Review
Review

Affiliation:
Instituto de Química Médica, Consejo Superior de Investigaciones Científicas (IQM-CSIC), 28006 Madrid, Spain
Email: diegonunez@iqm.csic.es
ORCID: https://orcid.org/0000-0002-1005-1464
Explor Drug Sci. 2024;2:6–37 DOI: https://doi.org/10.37349/eds.2024.00034
Received: September 28, 2023 Accepted: November 10, 2023 Published: February 01, 2024
Academic Editor: Jean-Marc Sabatier, Aix-Marseille University, France
310-Helices represent the third most abundant secondary structure proteins. Although understandably overshadowed by α-helices for decades, the 310-helix structure is slowly regaining certain relevance in protein science. The key role of this secondary structure in biological processes has been highlighted in reports over the last decade. In addition, 310-helices are considered key intermediates in protein folding as well as a crucial structure for the antimicrobial activity of naturally occurring peptaibols. Thus, it is clear that 310-helices are relevant scaffolds to take into consideration in the field of biomimetics. In this context, this review covers the strategies developed to stabilize the 310-helix structure in peptide chains, from the incorporation of constrained amino acids to stapling methodologies. In the last section, the use of 310-helices as scaffolds of interest in the development of bioactive compounds, catalysts for enantioselective reactions, supramolecular receptors, and membrane-embedded signal transducers are discussed. The present work aims to highlight the relevance, sometimes underestimated, of 310-helices in chemical biology and protein science, providing the tools to develop functional biomimetics with a wide range of potential applications.
Peptide secondary structures are essential to mediate how proteins function. There is a close relationship between the number and types of secondary structure elements and the protein three-dimensional (3D) structure [1]. Relevant residues in protein-protein interactions and protein recognition, known as “hot spots”, are usually embedded in sequences forming secondary structures [2, 3].
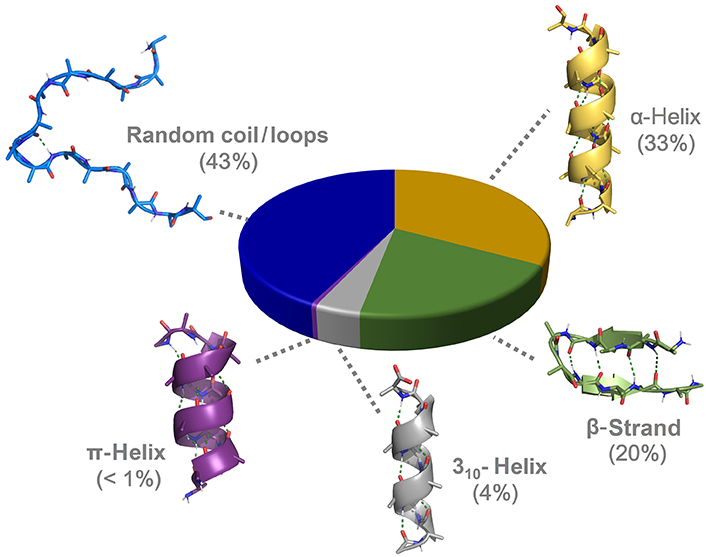
α-Helices and β-strands are the main structural features of proteins (Figure 1). Around a third of protein residues are present in α-helical segments. The H-bonding pattern in α-helices involves the carbonyl group of residue i and the nitrogen from the amide group in residue i + 4, giving rise to a helical arrangement with 3.6 residues per turn and a helical pitch of 5.4 Å. It is therefore composed of a repetitive pattern of α-turns. α-Helices play fundamental roles in a multitude of protein-protein and protein-nucleic acid interactions [4]. β-Strands are the second most abundant secondary structure, accounting for about 20% of amino acid secondary structure states [5]. β-Strands are linear arrangements of residues with almost coplanar amide bonds and side chains alternating above and below the plane of the peptide backbone [6]. They can interact with other β-strands via H-bonds to form the β-sheet secondary structure, a key structural element in proteins also involved in recognition events. β-Hairpins consist of two antiparallel β-strands joined by a short loop region, normally stabilized by an intramolecular H-bond between the carbonyl group of residue i and amide nitrogen in residue i + 3, thus forming a β-turn [7].
310-Helices represents the third principal secondary structure element occurring in proteins, involving about 4% of protein residues [9]. In 310-helices, a regular and repetitive H-bonding network is established between the carbonyl group in residue i and the amide nitrogen in residue i + 3, giving rise to a helical structure with three residues per turn and a helical pitch of 5.8–6 Å. It is formed therefore by a succession of β-turns, yielding a thinner and more elongated structure than the α-helix [10]. The third helical structure found in proteins is the π-helix, although with a low frequency of occurrence. It is stabilized by an intramolecular i/i + 5 H-bonding network. Despite the low abundance, π-helices contribute to protein folding and are relevant as ligand-binding site contributors [11]. Apart from the above-mentioned regular structural elements, a significant population of amino acids is located in unstructured regions and loops connecting secondary structure elements, lacking regular H-bonding patterns.
The 310-helix structure was first proposed as a reasonably stable peptide secondary structure by Taylor [12] in 1941, a decade before Pauling and Corey [13] described the α-helix. Shamala et al. [14] provided the first crystallographic evidence of a 310-helix in 1978 from a model homo-pentapeptide formed by α-aminoisobutyric acid (Aib). However, for decades, 310-helices have been understandably overshadowed by a huge interest in α-helices and β-structures due to their abundance and pivotal role in protein science. Little attention has been paid to the role of 310-helices in protein structure and function despite their nonnegligible population. This review intends to revisit the 310-helix structure and provide an updated overview of its relevance in protein science, covering the biological significance of 310-helices, strategies toward the development of biomimetics, and their use as scaffolds in medicinal and supramolecular chemistry as well as in protein engineering.
The α-helix is highly restricted in terms of conformation freedom, accepting little variation in the range of backbone torsions. It is energetically favorable due to the network of intrahelical backbone H-bonding and the staggering of the side chains, which minimizes steric and electronic clashes (Figure 2A). By contrast, side chains in 310-helices are disposed in ridges along the helix, making them thermodynamically less stable (Figure 2B). This has been postulated as the reason behind the lower abundance and shorter length of 310-helices in comparison to α-helices. While α-helices are on average ten residues long in natural proteins, 310-helices are significantly shorter and often involve a single helical turn comprising just three residues [8]. Indeed, the longest 310-helices in natural proteins are formed by three helical turns with less than ten residues, the average length found in α-helices. Longer 310-helices have been reported in natural fungi-derived and synthetic peptides with a high content of Aib [15].
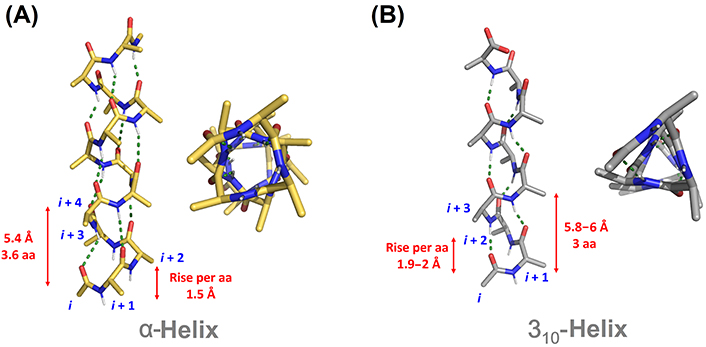
Comparison of canonical α-helical (A) and 310-helical (B) structures. aa: amino acid
Despite the differences in the disposition of the side chains between α-helix and 310-helix, both arrangements present three distinct faces with particular physicochemical properties. While hydrophobic faces are generally involved in protein-protein interactions, hydrophilic ones assist in the stabilization of protein tertiary structure and positively contribute to the solubilization of proteins in aqueous media [8, 16].
In terms of electronic properties, the carbonyl groups in α-helices are placed parallel to the helix axis, generating a dipole moment coinciding with this helix axis. In 310-helices, the carbonyl groups are tilted off the axis so the dipole moment is not as strong as in α-helix [17, 18]. The existence of such dipole in both helical arrangements generates an electrostatic positive charge on the N-terminal end and an electrostatic negative charge on the C-terminus. Negatively charged side chains or anionic ligands are often found near the N-terminal end, while positively charged side chains tend to stabilize the helical structure in the C-terminal end [19].
The main difference between the amino acid distributions in 310-helices and α-helices is the high aspartate frequency at the N-terminal position of 310-helices. This suggests that the interaction of aspartate residues with the electrostatic positive charge on the N-terminal is essential for the initiation of 310-helices [20]. 310-Helix propagation is normally mediated by favorable interactions between hydrophobic residues at positions i and i + 3 [19]. In contrast, these amino acid distributions are less critical for the initiation and propagation of α-helices, where H-bond cooperativity is the main factor for helix stabilization.
