. Several monoclonal antibodies (mAbs) anti-CD81 have been developed, such as the mAb 5A6 active against the invasion and metastasis of triple-negative breast cancer (TNBC) [30]. In contrast, small molecules targeting CD81 are extremely rare, limited to a few synthetic benzothiazole-quinoline derivatives [31] and isolated compounds like Hz-A and -B. These two rare natural products emerged from our recent comprehensive analysis of small molecules targeting CD81 [32].
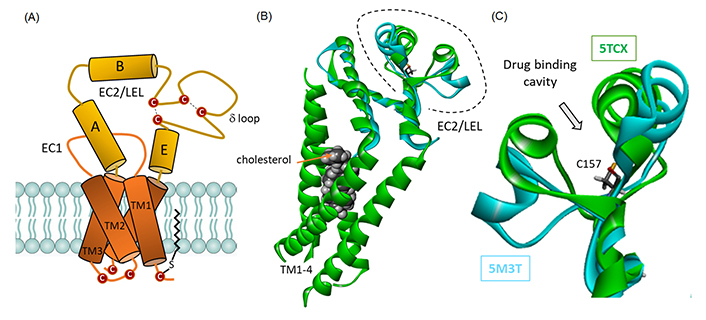
CD81 domain organization and structure. (A) Schematic representation of CD81 architecture, with four transmembrane (TM) segments embedded in the membrane bilayer and sequestering a molecule of cholesterol, and the extracellular loop. The disulfur bridges between cysteine residue in EC2 are represented, as well as a palmitoylated cysteine residue contributing to the anchoring the protein into the membrane bilayer; (B) a model of cholesterol-trapped CD81 full protein [in green, from Protein Data Bank (PDB: 5TCX)] and overlapped model of the EC2/large extracellular loop (LEL) protein (in cyan, from PDB: 5M3T); (C) a close-up view of the EC2 loop to figure out the location of the drug-binding cavity, with the central Cys157 residue. The close/open configuration of the loop can be clearly seen
These considerations prompted us to investigate the interaction of the two compounds with the tetraspanin CD81, for which there are several crystallographic structures available. The structures of the full-length, cholesterol-bound, CD81 protein (PDB: 5TCX) and the LEL (LEL or EC2; PDB: 5M3T) are available from the PDB. They provide useful models to investigate the mode of binding of the compounds to CD81. The two structures can mimic the equilibrium between the open and closed conformational states of the tetraspanin [33]. The tetraspanin is known to exchange between an open (cholesterol-unbound) and a closed (cholesterol-bound) conformation [34]. We have analyzed and compared the binding of Hz-A and -B to the open/closed forms of CD81. We also extended our investigations to the related tetraspanin CD9 to analyze the binding selectivity, and finally, we tested two small structural analogues, the natural products grandisol and fragranol, to offer drug design information.
Materials and methods
Molecular structures and software
The three-dimensional (3D) structures of tetraspanin CD81, both the full-length protein (PDB: 5TCX) and the truncated protein corresponding to the LEL/EC2 domain (PDB: 5M3T), were retrieved from the PDB (https://www.rcsb.org/). They refer both to high-resolution structures (2.96 Å and 2.02 Å resolution) obtained by X-ray diffraction [35, 36]. The GOLD 5.3 software (Cambridge Crystallographic Data Centre, Cambridge, UK) was used to perform the docking analyses. The Biovia 2020 platform (Dassault Systèmes BIOVIA Discovery Studio Visualizer 2020; Dassault Systèmes, 2020) was used for molecular graphics and analyses. The web server Computed Atlas of Surface Topography of proteins (CASTp) 3.0 was used to identify potential ligand-binding sites on proteins. The molecular modeling software Chimera 1.15 was used for visualization [37].
