Review
Review

Affiliation:
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of Ministry of Education and College of Life Sciences, Nankai University, Tianjin 300071, China
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0005-8395-5244
Affiliation:
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of Ministry of Education and College of Life Sciences, Nankai University, Tianjin 300071, China
†These authors contributed equally to this work.
ORCID: https://orcid.org/0009-0003-9300-0965
Affiliation:
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of Ministry of Education and College of Life Sciences, Nankai University, Tianjin 300071, China
ORCID: https://orcid.org/0009-0000-1623-2285
Affiliation:
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of Ministry of Education and College of Life Sciences, Nankai University, Tianjin 300071, China
Email: chenq@ioz.ac.cn
ORCID: https://orcid.org/0000-0001-7539-8728
Affiliation:
State Key Laboratory of Medicinal Chemical Biology, Key Laboratory of Bioactive Materials of Ministry of Education and College of Life Sciences, Nankai University, Tianjin 300071, China
Email: zhangcq@nankai.edu.cn
ORCID: https://orcid.org/0000-0001-8674-880X
Explor Drug Sci. 2024;2:154–189 DOI: https://doi.org/10.37349/eds.2024.00041
Received: October 17, 2023 Accepted: January 10, 2024 Published: April 18, 2024
Academic Editor: Xuechen Li, The University of Hong Kong, China
The article belongs to the special issue Bioactive Peptides discovery and development
Protein-protein interactions (PPIs) impersonate a significant role in many biological processes and are potential therapeutic targets in numerous human diseases. Stapled peptides, as the most promising therapeutic candidate for interfering with PPIs, have a higher degree of α-helicity, improved binding affinity, more resistance to proteolytic digestion, longer serum half-life, and enhanced cell permeability, which exhibits higher pharmacological activity compared with small molecule drugs and biologics. This review outlined the continuous progress of stapled peptides mainly concerning the design principle, structural stability, bioactivity, cell permeability, and potential applications in therapeutics, which is aimed at providing a broad reference for the design and exploration of stapled peptides with enhanced biological and pharmacokinetic properties as the next-generation therapeutic peptide drugs targeting various diseases.
Protein-protein interactions (PPIs) represent important nodes in cellular signaling networks, mediating numerous vital biological processes, such as molecular processes, signal pathways, and cellular functions, and their misregulation is often correlative with human diseases as an important consideration of therapeutic interventions against diseases [1–3]. Emerging pieces of evidence vindicate that targeting PPIs is a promising strategy for detecting, modifying, and orchestrating cellular functions, accompanied by various approaches to drug discovery [4]. Currently, PPI drugs that have been approved for clinical use are mainly divided into small-molecule drugs and biologics (including proteins and antibodies), for example, navitoclax and its analogs that prevent the interaction between B-cell lymphoma-2 (BCL-2) and proapoptotic proteins BCL-2 homologs antagonist/killer (BAK) and BCL-2-associated X-protein (BAX) [5] and the antibody trastuzumab that are used to block the human epidermal growth factor receptor-2 (HER2) receptor [6]. Small molecule drugs have always been the preferred molecules for the development of intracellular target drugs due to their good stability, high oral bioavailability, and enhanced cell penetrability. One of the enormous advantages of small molecule drugs is the identification of the “hot spot” residues that are essential to the interaction domain, which leads to precise target orientation. However, small molecule drugs have the problem of relatively low protein binding affinity and poor drug compatibility, due to the large and discontinuous interfaces of most PPIs [5, 7–12]. Besides, biologics have a high degree of specificity and affinity for therapeutic receptors owing to the extensive area of interaction with targets through established motifs, but they suffer from low cellular membrane permeability, poor oral bioavailability, and metabolic instability, resulting in limited intracellular applications [13, 14]. The limitations of the above therapeutics make it urgent to develop the next generation of therapeutic modalities.