For decades, molecular dynamics simulations have represented the computational method of preference for the prediction of the conformational properties of peptides and proteins, mainly due to its good balance between accuracy and computational cost [21]. For 310-helices, Patapati and Glykos [22] analyzed three popular force fields such as Chemistry at Harvard Macromolecular Mechanics (CHARMM), Optimized Potentials for Liquid Simulations (OPLS), and Assisted Model Building with Energy Refinement (AMBER), finding that the latter outperforms in the prediction of the conformational preferences of a 310-helical peptide.
In the last years, deep learning approaches have revolutionized protein structure prediction. AlphaFold 2 has excelled at solving the 3D structures of complex proteins. Stevens and He [23] reported that AlphaFold 2 slightly over-predicts regular secondary structure elements, including 310-helical segments, in loop regions.
Several analytical techniques are available to determine secondary structural information in peptides and proteins. X-ray crystallography proved to be very useful in studying helical segments in many peptide models, although it does not provide information about the peptide conformational preferences in solution. Cryogenic electron microscopy (Cryo-EM) has become a powerful technique to ascertain protein 3D structures of different functional states, which were usually elusive to X-ray crystallography. The expansion of this technique along with the improvements in detection and crystallization methods has significantly enlarged protein databases, requiring specific software packages for secondary structure assignments based on 3D coordinates. Popular tools for the correct assignment of 310-helices in proteins are Define Secondary Structure of Proteins (DSSP) and Structural Identification (STRIDE) [24, 25].
Spectroscopic techniques can provide reliable information about the helix type and population, with infrared spectroscopy (IR), circular dichroism (CD), and nuclear magnetic resonance (NMR) being the most widely used. The structural difference between α- and 310-helices is generally translated into distinct signal patterns.
In IR, the most sensitive signal related to protein secondary structure is the amide I band. 310-Helical model peptides containing quaternary amino acids have an amide I band at higher frequencies (≈ 1,662 cm–1) than α-helices (≈ 1,654 cm–1) in organic solvents [26, 27]. However, this small frequency shift can be due to other factors such as structural distortions as well as sequence, length, and solvent effects. In the case of CD, both structures exhibit two negative bands at around 208 nm and 222 nm and a positive maximum at 195 nm [28, 29]. The relative intensity ratio between the two negative bands, expressed as [θ]222/[θ]208, has been proposed by Silva et al. [30] as a strategy to differentiate both helical arrangements. A smaller ratio is observed for 310-helices, with values of 0.3–0.4, while α-helices display a larger ratio of around 1 [30, 31]. In addition, the positive band at 195 nm is reduced in 310-helices when compared with α-helices, which contributes to an increased negative CD at 207 nm.
NMR has been extensively used to characterize helical peptides, although distinguishing helical conformations was proved to be challenging, especially in short linear peptides. Wüthrich [32] first described the nuclear Overhauser effect that distinguish the 310-helix from the α-helix in 2D NMR spectra. Hydrogen/deuterium-exchange kinetics, 3J coupling constants, heteronuclear multiple bond connectivity (HMBC), and rotating frame Overhauser effect spectroscopy (ROESY) experiments have been also reported as useful tools to determine the relative 310- and α-helix content using NMR [33–35]. The incorporation of nitroxide spin labels in the side chain enables the use of electron paramagnetic resonance (EPR) as a complementary technique to distinguish the α-helical conformation from 310-helices. Measurements of biradical J-coupling and dipolar interactions between the labels allow the determination of the peptide geometry in different solvents [36].
In the last 30 years, many theoretical and experimental works have suggested that the 310-helix is an intermediate in the folding process of peptide chains into α-helices [37, 38]. Short peptide sequences are prone to stabilize a 310-helix rather than an α-helix. In the absence of stabilizing effects from H-bonding cooperativity and destabilizing effects from side-chain steric and electronic clashes, a short 310-helix is likely to be more stable than the corresponding α-turn due to the increasing intramolecular H-bonds interactions [39–42]. The same alanine (Ala)-based pentapeptide model displaying a canonical single α-turn and a 310-helix structure is shown in Figure 3A and B, respectively. The latter is stabilized by two intramolecular H-bonds (two consecutive β-turns) in contrast to the single H-bond for the α-turn.
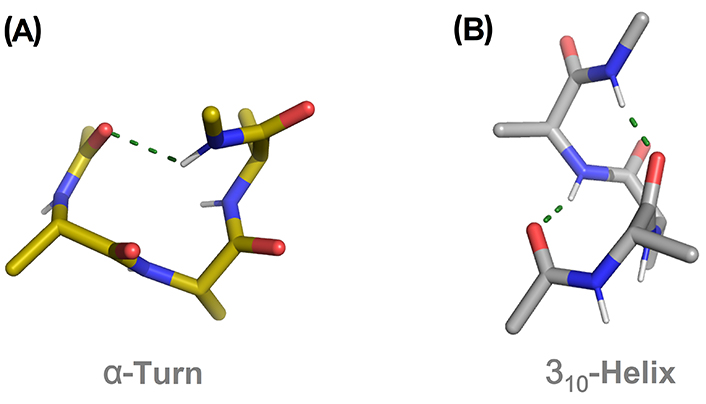
Comparison of an α-turn (A) and 310-helix (B) in the pentapeptide model Ac-(Ala)3-NHMe. H-bonds are displayed as green dashed lines. Ac: acetyl
In longer segments, the α-helix dominates due to the staggering of the side chains and H-bond cooperativity [16, 41, 43, 44]. It has also been postulated that the H-bond length in 310-helix is on average longer and therefore weaker than in α-helix [45]. According to crystallographic data, the length, concentration, and capping groups in the peptide chain also play an important role in the stabilization of the helical arrangement [39, 46–50]. In solution, the nature of the solvent is relevant in the equilibrium between the two helical states. It is generally accepted that the α-helix dominates in a polar environment while non-polar solvents tend to stabilize 310-helices [51–53]. Facile exchange between the two helical types was observed for helical peptides incorporated into a self-assembled monolayer (SAM) as a function of the polarity of an applied electric field [18].
The folding of peptide segments into helical structures in solution is not a two-state equilibrium between random coil and helix. Instead, partially folded states are formed as intermediates, requiring the use of a helix/coil theory considering every possible conformation of the peptide chain affecting the formation of the helix [54, 55]. Computational studies suggest that 310-turns are a common part of helix initiation and often persist at the end of α-helical segments as they require fewer torsional constraints than the α-turn angles [56–59]. The parametrization of a new form of the Zimm–Bragg theory to include 310-helices demonstrated that they are plausible folding intermediates [60]. Similar conclusions were obtained when considering the Lifson–Roig helix-coil theory [61]. In the interface of water and a non-polar solvent or membrane, theoretical and experimental studies suggest that peptides are more promiscuous, stabilizing a mixed 310-/α-helix conformation [59, 62]. These systems serve as models to study the folding of membrane proteins where both electrostatic forces and lipophilic interactions can play a role in stabilizing 310-helical intermediates [63, 64].
Peptaibols are a family of amphipathic linear peptides, typically between 4 and 20 residues, biosynthesized by fungi of the genre Trichoderma and Emericellopsis [65]. These peptides are rich in non-standard residues, mainly Aib, which favors the adoption of helical secondary structures. They are usually acetylated at the N-terminal end and reduced at the C-terminus, containing therefore a terminal hydroxyl group [66]. They possess antimicrobial activity derived from their ability to form pores in microbial lipid membranes, leading to a loss of osmotic balance and subsequent cell death [67]. The strong tendency of peptaibols to be embedded in the hydrophobic core of the membrane bilayer is driven mainly by their high tendency to stabilize helical conformations and amphipathicity [68]. It has also been suggested that the terminal hydroxyl group forms H-bonds with the glycerol and phosphate headgroups, contributing to the stabilization of ion channels [69].
Peptaibols have been classified according to their length in three categories: short (4 to 10 residues), medium (14 to 16 residues), and long (17 to 22 residues) peptaibols. The abundance of Aib residues in the sequence as well as the length of peptaibols determine the type of helical arrangement adopted [70]. In general, short and medium peptaibols tend to favor 310-helices over α-helices while long peptaibols stabilize mixed or distorted helical conformations [71]. Other peptaibol classifications consider residue similarity or the polarity of the N-terminal end [66].