In silico molecular docking procedure
Prior to docking, the protein and the ligand are independently prepared. The eventually missing atoms in the protein according to the PDB file are added and the protein structure is minimized using the Spectroscopic Potential Algorithm for Simulating Biomolecular conformational Adaptability (SPASIBA) force field (taking into account all parameters and atomic charges). A Monte Carlo (MC) conformational searching is performed for the free ligand. The final conformer is the one used in the induced fit docking procedure. The multi-step docking procedure includes an MC conformational analysis of the ligand, performed using the BOSS software. We rely on a previously described MC conformational search procedure with BOSS [38] to optimize the ligand structure. The conformational analysis serves to define the starting geometries for the ligand. With this procedure, all minimum-energy conformers for the ligand are characterized and then the energy minimization defines a unique conformer for the free ligand. The MC simulations were performed within the constant number of particles, constant-pressure, constant-temperature ensemble (NPT), as defined in BOSS. Then, an evaluation of the free energy of hydration (ΔG) is performed for the defined structure of the ligand, using the molecular mechanics (MM)/generalized Born surface area (GBSA) procedure [39]. The calculation of ΔG values was performed with the MC search, within BOSS using the xMCGB script, as previously described [39, 40]. According to this process, the best ligand structure is defined and then used for the docking analysis. The next step corresponds to a definition of the sites of interaction between the tetraspanin and the ligand, using the CASTp 3.0 program. A sphere of 10 Å radius is superposed to the CASTp region in order to define the active site. Within the sphere, the amino-acids side chains are considered fully flexible together with the ligand. This allows a mutual conformational adaptability of both the ligand and the protein. Once the docking is completed, the ligand in its environment is the subject of an MM/GBSA calculation to evaluate the hydration-free energy using the MC BOSS program.
The flexible amino acids are: (i) Thr161, Leu165, Ser168, Asn172, Ser179, Ile181, Asn184, Leu185, Asp189, and His191 for CD81 full-length (PDB: 5TCX), (ii) Lys148, His151, Glu152, Ser160, Leu170, Leu174, Asn184, Leu185, Lys187, and Asp189 for the LEL fragment of CD81 (PDB: 5M3T). The docking grid is centered in the volume defined by the central amino acid (C157 for CD81/LEL) and within the binding site, the side chains of these amino acids are fully flexible during docking. The docking procedure is performed using GOLD. Our typical process considers 100 energetically reasonable poses [defined with the Chem Piecewise Linear Potential (PLP) scoring function] which are screened to search for the optimized ligand binding mode. The PLP fitness scoring function of GOLD v5.3 is used to define and rank the binding poses [41]. In general, 6 poses are selected per analysis. Then the ranking leads to the evaluation of the empirical potential energy of the interaction (ΔE), defined using the expression ΔE(interaction) = E(complex) – [E(protein) + E(ligand)]. The SPASIBA spectroscopic force field is used to calculate the final energy. The required parameters are derived from vibrational wavenumbers obtained in the infrared and Raman spectra of a large series of compounds of diverse chemical nature (organic molecules, amino acids, saccharides, nucleic acids, and lipids). The last step corresponds to a validation using the SPASIBA force field, an essential step to determine the best protein-ligand structure. This force field has been specifically developed to provide refined empirical MM force field parameters [42]. SPASIBA (integrated into CHARMM) empirical energies of interaction are calculated as described [43–45]. It is an excellent system for reproducing crystal phase infrared data. We used similar operational procedures to construct molecular models for CD81 and CD9.
Results
Structural models of the LEL domain of tetraspanin CD81
Two crystallographic structures of CD81 were considered and an overlapping model of the paired structure is given (Figure 2B). 5TCX is a full-length structure of CD81 (246 amino acids) with a molecule of cholesterol bound to the four TM helices [35]. In this case, the LEL extracellular portion of the tetraspanin adopts a compact (closed) configuration that restricts access to the central cavity of the loop, defined as the drug binding site (see below). 5M3T corresponds to the crystal structure of the LEL fragment of the truncated protein comprising 101 amino acids [36]. In this case, the LEL adopts a more extended (open) conformation, allowing wider access to the central cavity. The superimposition of the two structures is good, with a satisfactory overlap between the non-EC2 portions (Figure 2). The superimposition between the two PDB structures 5TCX and 5M3T is excellent with an average C-alpha atom root-mean-square deviation (RMSD) of 0.118 Å. But the subtle deviation is marked at the level of the loop positions 154–190 where the calculated RMSD value amounts to 2.85 Å, with a significant local opening of the loop. The central cavity centered around residue Cys157 is well-defined, with two surrounding walls, more or less open, as shown in Figure 2C.