Among short peptaibols, peptaibolin and trichogin are prototypic and widely studied examples. Peptaibolin is an unusually very short peptaibol, with only five amino acids including phenylalaninol (Phol) as a C-terminal cap [72]. It has moderate antimicrobial activity against Gram-positive bacteria and yeasts. In the solid-state structure, peptaibolin adopts a fused β-/α-turn at the N-terminus, with a second α-turn stabilized by an additional H-bond between the Phol OH group and an Aib carbonyl group (Figure 4A) [73]. Interestingly, the capping of the hydroxyl group at the C-terminus leads to the formation of a regular 310-helix, the most common structure found in short Aib-rich peptides. Trichogin is another relevant short peptaibol, containing 11 residues [74]. It has been widely studied as the prototype of lipopeptaibol, which is characterized by an N-terminal lipophilic chain [75]. Trichogin possesses a remarkable membrane-perturbing activity, superior to longer non-lipidated peptaibols, demonstrating that N-terminal lipidation is therefore relevant for the antimicrobial activity. This peptaibol displays a distorted, right-handed 310-helix combined with a segment of irregular, right-handed α-helix [76].
X-ray structures of representative peptaibols. (A) Peptaibolin [Cambridge Crystallographic Data Centre (CCDC) 160328]: Ac-L-Leu-Aib-L-Leu-Aib-L-Phol; (B) Trichovirin I-4A [Protein Data Bank (PDB) ID: 3SBN]: Ac-Aib-L-Asn-L-Leu-Aib-L-Pro-L-Ala-L-Val-Aib-L-Pro-Aib-L-Leu-Aib-L-Pro-L-Leuol; (C) Gichigamin A (PDB ID: 4Z0W): Ac-(Aib-L-Pro)2-L-Phe-D-Iva-L-Pro-L-Ala-Aib-βAla-L-Ala-D-Iva-βAla-L-Leu-Aib-βAla-L-Leu-(Aib)2-L-Leu-βAla-Glyol. Two views are shown. Asn: asparagine; Glyol: glycinol; Iva: isovaline; Leu: leucine; Leuol: leucenol; Phe: phenylalanine; Pro: proline; Val: valine
Some interesting medium-size peptaibols are harzianins (14 residues) [77], trichovirins (14 residues) [78], pentadecaibins (15 residues) [79], antiamoebins (15 residues) [80], heptaibins (15 residues) [81] and zervamicins (16 residues) [82]. Depending on the content and position of restricted amino acids, mainly Aib and Pro, these medium-length peptaibols stabilized 310-helices or mixed 310-/α-helices. The X-ray structure of trichovirin I-4A is shown in Figure 4B, displaying a long 310-helix with four complete helix turns [83]. In this case, a repetitive Aib-Pro pattern is the driving force for the adoption of a 310-helix over the α-helix.
In the category of long peptaibols, alamethicin has been intensively studied as a model peptaibol to ascertain the mechanism of membrane channel formation [84]. Alamethicin is a 20-residue peptide able to form a stable amphipathic α-/310-helical structure in membranes and membrane-mimetic environments. Recently, the longest peptaibol from a natural source, gichigamin A, was isolated and characterized [85]. The gichigamins are rich in β-Ala, a very rare residue in peptaibols, and possess a repeating α-residue/α-residue/β-residue motif creating a 311-P-helix (Figure 4C) [86]. This is an unusual secondary structural element among natural peptides but its spectroscopic features are similar to 310-helix. This structural motif is essential for the unexpected high penetrability of gichigamin A through cell membranes, leading to a potent in vitro cytotoxicity by direct disruption of mitochondrial function.
310-Helices have been postulated to play a relevant role in the mechanism of some ionic and water channels. Aquaporin-4 (AQP4) is the predominant water channel in brains and plays an important role in the regulation of water homeostasis in brains and in the pathophysiology of brain edema. Tani et al. [87] demonstrated that the Pro138-glycine (Gly) 144 segment of AQP4, located in an extracellular loop, adopts a 310-helix, which mediates the adhesive interactions between AQP4 tetramers.
Long 310-helices have been identified in the structure of three voltage-gated potassium channels: the bacterial cyclic nucleotide-regulated potassium (MlotiK1), the human Kv1.2- and Kv2.1-chimeric channels [88–91]. In particular, 310-helices were found in the positively-charged S4 helix domain, one of the six transmembrane helices functioning as the voltage sensor. The structure of the transmembrane regions of MlotiK1 structure is shown in Figure 5, with a zoom on the S1–S4 domain where the 310-helical segment is present (highlighted in orange) [88]. It has been postulated that an α- to 310-helix transition in S4 is responsible for the gating process [90].
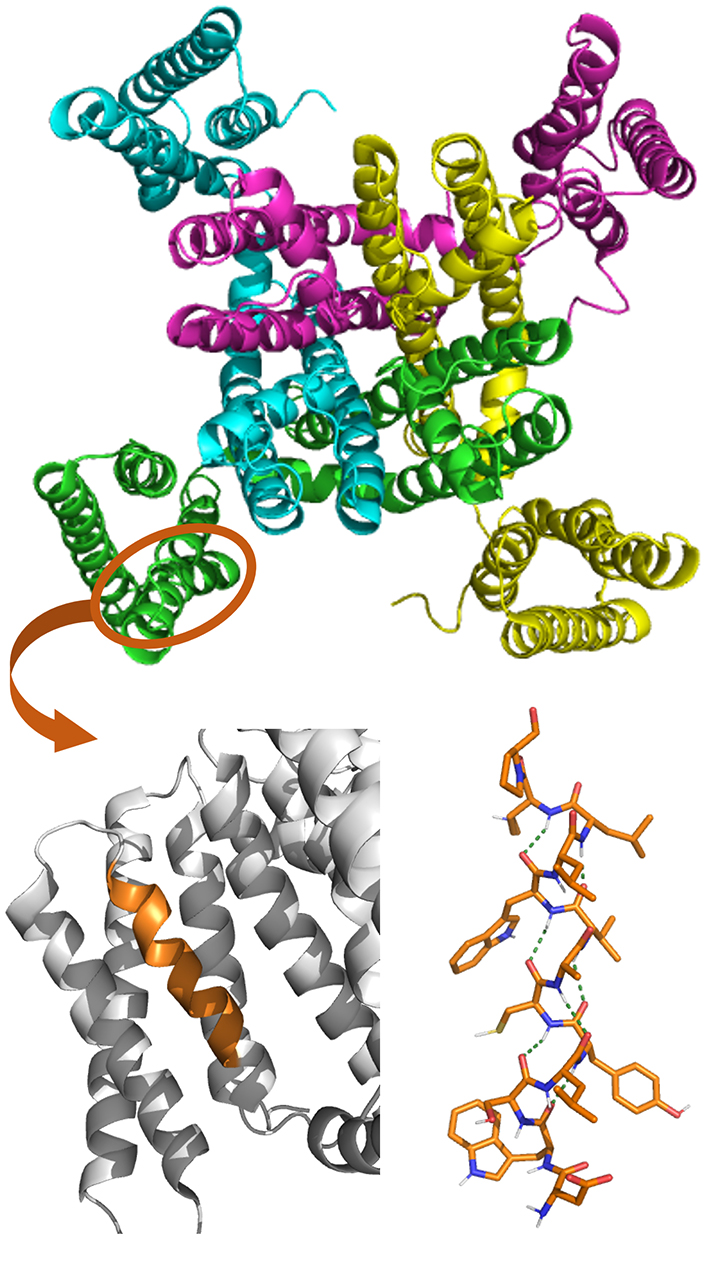
Crystal structure of MlotiK1 (PDB ID: 2ZD9) potassium channel (top). The S1–S4 domain is zoomed at the bottom, with the 310-helix segment in orange. The molecular structure of this 310-helix is also shown, displaying the arrays of H-bonding stabilizing the helical conformation as a green dotted line
Vieira-Pires and Morais-Cabral [91] performed a structural analysis of the transmembrane proteins deposited in the PDB, finding a relatively high prevalence of long 310-helices in membrane proteins when compared with soluble proteins. These results suggest that, in general, the 310-helix structure is somewhat relevant in the function and structure of some membrane proteins.
Long 310-helices have been identified in other proteins such as dienelactone hydrolase (key in chlorocatechol degradation), glycogen phosphorylase (important allosteric enzymes in carbohydrate metabolism), cellulase celC (relevant in cellulose, lichenin, and cereal beta-D-glucan hydrolysis), hemoglobin I (oxygen transport), transducin-α (vertebrate phototransduction), and thymidylate synthase (crucial in early stages of DNA biosynthesis) [92]. The crystal structure of glycogen phosphorylase is shown in Figure 6A, with the long 310-helical segment highlighted in pink [91]. However, the role of such long 310-helix segments in protein function or structure has not been investigated.
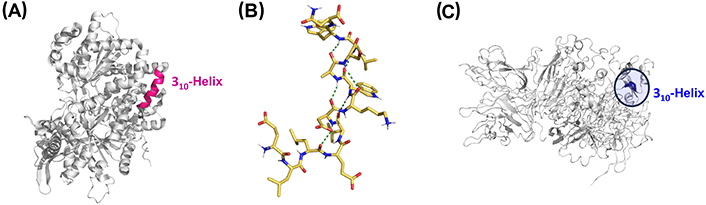
Representative 310-helices involved in biological and pathological processes. (A) Crystal structure of glycogen phosphorylase with the 310-helix colored in pink (PDB ID: 1GPB); (B) NMR structure of the peptide gp41659–671, which contains the epitope of the HIV-1 neutralizing antibody 2F5 (PDB ID: 1LB0); (C) crystal structure of coagulation factor VIII (FVIII) with the key 310-helical segment highlighted (PDB ID: 2R7E). gp41659–671: residues 659–671 of the envelope glycoprotein 41 of human immunodeficiency virus type 1 (HIV-1)
Biron et al. [93] reported the NMR structure of the peptide gp41659–671, from the transmembrane glycoprotein gp41 which mediates the fusion of the HIV-1 with host cells. Within the C-terminal region involved in viral fusion, gp41659–671 contains the entire epitope of the HIV-1 neutralizing antibody 2F5 and stabilizes a 310-helix in water (Figure 6B). The existence of an i/i + 3 salt bridge and the presence of a Leu-Leu sequence along with α-helix destabilizing aspartic acid and tryptophan residues favor the formation of the 310-helix over the α-helix. This monomeric 310-helix facilitates the exposure of the hydrophobic face to the immune system while the hydrophilic residues form the opposite face.
Hepatitis C virus is responsible for chronic hepatitis C liver disease and can disrupt the signaling pathways responsible for the activation of cellular antiviral defenses, leading therefore to persistent infections. This evasion takes place through a viral protease, which promotes the proteolysis of Toll-interleukin-1 (IL-1) receptor domain containing adaptor-inducing interferon-β (TRIF). NMR chemical shift perturbation experiments demonstrated that the recognition of TRIF by the viral protease involves a 310-helical segment in close proximity to the protease active site and is composed of hydrophobic residues [94].
NMR studies have provided evidence of a conformational equilibrium between a β-turn and a 310-helix in an aggregation-prone segment of the β2-α2 loop of prion proteins, which causes neurodegenerative diseases via aggregation [95]. This fragment remains buried when it adopts a 310-helical conformation, so it is not available for self-assembly, and aggregation is therefore prevented. A similar amyloid aggregation profile was observed in an α-helical peptide bundle, which undergoes concentration-dependent conversion to β-sheet fibrils via the formation of 310-helical intermediates [49].
A change in pitch and partial unwinding of a 310-helix is key in the interaction between the major histocompatibility complex II with human leukocyte antigen dermatomyositis (DM), a relevant process in the mechanism of antigen presentation of the immune system [96]. In a different example, Zhang et al. [97] performed density functional calculations on a 310-helical peptide suggesting that it can serve as novel relay elements in charge-transfer reactions in proteins, vital in biological processes such as photosynthesis or nitrogen fixation. Another study reported that the proliferating cell nuclear antigen (PCNA) interacting protein (PIP), relevant in DNA replication, forms a 310-helix that enters into the hydrophobic groove of the PCNA, representing a key structural motif controlling PCNA-protein interactions [98]. Similarly, a 310-helical turn is essential for the proliferation-inhibiting properties of macrophage inflammatory protein-1 alpha, a chemotactic chemokine involved in stem cell inhibition, wound healing, and maintaining the effector immune response [99].
It has been also reported that a 310-helix has implications for the development of hemophilia, a bleeding disorder caused by a deficiency of coagulation FVIII. Hemophilia A mutations near the unused N-glycosylation site of the A2 domain (N582) of FVIII affect protein conformation and intracellular trafficking. N582 is located in the middle of a short 310-helical turn, which was found to be critical for proper biogenesis of the A2 domain and FVIII, and therefore implicated in the progress of hemophilia A (Figure 6C) [100].
Another relevant example involves cyclin-dependent kinases (Cdks), key cell cycle regulators, which are considered important therapeutic targets for cancer treatment. Experimental evidence determined that a 310-helical region in reported Cdk inhibitors is critical in the inhibitory activity and, in consequence, in their growth-suppressing function [101]. A-kinase-anchoring protein (AKAP) 79/150 is an AKAP with a key role in synaptic long-term depression. Ca2+ directly regulates AKAP79 through its effector calmodulin (CaM), which adopts a compact conformation able to recognize a mixed α-/310-helix in AKAP79 [102].
In a different context, polyamines are involved in important biological functions. A unique 310-helix provides the steric constriction that directs the polyamine substrate specificity of histone deacetylase 10 (HDAC10), a biomarker and target involved in the prevention of autophagic responses to cancer chemotherapy [103]. Kinesin family member 1A (KIF1A), a critical cargo transport motor within neurons, is also a relevant therapeutic target. A key mutation in KIF1A, associated with neurological disorders, is located in a 310-helix adjacent to a loop involved in microtubule association. From these studies, it was postulated that the 310-helix facilitates a specific loop conformation that is critical for protein function [104].
In this section, the variety of approaches reported over the last decades toward the stabilization of 310-helical conformations in peptides is presented. These methods involve both the incorporation of conformationally restricted unnatural amino acids into peptide sequences combined with rational design of 310-helix-prone sequences and well-established methodologies such as peptide stapling and hydrogen bond surrogates.
The use of constrained quaternary amino acids is one of the most common strategies to induce the folding of a peptide chain into secondary structure elements. Cα-Tetrasubstitution imposes a significant restriction on the conformation space of a peptide chain by the Thorpe-Ingold effect, bringing the nearby atoms on both sides of the substituted carbon in close proximity.
Among quaternary amino acids, achiral Aib is the simplest and by far the most utilized for the induction of helical conformation in peptides (Figure 7A). Aib homo-oligomers show a great tendency to display 310-helices from the shortest tripeptide to the undecapeptide, the longest observed at atomic resolution (Figure 7B) [15, 105, 106]. The 310-helical induction by Aib is especially strong in non-polar solvents [52]. From 12 residues, there is a switch from 310- to α-helical structures with increasing peptide length, which translates into different supramolecular aggregations [107]. Short 310-helical homo-oligomers form globular structures, while micrometric filaments were predominantly imaged for α-helical oligomers [108, 109].
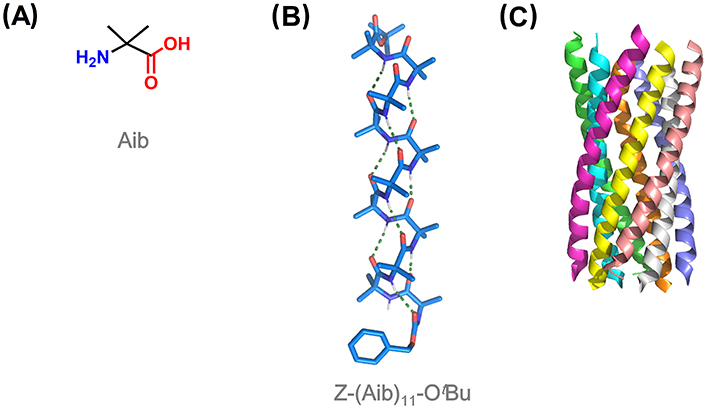
310-Helices adopted by Aib homo-oligomers show an equal preference for both right- and left-handed screw sense, with rapid interconversion between the two states at room temperature [110]. The relative population of these two helical states is sensitive to chiral influences. Thus, the incorporation of a single chiral amino acid at the N-terminus induces a screw-sense preference. N-terminal Ac or benzyloxycarbonyl (Cbz)-protected L-amino acids induce a left-handed helix, switching to a right-handed helix when a chiral quaternary amino acid is used instead [111–113]. Chiral induction was also accomplished with chiral ligands covalently or non-covalently bound to one terminus of the Aib foldamer, which is propagated throughout the entire foldamer from the N- to the C-terminus [114–116].
Aib maintains the ability to induce helical conformations when combined with non-quaternary proteinogenic amino acids, although the increase in conformational flexibility leads to a variety of helical arrangements including 310-, α-, and distorted or mixed 310-/α-helices [106]. Many studies have analyzed the conformational preferences of Aib in Ala-based peptides, in order to determine the critical main-chain length for the 310- to α-helix transition [39, 117–119]. In -(Aib-Ala)n- oligomers, it was found that the 310-helix is preferentially adopted in systems containing 6 or fewer residues, while the α-helix dominates in longer systems [120]. However, other factors influence the stabilization of one or the other type of helix, such as the solvent, terminal protecting groups, whether the N-terminal residue is Ala or Aib, packing motifs, and co-crystallized solvent molecules.