We then used these two structures to perform a binding site analysis using the web server CASTp 3.0 well-adapted and convenient to predict the position of drug binding areas (Figure 3) [37]. The analysis with the full-length protein (5TCX) revealed a major interaction site in the TM part of the protein, evidently corresponding to the cholesterol-binding site (area in red in Figure 3A). Another much smaller site is detected in the LEL portion. It is a small site (purple area in Figure 3A) with a volume of 11.30 Å3 and a surface of 43.55 Å2. There is also a cavity toward the intracellular portion of the protein (areas in green/blue/orange in Figure 3A) which was not considered for drug binding because it has been shown that Hz-A and -B bind to the LEL domain of CD81 [22]. The situation is significantly different when considering the open conformation of the LEL domain (5M3T). In this case, the LEL-binding area is much larger, with a volume of 81.17 Å3 and a surface of 127.90 Å2. The volume of the central cavity suitable to accommodate a small molecule has expanded considerably. It is now 7-time bigger with the open cavity compared to the closed one (Figure 3B). It can be appreciated also by comparing the distance between two residues on each wall of the cavity, Leu165, and Leu185, as depicted in Figure 3C. The L165-L185 distance increases from 6.70 Å to 17.50 Å in the closed (5TCX) and open (5M3T) structures, respectively. Similarly, the distance between residues Ile181 and Val169 increased from 6.38 Å to 18.85 Å, in the closed and open structures, respectively. The local widening of the cavity is significant. The structural variation is very localized and confined to a portion of the LEL domain. This local variation is exploited for drug binding.
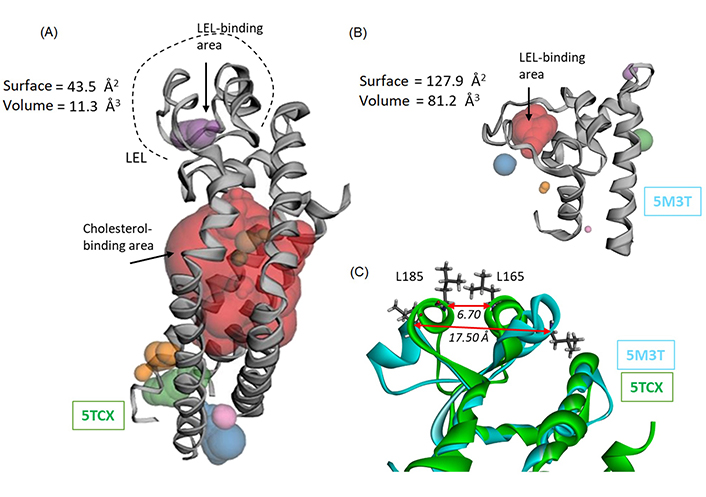
Binding site analysis of CD81 using web server CASTp 3.0. (A) The analysis of the full-length protein (5TCX) points essentially to the cholesterol-binding site located in the TM portion of the protein (in red). Only a small site can be identified within the LEL portion, with the indicated surface and volume; (B) Computed Atlas of Surface Topography (CAST) analysis of the LEL protein, with the identification of a binding cavity within the LEL ore (in red). Very minor areas are identified around the protein (small areas colored in blue, green, and orange); (C) an overlapping model of the loop area of CD81 and LEL proteins, to figure out the conformation difference between the two proteins. The distance between the two facing amino acids Leu165-Leu185 is indicated, to illustrate the opening of the LEL and the marked difference between the closed (5TCX) and open (5M3T) configuration of the loop
Binding of Hz to CD81: docking study
The two conformations of the LEL domain were considered for the binding study with compounds Hz-A and -B. With closed structure 5TCX, the docking was performed with the cholesterol molecule in place, leaving free access of the test molecule to the small cavity centered around position Cys157. The same drug binding site (C157) was used with the open structure 5M3T. In both cases, energies of interaction were calculated (Table 2). The calculated empirical energy of interaction (ΔE) and free energy of hydration (ΔG) are much more negative with the LEL structure compared to the full-length protein. The open conformation of the EC2 loop allows easier access of the natural product to the binding cavity compared to the closed conformation. The more compact structure of the loop in the full protein does not permit drug binding; the energies are weak (ΔE < 40 kcal/mol). In contrast, the LEL alone presents a wider groove to which the natural product can bind. The best compound is Hz-B with a ΔE value a little more negative than that calculated with Hz-A. Our analysis is essentially based on the calculated ΔE values; the predicted molecule’s solvation-free energy (ΔG) provides essentially complementary information to verify the conformity of the calculations and the lack of major difference between the two test compounds. ΔG represents the solute−solvent interactions and no large variations have been observed in the present cases.