In general, peptides containing four or more Aib residues adopt a stable 310-helix conformation in the solid state and in solution, especially when non-polar solvents are used [121]. Other factors to take into account are the sequence and length of the peptides, as well as the intermolecular helix-helix self-association, packing efficiency, and interactions with helix macrodipoles [106]. Longer peptides in polar solvents tend to stabilize α-helices, especially if Gly is present in the sequence [110, 120].
Recently, Kumar et al. [8] have reported the de novo design of discrete and stable 310-helical peptide assemblies (Figure 7C). They described the rules and principles behind the rational design of these assemblies, based on the use of 310-helix-prone six-residue repeats identified in known protein structures along with the incorporation of Aib residues.
Many quaternary amino acids bearing more complex side chains than Aib have been reported in the literature. They can be classified into 4 categories, as illustrated in Figure 8. The first distinction can be drawn regarding if the side chain is linear or cyclic. Then, acyclic quaternary amino acids can be divided into achiral (Figure 8A), bearing two equal substituents at the α-carbon, and chiral ones (Figure 8B), with two different substituents at such position. On the other hand, cyclic quaternary amino acids can be divided into carbocyclic ones (Figure 8C), where the cyclic side chain is composed only of carbon atoms, and heterocyclic (Figure 8D), containing at least one heteroatom in the cycle.
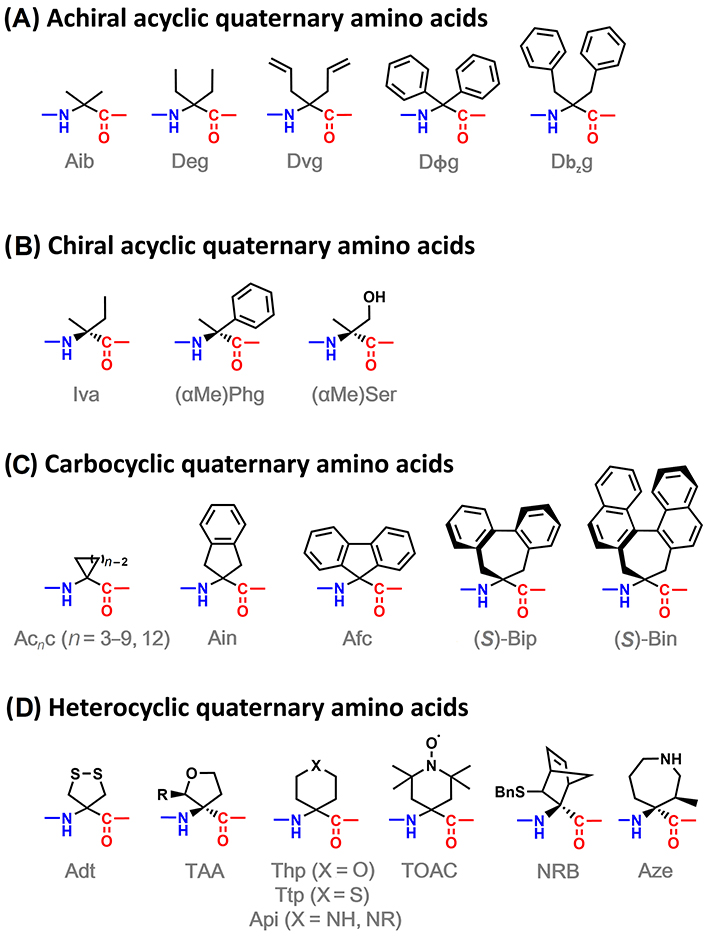
α,α-Disubstituted amino acids able to induce 310-helices: achiral acyclic (A), chiral acyclic (B), carbocyclic (C), and heterocyclic (D) amino acids. Adt: 4-amino-1,2-dithiolane-4-carboxylic acid; Afc: 9-amino-9-fluorenecarboxylic acid; Ain: 2-aminoindane-2-carboxylic acid; Api: 4-aminopiperidine-4-carboxylic acid; Aze: (3R,4S)-4-amino-4-carboxy-3-methylazepane; Bin: 4,5-dihydro-4-amino-3H-cyclohepta[2,1-a:3,4-a’]dinaphthalene-4-carboxylic acid; Bip: 2’,1’:1,2;1’’,2’’:3,4-dibenzcyclohepta-1,3-diene-6-amino-6-carboxylic acid; Dbzg: Cα,α-dibenzylglycine; Deg: Cα,α-diethylglycine; Dvg: Cα,α-divinylglycine; Dϕg: Cα,α-diphenylglycine; NRB: 3-sulfanyl-norbornene amino acid; Phg: phenylglycine; Ser: serine; TAA: tetrahydrofuran Cα-tetrasubstituted amino acid; Thp: 4-aminotetrahydropyran-4-carboxylic acid; TOAC: 2,2,6,6-tetramethyl-N-oxyl-4-amino-4-carboxylic acid; Ttp: 4-aminotetrahydrothiopyran-4-carboxylic acid
Achiral acyclic quaternary amino acids are Aib analogues showing a similar restriction of the conformational space [122–125]. However, branching at the β-carbon has been reported to favor extended conformations [126–128]. Regarding the chiral counterparts, the effect of Iva on a peptide chain is similar to Aib, promoting 310-, α-, or mixed-helical conformations [129]. More complex chiral residues, like α-methylphenylglycine and α-methylserine, present a higher tendency to induce β-turns and 310-helices, the latter with defined screw-sense inherent to the chirality of the quaternary residue [130–132].
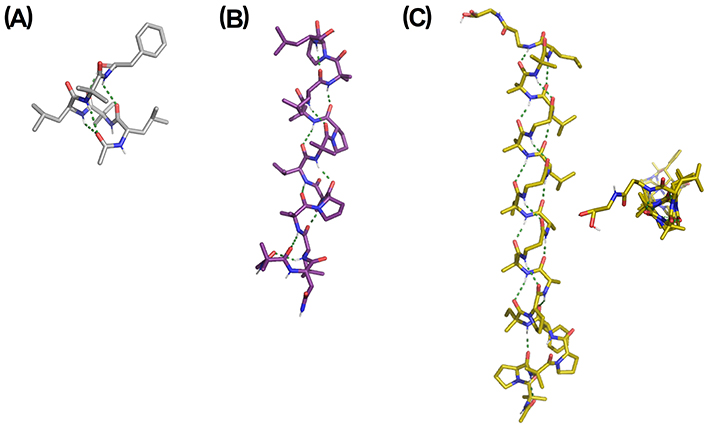
Cyclic quaternary amino acids have received considerable attention as they impose higher conformational rigidity compared with their linear analogues. Among carbocyclic analogues, 4- to 12-membered 1-aminocycloalkane-1-carboxylic acid (Acnc) residues (n = 4–12) were found to promote type III β-turns in short peptides and regular 310- or α-helices in longer systems, while Ac3c favors type I or II β-turns and distorted 310-/α-helices [106, 125, 133–139]. A chiral version of Ac4c developed as a cyclobutane analogue of Ser was reported to be an effective 310-helix inducer in Ala-based pentapeptide models [140]. 5-Membered aromatic analogue Ain has a similar effect as Ac5c, while Afc can be considered a combination of Ac5c and Dϕg able to promote helical or extended conformations depending on the sequence, length, and solvent [141–143]. Chiral Ac5c and Ac6c analogues have also been described as inducers of 310-helices with defined screw-sense (Figure 9A) [15, 139, 144]. 7-Membered biphenyl-based amino acids such as Bip and Bin are analogues to Ac7c but possess axial chirality, which provides interesting features to these systems [145–148]. They are able to induce 310-helical conformations in peptide chains, although extended conformations have also been reported in short peptides.
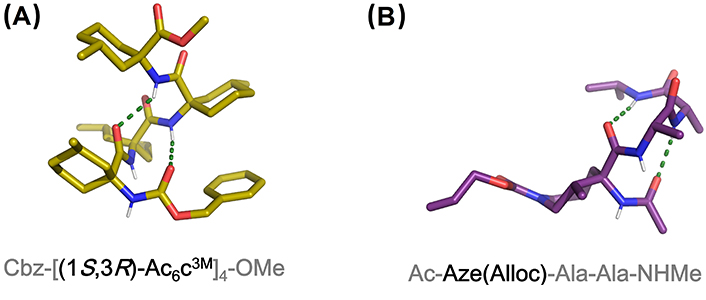
X-ray structures of (A) Cbz-[(1S,3R)-Ac6c3M]4-OMe containing four consecutive residues of a chiral analog of Ac6c (CCDC 1036631) and (B) Ac-Aze(Alloc)-Ala-Ala-NHMe. Aze: residue at the i + 1 position of a pentapeptide model (CCDC 887956). Alloc: allyloxycarbonyl; OMe: methoxy
Examples of quaternary heterocyclic amino acids are scarce as they are synthetically challenging building blocks. Nevertheless, the presence of heteroatoms in the side chains provides interesting features to the peptides incorporating them, serving as anchor points to engineer additional functionalities and to facilitate their solubility in polar media. Achiral 5-membered 1,2-dithiolane Adt has been incorporated into tetrapeptide models showing a great ability to stabilize β-turns, although the stabilization of 310-helices in longer systems could be prevented due to unfavorable interactions between dithiolane rings [149–151]. The tetrahydrofuran-derived amino acid TAA is obtained as a racemic mixture but only the S-diastereoisomer is able to induce 310-helices when incorporated into peptide sequences [152, 153].
Thp and Ttp derivatives were studied as reverse turn and 310-helix inducers in peptide models combined with other restricted amino acids such as Aib and Pro [154–156]. Piperidine-derived quaternary amino acids (Api) have attracted more attention than oxygen and sulfur counterparts due to their potential for incorporating substituents in the heterocyclic nitrogen. Their ability to induce helical conformations in peptide sequences has been reported although always in combination with Aib [157, 158]. Api residues allow the reinforcement of 310-helical conformations in peptides though covalent and non-covalent bonds involving the piperidine ring, either via metal chelation, salt bridges, or stapling, a feature covered in the section entitled “310-Helix stabilization via stapling and related methodologies” [159–161]. The N-oxide piperidine-derived amino acid TOAC has been widely used in peptide conformational studies. Along with its ability to provide conformational restriction to peptide chains, it is a paramagnetic residue enabling the use of EPR for conformational studies [162–164]. As discussed in the section “Experimental techniques for 310-helix characterization”, EPR can clearly determine if a peptide adopts a 310- or an α-helix because it allows the quantitative measure of distances between paramagnetic residues. In the absence of Aib residues, commonly used in combination with TOAC, this 6-membered amino acid is able to induce helical structures in peptides of 6–8 residues long [36, 163, 165, 166]. NRB residue has been described as a conformationally constrained cysteine (Cys) analogue and is able to induce 310-helices in model peptides, although in combination with Aib residues [167].
A seven-membered diastereopure azepane amino acid has been reported as an effective β-turn and 310-helix inducer in Ala-based peptide models (Figure 9B) [168, 169]. This amino acid is obtained by intramolecular ring opening of an ornithine-derived β-lactam in a diastereoselective manner [170].
In addition to α,α-disubstituted amino acids, other constrained residues have been described as 310-helix inducers when incorporated into peptide sequences. Among them, 4-carboxy-5-substituted-oxazolidin-2-ones have been described as a new class of pseudo-Pros, able to adopt a subtype of the 310-helix in combination with Ala [171]. α,β-Dehydroamino acids are non-coded amino acids found in a variety of natural products. They contain a C-C double bond between the α- and β-carbon, which limits the conformational freedom of peptides incorporating such residues. In contrast, they are achiral molecules unable to discriminate themselves alone the screw sense in helical structures, so they are usually combined with chiral proteinogenic amino acids. In particular, the alkene residues from α,β-dehydrophenylalanine (ΔPhe) and α,β-dehydroleucine (ΔLeu) were found to promote the formation of β-turns and 310-helices (Figure 10A) [172–178]. Recently, a systematic study by Joaquin et al. [179] was published aiming to compare the effect of small (α,β-dehydroalanine; ΔAla), medium-sized (α,β-dehydro-2-aminobutyric acid; ΔAbu), and bulky dehydroamino acids (α,β-dehydrovaline; ΔVal) on pentapeptide models containing a Pro residue at the i + 2 position. The incorporation of any of these residues at the i + 1 position favors the adoption of a 310-helical shape, whereas a β-sheet-like orientation is observed when incorporated at the i + 3 position.
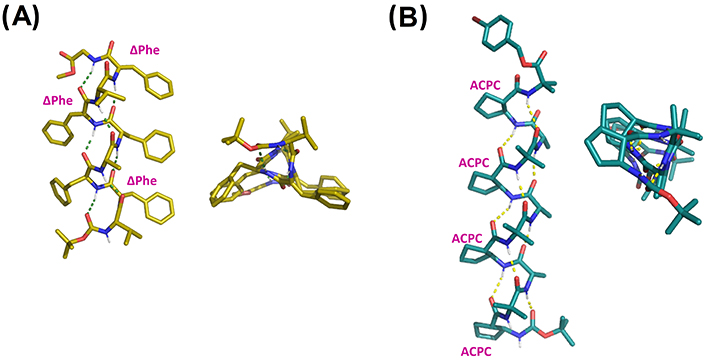
X-ray structures of (A) Boc-Val-ΔPhe-Phe-Ala-Phe-ΔPhe-Val-ΔPhe-Gly-OMe (CCDC 1201496) and (B) Boc-ACPC-(Aib-Ala-ACPC)3-Aib-OPBB (CCDC 685821). ACPC: trans-2-aminocyclopentanecarboxylic acid; Boc: tert-butyloxycarbonyl; PBB: 4-bromobenzyl
Depsipeptides have been also described in the context of 310-helices. They contain one or several ester bonds replacing amide linkages, so they are useful for investigating the contribution of individual H-bonds in the formation of specific secondary structures. A depsipeptide composed of a peptide unit (-Leu-Leu-Ala-)2 and a depsipeptide unit (-Leu-Leu-Lac-)3, with Lac being a lactic acid residue, displaying a 310-helical structure at the connective part between the peptide and the depsipeptide unit [180]. This is due to the disruption of the α-helix H-bond network by the presence of ester bonds.
β-Peptides are derived from β-amino acids. Depending on the substitution pattern at positions 2 and 3, they can adopt helical conformations such as 8-, 10-, 12-, 14-, and 10-/12-helices [181]. The synthesis and conformational studies of hybrid peptides combining α- and β-amino acids have received particular attention in the past few years. Choi et al. [182] obtained X-ray structures of several 1:1 and 1:2 α-/β-peptides, which show a folding pattern comparable to 310-helices, i.e., the formation of an i–i + 3 H-bond pattern (Figure 10B). Other α-/β-, α-/γ-, and γ-peptides have been described to adopt related helical structures [183–186].
Peptide stapling is one of the most established methods for inducing helicity to peptide chains and has been widely used to study protein-protein interaction interfaces [187]. In general, a wide range of covalent linkages have been developed to connect two turns of the same face of the α-helix structure (i and i + 4 or i + 7) [188].
In 310-helices, the side chains displayed on the same face are those at positions i/i + 3/i + 6 so the stapling strategies to stabilize 310-helices have mainly focused on the covalent connection of residues at i and i + 3 positions. The first and simplest example of 310-helix stabilization via stapling involved a lactam bridge from the amide coupling between a glutamic acid and lysine side chains with the referred spacing [189, 190]. Stapling via lactam bridges has been also described in sequences containing the piperidine-derived quaternary amino acid Api (Figure 8). An activated diester linker was used to form a bis-lactam bridge between the piperidine nitrogen of two Api residues at i and i + 3 positions, leading to an increase in the stability of the 310-helix structure [160]. Similarly, a chiral disulfide bridge was developed to connect two Api residues in an achiral peptide. Interestingly, the chiral staple induces the total helical-sense bias of the achiral peptide main chain (Figure 11A) [161].