Calculated potential energy of interaction (ΔE) and free energy of hydration (ΔG) for the interaction of Hz with CD81
| Compounds | Full-length protein CD81 (5TCX) | LEL fragment of CD81 (5M3T) | ||
|---|---|---|---|---|
| ΔE (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | ΔG (kcal/mol) | |
| Hz-A | –37.50 | –14.60 | –57.80 | –21.00 |
| Hz-B | –38.80 | –13.50 | –61.40 | –21.50 |
A molecular model of Hz-B bound to the LEL structure is presented in Figure 4A. The compound fully enters the cavity in the center of the structure, approaching the central Cys157 residue (Figure 4B). The cavity presents a relatively large solvent-accessible surface (SAS) and the compound engages its elongated carboxypentenyl side chain deep into the cavity (Figure 4C). The ligand-protein complex is stabilized by a set of H-bonds, alkyl/π-alkyl interactions, and van der Waals contacts (Figure 5). There are two key H-bonds with residues Asp155 and Lys187 common to Hz-A and Hz-B. A third H-bond with Cys175 is specific to Hz-B and explains the higher affinity of this compound for the LEL structure compared to Hz-A. Altogether, we identified 14 possible contact points between Hz-B and the LEL protein. This is significant to maintain the product in place in the cavity. The three side chains attached to the 2-methylcyclobutane core of Hz-B contribute to the protein interaction, notably the 3-[(1S)-1-carboxy-1-hydroxyethyl] side chain which is specific to Hz-B, engaged into an H-bind with Cys175 (Figure 5).
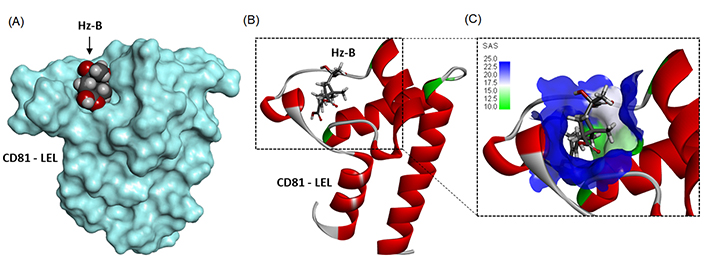
Molecular model of Hz-B bound to CD81-LEL (PDB: 5M3T). (A) A surface model of the protein with the compound bound to the loop; (B) a ribbon model of CD81-LEL with bound Hz-A, with the α-helices (in red) and β-sheets (in cyan); (C) a close-up view of Hz-B bound to the loop cavity. The SAS around the drug binding zone is represented with the indicated color code
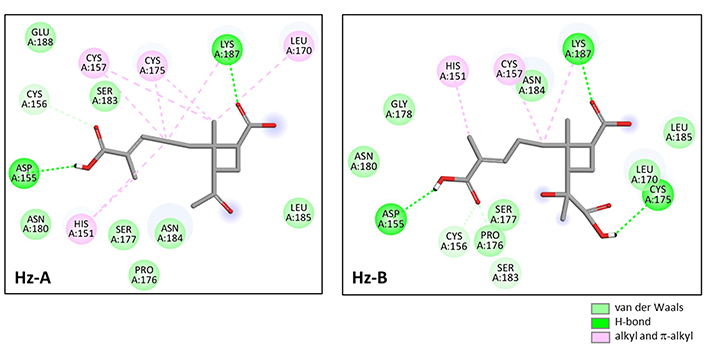
Tetraspanin binding selectivity
There are 33 tetraspanins in humans, plus non-conventional forms generated by alternative splicing [46, 47]. The majority of them present an organization similar to that of CD81, with an intracellular portion, a TM-4 domain, and a LEL. Several tetraspanins are involved in cancers, notably CD9, CD37, CD63, CD81, CD82, and CD151 [27]. Among them, the tetraspanin CD9 is of prime interest because it plays a major role in cell motility and adhesion, and contributes to head and neck cancer and other advanced solid tumors [48–50]. Like CD81, CD9 is considered as a target for cancer therapy [51]. The 3D structures of both the full-length CD9 protein (6K4J) [52] and the LEL portion (6Z1V) [53] are available from the PDB (https://www.rcsb.org/). The protein organization of CD9 is very close to that of CD81 [54]. These two tetraspanins are often considered surface markers for extracellular vesicles [55, 56]. For these different reasons, we investigated the potential binding of Hz-A and -B to CD9, in comparison to CD81.