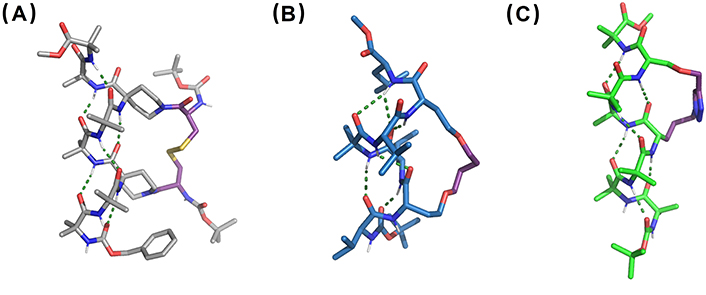
X-ray structures of (A) disulfide-bridged stapled peptide from linear Cbz-(Aib)2-Api(Boc-L-Cys)-(Aib)2-Api(Boc-L-Cys)-Aib2-OMe, Api: CCDC 733323, (B) RCM-stapled peptide derivative from linear Boc-Val-Hse(allyl)-Leu-Aib-Val-Hse(allyl)-Leu-OMe, Hse: homoserine (CCDC 101810), and (C) CuAAC-stapled peptide derived from linear Boc-(Aib)3-Nva(N3)-(Aib)2-Ser(propargyl)-Aib-OMe, Nva(N3): δ-azido-L-norvaline (CCDC 770132). CuAAC: copper-catalyzed azide-alkyne cycloaddition; RCM: ring-closing metathesis
RCM has also been reported to connect two side-chains toward the stabilization of 310-helices [191]. The reaction between two allyl-Ser residues on the same helical face led to cyclic 310-helical peptides, able to retain such conformation both in solution and in the solid state while the acyclic analogue shows a 310- to α-helix transition (Figure 11B) [192]. Click chemistry has also been employed in 310-helix stapling. CuAAC between a propargyl Ser and an ornithine-derived azide with i/i + 3 spacing led to an almost ideal 310-helix structure (Figure 11C) [193]. In general, stapling proved to be an efficient tool to direct the folding of peptide chains into 310-helix conformations. Nevertheless, it is worth mentioning that all the examples here described contain Aib residues, so the starting acyclic peptides have already a certain 310-helical tendency.
A close related methodology for the stabilization of helical conformations is the use of H-bond surrogates (HBS), based on the replacement of the first H-bond for a covalent linkage [194]. In the context of 310-helices, a disordered pentapeptide lacking helicogenic residues is able to display a 310-helix structure when a propyl linker replaces the potential i/i + 3 H-bond [195]. This HBS peptide retains the 310-helix structure at different conditions of temperature and pH, as assessed by NMR and CD. In another work by Banerji et al. [196], RCM was used to connect a pentenoyl and an allyl group at the terminal positions of a tripeptide derivative. This connection leads to the formation of a linker surrogate fourth amino acid and the obtained cyclic peptide displays a pseudo-310-helical structure.
This section covers the use of 310-helical systems in different research fields. The alignment of the i/i + 3 side chains with a spacing of 5.8–6 Å provides a molecular scaffold of interest for the development of functional 310-helices with applications in chemical biology, catalysis, host-guest chemistry, and nanomaterials.
A range of synthetic 310-helical peptides have been developed to target therapeutically relevant receptors. For example, stapling of linear peptides to stabilize 310-helical conformations was successfully applied for the rational design of potential inhibitors of the human sliding clamp (PCNA), a promising anti-cancer target involved in the modulation of DNA replication and repair [197]. A short, conserved peptide motif known as the PIP-box stabilizes a 310-helix structure, key for PCNA binding. Lactam-based stapling was used to constrain this peptide sequence in a 310-helix structure, leading to a peptidomimetic with higher affinity for PCNA than most PIP-box peptides.
Bicycle Therapeutics is a biotechnological company founded by Sir Greg Winter, the 2018 Nobel Prize winner in Chemistry for the pioneering phage-display screening technology. Bicycle Therapeutics pursues the de novo discovery of highly constrained and bioactive bicyclic peptides via phage-display screening [198]. One of their drug candidates in clinical-phase trials is BT5528, which targets ephrin type-A receptor 2 (EphA2) [199]. This receptor is overexpressed in cancer and is linked to malignant progression and poor prognosis. BT5528 is composed of a bicyclic peptide targeting EphA2 and a cytotoxic payload connected by a labile linker. A 310-helical segment, stabilized in the middle of one of the loops of the bicyclic peptide, is the key for the high binding affinity of BT5528 to the target receptor (Figure 12A).
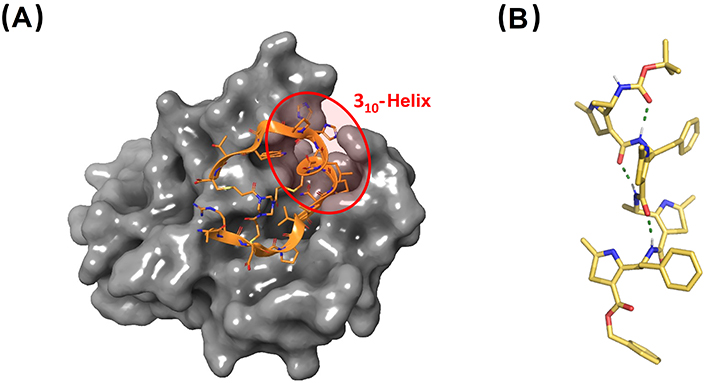
X-ray structures of therapeutically relevant 310-helical peptides. (A) Co-crystal between the bicyclic peptide component of BT5528 and EphA2. The central 310-helix which is key in the interaction is highlighted (PDB ID: 6RW2); (B) γ-peptide foldamer composed of 4-amino(methyl)-1,3-thiazole-5-carboxylic acids (ATC). The structure shown corresponds to the tetramer Boc-(ATC)4-OBn adopting a 9-helix, closely related to the 310-helix conformation (CCDC 922304). Bn: benzyl
γ-Peptides composed of 4-amino(methyl)-1,3-thiazole-5-carboxylic acid residues adopt well-defined 9-helix structures in the solid state and in solution, as shown in Figure 12B [186]. The 9-helix arrangement displays similar topographical features to those found in canonical 310-helices. The helical foldamers were found to act as dual inhibitors of amyloid-β peptide and human islet amyloid polypeptide oligomerization and fibrillization, a process involved in the development of Alzheimer’s disease and type 2 diabetes respectively [200].
D’Addona et al. [201] described the synthesis and binding studies of octreotide analogues to different somatostatin (sst) receptors, which are known to be expressed in human tumors. Octreotide is a mimetic of natural sst used to treat carcinoid syndrome. The replacement of the disulfide bridge of octreotide for an ethylene group derived from RCM cyclisation and reduction led to octreotide analogues with good affinity and high selectivity for the sst5 receptor. Interestingly, their 310-helical propensities correlate with the sst5 selectivity and the suppression of the affinity for sst2.
Several studies have demonstrated that synthetic 310-helical Aib-rich peptides are ionophores when interacting with membranes [202–204]. The mechanism of action of these synthetic foldamers is analogous to the one described for peptaibols, which has been discussed in the previous section entitled “310-Helices in nature”. It has been reported that the foldamer length is more important for the ionophoric activity than their self-assembly tendency. This is the case for Aib homopeptides of 10 residues or longer, which can span the membrane displaying a relevant pore-forming behavior [202–204].
In the last decades, the field of peptide-based organocatalysis has grown significantly with the aim of developing efficient metal-free asymmetric reactions. Among them, the use of peptide catalysts with a defined secondary structure has opened new avenues in the field. In particular, Metrano et al. [205], Metrano and Miller [206] have developed short peptides able to stabilize β-turns as efficient catalysts for asymmetric reactions.
Regarding 310-helices in particular, Rossi et al. [207] reported an azacrown-functionalized Aib-based heptapeptide, able to complex Zn(II) ions at i and i + 3 positions while displaying a 310-helix structure. This complex is active in catalyzing the intramolecular transphosphorylation of an RNA model substrate. The spacing between the two metal centers is determined by the pitch of the helix, which matches the distance between two adjacent phosphate groups [207–209]. In addition, 310-helical peptides have been used as catalysts in enantioselective epoxidation of α,β-unsaturated ketones. In one example, RCM-stapled L-Leu heptapeptides were successfully used as chiral catalysts. Among them, high enantioselectivities of up to 99% enantiomeric excess were obtained with a peptide displaying a right-handed (P) 310-helix in solution (Figure 13A, top channel) [210]. A related example involves a L-Leu hexamer. This peptide is also able to stabilize a 310-helix structure in dimethyl sulfoxide solution, which is again key for the catalytic action in the asymmetric epoxidation of α,β-unsaturated ketones. In the proposed mechanism, the N-terminus acts as an oxyanion hole that interacts with the β-hydroperoxyenolate intermediate via H-bonding (Figure 13A, bottom channel) [211, 212].
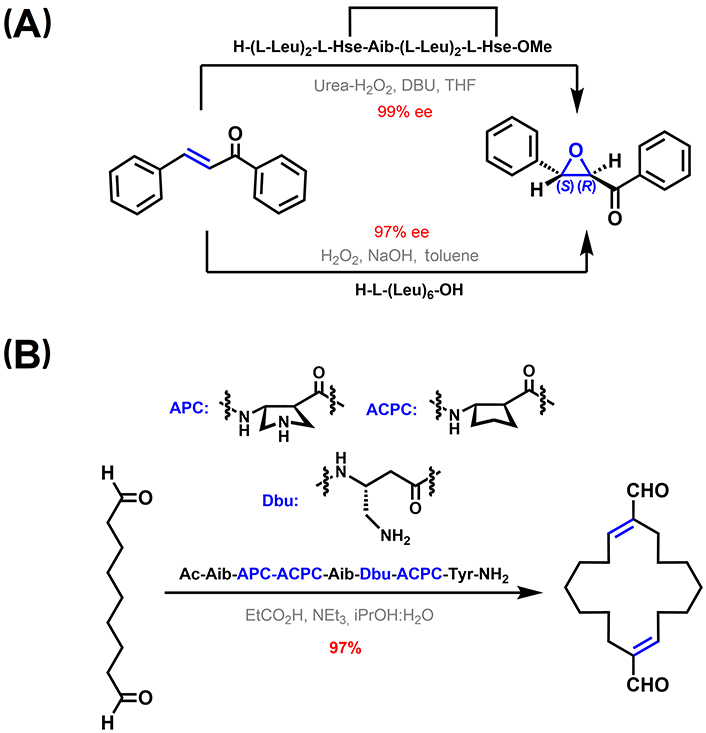
310-Helical peptide-based catalysis. (A) Asymmetric epoxidation of α,β-unsaturated ketones [210–212]; (B) catalytic templated macrocyclization of a dialdehyde using a 310-helical foldamer [213]. APC: trans-4-aminopyrrolidine-3-carboxylic acid; DBU: 1,8-diazabicyclo[5.4.0]undec-7-ene; Dbu: 3,4-diaminobutyric acid; THF: tetrahydrofuran; Tyr: tyrosine
Girvin et al. [213] reported a catalytic covalent template-directed aldol macrocyclization of a linear dialdehyde (Figure 13B). The catalytic template is a hybrid α-/β-peptide foldamer with an αββ backbone pattern containing constrained 5-membered residues. As featured in the previous section entitled “Stabilization of 310-helices in peptides”, this type of peptide foldamers can adopt 310-helix-like conformations, critical for the formation of an enamine and an iminium adduct with the substrate displaying the right geometrical constrains to favor the macrocyclization reaction over potential linear polymerization pathways [213].
Another example, reported by Ghosh et al. [214], is based on a 310-helical peptide dimeric assembly obtained via Cys disulfide bond formation. This peptide scaffold contains an N-terminal tris-(2-pyridylmethyl)amine group, able to form a dimeric Fe(II) complex with modest catalytic activity in the C-H oxidation of cyclohexane.
Aib-derived 310-helical peptides have been also exploited in host-guest chemistry. Of particular relevance is a system based on a nonapeptide with two pendant-substituted hydrophobic Tyr residues separated by two helical turns (i and i + 6 positions). This peptide folds into a 310-helix and is able to selectively recognize grafted fullerenes in the cavity formed in between these hydrophobic residues [215]. When the Tyr residues incorporate ferrocenoyl groups, the peptide is capable of binding N-methylfulleropyrrolidine in non-polar solvents, which suffers rapid deactivation upon photoexcitation (Figure 14A) [216].
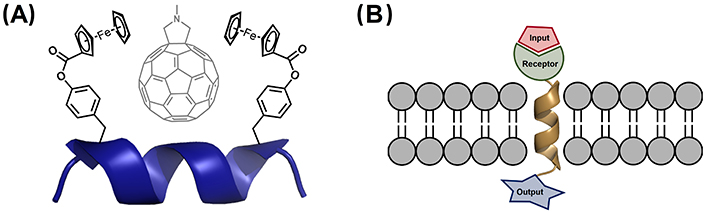
310-Helical scaffolds used in supramolecular chemistry. (A) Complexation of N-methylfulleropyrrolidine by the 310-helical peptide Bz-Aib-L-Tyr(Fc)-(Aib)2-Gly-(Aib)2-L-Tyr(Fc)-Aib-OMe, with Fc being the ferrocenoyl moiety incorporated in the Tyr residue [216]; (B) schematic representation of membrane-bound artificial receptors based on a 310-helical Aib homo-oligomers containing a receptor for binding of the input and a reporter, which provides the output signal across the membrane [217–219]. Bz: benzoyl; Fc: ferrocene
As mentioned in the previous section entitled “Stabilization of 310-helices in peptides”, Aib homo-oligomers have two conformational states, in which the foldamer adopts a global left- or right-handed 310-helix. 217. Solà et al. [217] have demonstrated that chiral N-terminal residues and non-covalent ligands bound to the N-terminus are effective inducers of a screw-sense preference, which is propagated through the entire foldamer length. These features have been exploited to develop membrane-bound synthetic foldamers able to communicate chemical signals across lipid membranes (Figure 14B). In particular, Jones et al. [202] synthesized Aib decamer foldamers, sufficiently long to span a bilayer. Light-induced and ligand-modulated conformational switching of this foldamer triggers end-to-end conformational communication, allowing signal transduction to happen across the membrane [218, 219].
In the field of nanotechnology, 310-helical Aib peptides have been grafted on gold nanoparticles through N-terminal thiol groups. The properties and stabilities of monolayer-protected clusters of gold nanoparticles depend on the nature and interactions of adsorbate molecules. In this context, Aib-peptides retain the 310-helical conformation once assembled on gold nanoparticles [220, 221].
310-Helices represent the third most abundant secondary structure element in proteins. For decades, α-helices have understandably monopolized the interest as the principal helical structure in the study of protein structure, folding, and recognition. Nevertheless, the abundance of 310-helices is not negligible and there is a growing interest in the study of their role in relevant biological processes. This review intends to revisit 310-helices and provide an update on their relevance in protein science. In this sense, it offers an overview of the strategies toward the stabilization of 310-helices, thus providing the tools for developing 310-helical functional biomimetics. The rational design of peptide foldamers using a combination of constrained amino acids and 310-helix-promoting proteinogenic residues seems the most successful approach to develop 310-helical foldamers. Convenient enantioselective synthetic routes are still needed to access more complex quaternary amino acids able to fine-tune the conformation restriction imposed on peptide chains.
310-Helices have been exploited in a wide range of applications, from the design of bioactive compounds to their use as a scaffold of interest in catalysis, supramolecular chemistry, and nanotechnology. The ultimate goal of the present work is to highlight the relevance, sometimes underestimated, of 310-helices in different research areas. The strategies to stabilize 310-helices enable access to a vast region of unexplored protein structure space of particular interest for the design of novel protein biomimetics and assemblies, with applications in biotechnology, cell biology, and synthetic biology.
Another field with growing and interesting future perspectives involving 310-helices is catalysis and template-directed synthesis. The development of small-molecule enzyme mimics with broad synthetic applicability and involving sustainable protocols offers an interesting solution to some of the current challenges in organic synthesis. Thus, this work aims to provide a set of tools for the development of functional 310-helical biomimetics with a broad range of biomedical, synthetic, and material applications.
3D: three-dimensional
Ac: acetyl
Acnc: 1-aminocycloalkane-1-carboxylic acid
Aib: α-aminoisobutyric acid
AKAP: A-kinase-anchoring protein
Ala: alanine
Api: 4-aminopiperidine-4-carboxylic acid
AQP4: aquaporin-4
Boc: tert-butyloxycarbonyl
Cbz: benzyloxycarbonyl
CCDC: Cambridge Crystallographic Data Centre
CD: circular dichroism
Cys: cysteine
EphA2: ephrin type-A receptor 2
FVIII: factor VIII
Gly: glycine
gp41659–671: residues 659–671 of the envelope glycoprotein 41 of human immunodeficiency virus type 1
HIV-1: human immunodeficiency virus type 1
Iva: isovaline
Leu: leucine
NMR: nuclear magnetic resonance
PCNA: proliferating cell nuclear antigen
PDB: Protein Data Bank
Phe: phenylalanine
Phol: phenylalaninol
PIP: proliferating cell nuclear antigen interacting protein
Pro: proline
RCM: ring-closing metathesis
Ser: serine
sst: somatostatin
TOAC: 2,2,6,6-tetramethyl-N-oxyl-4-amino-4-carboxylic acid
Tyr: tyrosine
ΔPhe: α,β-dehydrophenylalanine
The author thanks Dr. Rosario González-Muñiz from IQM-CSIC for helpful advice and discussions.
DNV: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Structural data shown in the Figures of this review article was obtained from publicly available repositories: RCSB Protein Data Bank (www.rcsb.org) and the Cambridge Structural Database (CSD) from the Cambridge Crystallographic Data Centre (www.ccdc.cam.ac.uk). These publicly available repositories allow direct use of the Figure it provides. CIF files were analyzed with Mercury and the images were created with PyMOL (https://pymol.org/pymol.html).
This work is supported by a ComFuturo fellowship from the Fundación General CSIC (European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement [101034263]). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.