A superimposition of the full-length CD81 and CD9 proteins and the corresponding LEL structures is shown in Figure 6A. The pairs of molecular models superimpose very well. The two tetraspanins display the same configuration, and the superimposition of the LEL portions is almost perfect (Figure 6B). Nevertheless, subtle differences can be seen at the level of the binding cavity in the EC2 loop, as shown in Figure 6C. These models were used to perform the docking of Hz-A and Hz-B, and the calculated energies are given in Table 3. The empirical energies of interaction (ΔE) calculated with CD9 are significantly weaker (less negative) than those measured with CD81. In other words, the two natural products exhibit a significant preference for CD81 over CD9, despite the similarity of the two protein structures. The difference is very important when comparing the two LEL structures. The ΔE values amount to –61.40 kcal/mol and –43.30 kcal/mol for Hz-B bound to CD81 and CD9, respectively. The difference in energy (–18.1 kcal/mol) is major, and it is even more pronounced (–21.8 kcal/mol) in the case of Hz-A (Table 3). There is no doubt that the two compounds are better adapted for binding to CD81 compared to CD9. The LEL portions of CD9 and CD81 are very similar (Figure 6B) but there are local variations that can be exploited for drug binding. Apparently, CD81 offers a suitable binding cavity for Hz-A but not CD9. This tetraspanin selectivity aspect warrants further examination.
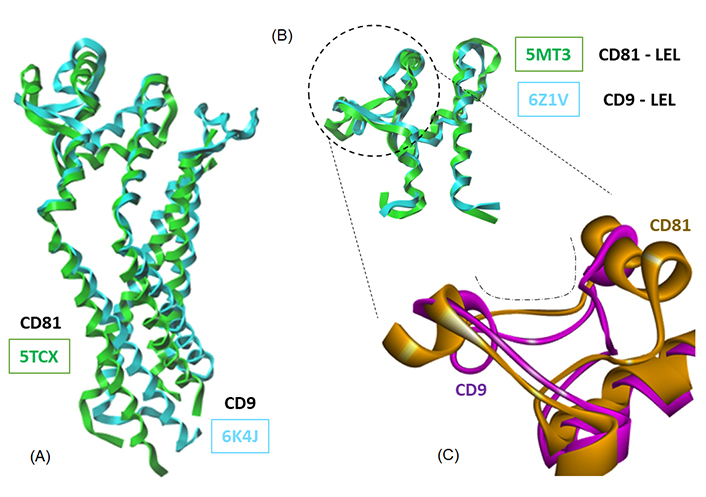
Overlapping models of tetraspanin CD81 and CD9. Models of (A) the full-length proteins and (B) the LEL domains (PDB codes indicated); (C) a close-up view of a portion of the LEL domain to illustrate the conformational difference between the two proteins at the level of the drug-binding cavity (dashed line)
Calculated potential energy of interaction (ΔE) and free energy of hydration (ΔG) for the interaction of Hz with CD9
| Compounds | Full-length protein CD9 (6K4J) | LEL fragment of CD9 (6Z1V) | ||
|---|---|---|---|---|
| ΔE (kcal/mol) | ΔG (kcal/mol) | ΔE (kcal/mol) | ΔG (kcal/mol) | |
| Hz-A | –37.00 | –16.90 | –36.00 | –18.30 |
| Hz-B | –39.20 | –19.00 | –43.30 | –18.20 |
Drug design perspectives
There are few small molecules known to bind to CD81. In the natural product class, the only two compounds for which binding to CD81 has been evidenced at the experimental level are Hz-A and -B, no other ones. Recent computational analyses have suggested the possible binding of the pentacyclic triterpene maslinic acid [57] and diverse flavonoids to the EC2 loop of CD81, such as puerarin, quercetin, and (–)-epicatechin [58]. This is entirely plausible, but these polyphenolic compounds tend to bind to many proteins in cells. We searched the natural products database for analogues of Hz, but we did not identify structurally close compounds. The only two products with a certain degree of similarity are the monoterpenes grandisol and the diastereomer fragranol (Figure 7A) which can be found in essential oils from the aerial parts of different plants, such as sagebrush (Artemisia spp.) [59] and the Mediterranean plant Achillea ligustica All. [60]. Their chemical synthesis has been described recently [61]. Fragranol is a rare monoterpene alcohol endowed with an anxiolytic action [62] but the compound has been little studied. Grandisol is a little more known, acting as a pheromone for curculionid beetles, such as Polygraphus punctifrons and Sternechus subsignatus (Coleoptera) [63, 64]. We have modeled the interaction of these two natural products with the LEL domain of CD81 but found only a weak interaction (ΔE = –40.35 kcal/mol and –35.50 kcal/mol for grandisol and fragranol, respectively; Figure 7B). The binding capacity of grandisol to CD81-LEL is modest but it relies on the same type of contacts as with Hz-B, notably the key H-bond with Lys187 and Cys175 (Figure 7C). The molecule is accessible chemically, with different total syntheses described [61, 65, 66]. Therefore, it is conceivable to design grandisol analogues with improved CD81-binding properties.
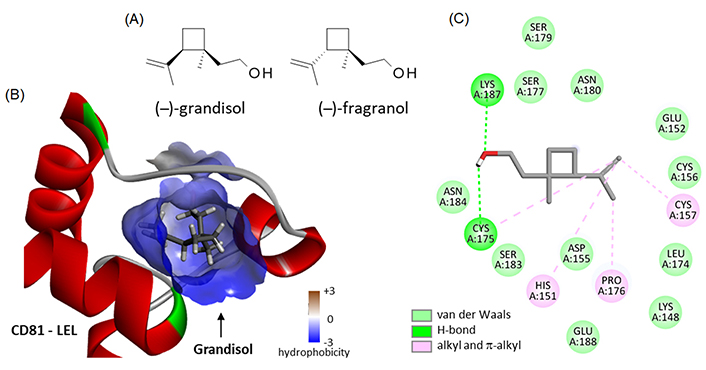
Binding of two monoterpenes to CD81. (A) Structures of grandisol (CID: 169202) and fragranol (CID: 6432285); (B) molecular model of grandisol bound to the LEL of CD81 with a detailed view of the hydrophobicity surrounding the drug binding zone (color code indicated); (C) binding map contacts for grandisol bound to CD81-LEL (color code indicated)
Discussion
CD81 is a prototypic tetraspanin, a member of the TM-4 superfamily, with major roles in cell-cell interactions and cellular trafficking. CD81 functions as a co-receptor for several viruses, notably HCV and HIV-1, and is considered an emerging antiviral target by New et al. [23]. This ubiquitous protein is also an anticancer target, implicated in the aggressivity of B-cell lymphoma [67], and contributing to the development of solid tumors, such as colorectal, liver, and gastric cancers [28]. For these reasons, several mAbs targeting CD81 have been developed and are being tested in experimental models. This is the case for the specific anti-human CD81 antibody 5A6 which recognizes a conformational epitope on the ectodomain of CD81 and has proved very efficient at inhibiting invasion of TNBC cells and the formation of metastases in xenograft models of TNBC [30]. Several antibodies targeting CD81 are being developed [68]. In contrast, there are no small molecules at present known to efficiently and selectively target CD81. Such small molecules could be extremely useful for oral therapy of cancer, viral diseases, and possibly other pathologies with an immune component. Indeed, CD81 is implicated in the regulation of interleukin-10 secretion in T regulatory cells [69]. But at present, there is only a handful of small molecules known to target CD81, including a few synthetic compounds and the natural products Hz-A and -B [32]. It is therefore important to better comprehend how these molecules bind to CD81, to identify the molecular determinants implicated in the interaction, and to guide the design of other molecules. Our analysis contributes to this objective, providing information to better understand the mode of binding of Hz-A/B to the tetraspanin.
For the first time, a modeling analysis compares drug binding to two conformational states of CD81, the closed form represented by the full-length protein and the open form represented by the LEL portion of the protein. Important differences have been identified between the two states, which translate into a distinct drug-binding process. In fact, the Hz molecules can target essentially the open structure (LEL), not really the closed form. To our knowledge, such a differential drug-binding process has never been pointed out before. CD81 is a highly dynamic protein, subject to conformational changes that affect its receptor function and its activity in cells. Cholesterol plays a role in the regulation of the conformational switch in the large extracellular domain of CD81 and this mechanism is important for HCV entry [34]. The structural flexibility of the EC2 loop (LEL) is important for virus entry and is essential for the selection of neutralizing antibodies targeting the flexible epitope [70]. Similarly, the flexible nature of the LEL moiety must be taken into account when designing molecules to target the receptor. Drug-binding studies are often performed with the truncated LEL protein, easier to produce and to handle than the full-length protein, but it may not represent the most common form of the protein embarked into tetraspanin-enriched microdomains. Both the open and closed forms of the LEL are important factors to consider for drug design.
The binding analysis of Hz-A/B to the LEL of CD81 and CD9 brings useful information (Figure 6 and Table 3). It provides a preliminary view of the tetraspanin-selectivity of the molecules and can give ideas to design CD9-binding compounds as well. There is no small molecule known to interact with CD9 at present. The approach used here, starting from CD81-LEL, can offer options to design CD9 ligands. We have recently identified bi-aryl molecules binding to CD9 and/or CD81 (Bailly C, Vergoten G, unpublished manuscript). CD9 is an important anti-cancer target from which novel anticancer agents can be designed [51].
At the natural product level, the work reported here provides a refined analysis of the interaction between CD81 and Hz. The initial work by Li and coworkers [22], using a distinct structure of the truncated CD81-LEL protein (PDB: 1G8Q, 90 amino acids), identified key interactions between Hz-A and the LEL protein, notably Asn184 and Lys187. Here we used a slightly longer LEL structure (PDB: 5M3T, 101 amino acids) but the identified contacts between the protein and the two compounds are essentially the same. However, the key observations are (i) the marked preference of the two compounds for the open versus closed conformation of the LEL and (ii) the noticeable selectivity for CD81 over the analogous tetraspanin CD9. The information is essential to guide drug design. The Hz scaffold is unique, relatively complex, and certainly not easy to modulate chemically. There are no direct analogues of Hz-A/B known at present, but a first option is offered to design derivatives, based on the monoterpene grandisol. This natural product, accessible chemically, may represent a starting point for a pharmacomodulation aimed at discovering novel CD81-binding compounds. Hz-A/B do not obey the “Lipinski’s rule of five” with log P values < 2 (n-octanol-water partition coefficient). There is a need to improve the compound molecular properties (notably log P) to facilitate the crossing of the blood-brain barrier, after oral delivery. New molecules could be designed from the grandisol scaffold.
Conclusions
In recent years, new insights have been gained into the structure and function of tetraspanin proteins and CD81 in particular. This TM protein is a target of interest for the treatment of viral diseases and cancers. The identification of small molecules binding to CD81 remains little studied and the design of compounds targeting selectively the LEL domain has been largely untouched. There is a need for new compounds and scaffolds interacting with CD81-LEL. Our study supports the use of the terpenoid Hz-A as a possible template to elaborate new CD81 binders. The study underlines the importance of considering the dynamic conformation of the LEL binding domain. The binding of Hz-B to CD81 varies markedly according to the open/closed conformation of the protein loop, and the nature of the flexible loop. Hz-B exhibits a net preference for binding to CD81-LEL over CD9-LEL, suggesting that a tetraspanin selectivity can be achieved. A cyclobutane-containing fragment (grandisol) has been identified as a possible starting element for drug design. The identification of the fungal sesquiterpene Hz-B as a ligand for CD81-LEL offers perspective to the synthesis analogues. The drug-based modulation of tetraspanin biology opens novel avenues for the treatment of advanced cancers and viral diseases.
Abbreviations
| BOSS: |
Biochemical and Organic Simulation System |
| CASTp: |
Computed Atlas of Surface Topography of proteins |
| HCV: |
hepatitis C virus |
| Hz-A: |
harzianoic acid A |
| LEL: |
large extracellular loop |
| mAbs: |
monoclonal antibodies |
| MC: |
Monte Carlo |
| MM: |
molecular mechanics |
| PDB: |
Protein Data Bank |
| SPASIBA: |
Spectroscopic Potential Algorithm for Simulating Biomolecular conformational Adaptability |
| TM: |
transmembrane |
| TNBC: |
triple-negative breast cancer |
Declarations
Author contributions
GV: Visualization, Software, Data curation. CB: Conceptualization, Investigation, Visualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
All relevant data is contained within the manuscript.
Funding
Not applicable.
Copyright
© The Author(s) 2023.