With the easily designed structure and appropriate molecular size between small molecules and large proteins, peptide therapeutics displayed a high binding affinity with the PPI interface and good cell penetrability, which seem to be promising candidates that combine the advantages of the above two therapeutic arsenals (Table 1) [15–20]. However, there has been a fatal problem that peptides cannot maintain structural and functional integrity due to the tendency to lose native conformation while lacking the structural reinforcement available from the other parts of the protein [21, 22]. As the most abundant protein secondary structure found in nature, α-helix represents the typical recognition motif at PPI interfaces, which is intrinsic to many PPIs that mediate important biological functions, in addition, approximately 60% of PPIs in Protein Data Bank (PDB) adopt the α-helix or similar to helical conformations [23, 24]. α-Helical structures endow peptides with the powerful capability of binding to target proteins involved in α-helix-mediated PPIs as the modulators of PPI, which is essential to pre-organize into the bound conformations. Based on the reasons above, it is vitally important and imperative to investigate methods for stabilizing the α-helix conformation of peptides and, thereby, restoring the binding affinity towards the targeted proteins. Through continuous exploration and development, various modified peptides with stable helical structures have developed as PPI modulators, among which stapled helical peptides (hereinafter called stapled peptides) have attracted much interest owing to their more stable structure and unique bioactive properties [25–36].
The comparison properties of three classes of therapeutic molecules
| Properties | Small molecules | Stapled peptides | Biologics |
|---|---|---|---|
| Molecular weight | < 1,000 | 1,000–5,000 | > 10,000 |
| Stability | High | High | Low |
| Binding affinity | Low | High | High |
| Specificity | Low | High | High |
| Cellular permeability | High | High | Low |
| Proteolysis resistance | High | High | Low |
| Toxicity/side effects | High | Low | Low |
| Ability to disrupt PPIs | Low | High | High |
| Manufacturing cost | Low | Low | High |
The concept of “stapled peptides” was first proposed by Schafmeister and co-workers [37] in 2000, which has been rapidly developed to become the main force for enhancing the stability of structures and resistance to hydrolysis of peptides over the past two decades. Initially, stapled peptides mainly referred to those α-helical peptides supported by all-hydrocarbon stapling [38]. More broadly, the concept of stapled peptides hitherto has been extended to incorporate all the α-helical peptides that can be constrained via various cross-linking strategies [26, 39–44]. The stapled peptides with pre-organized stable α-helical conformations usually have a thermodynamically favorable process when interacting with molecular targets, giving rise to enhanced binding affinity [45–60]. Additionally, stapled peptides also represent a significant improvement in pharmacologic performance, which is manifested in more resistance to proteolytic digestion, longer serum half-life, and higher levels of cell permeability [61–68]. Furthermore, there have been reports that the side chains of cross-linkers may also interact with the interfaces of PPIs, which can provide additional binding interfaces and improve binding affinity [69, 70]. Up to now, stapled peptides have shown immense potential in therapeutic drug discovery.
Previous reviews have mostly focused on the rational design of different anchoring residues or the specific application in drug discovery. This review will systematically discuss the recent advances of the stapled peptides with various forms of constraints as therapeutic candidates with respect to the design principles, structural stability, bioactivity, cell permeability, and potential applications in therapeutics. This work aims to provide references for the rational and innovational design of stapled peptides, including but not limited to the optimization of cross-linkers and the introduction of additional functional groups, with the hope of exploring the therapeutic stapled peptides with enhanced biological and pharmacokinetic properties.
Obtaining the right amino acid sequences is a primary and fundamental step in the design of stapled peptides [42]. Firstly, the amino acid sequence is crucial for the functionality and structure of stapled peptides. Accurate design of the amino acid sequence allows stapled peptides to fold into an ideal spatial conformation, ensuring effective interactions with specific receptors and achieving therapeutic effects [38]. Secondly, the amino acid sequence also determines the position of key residues and functional groups on the stapled peptide, influencing bioactivities such as the binding affinity and selectivity towards targeting molecules [26]. Lastly, precisely designing the amino acid sequence may enhance the therapeutic effect while reducing potential toxicity by minimizing interactions with non-target molecules [24]. This section delves into classic cases and recent advances in the design of helical stapled peptides, providing a summary of the techniques employed to acquire these essential amino acid sequences.
The most direct approach to obtaining an amino acid sequence for the early development of stapled peptide-based PPI inhibitors is by excising a short segment from the natural PPI interface. This segment should be as short as possible while still maintaining sufficient binding affinity to compete with the natural ligand for the receptor binding site. For instance, forkhead box protein P3 (FOXP3) homodimer serves as an essential PPI for the development, preservation, and suppressive function of regulatory T (Treg) cells. Guided by the crystal structure of the FOXP3 homodimer, Hawley and colleagues [68] devised a set of hydrocarbon-stapled (HCS) α-helical (SAH) peptides. These peptides, resembling a segment of the native FOXP3 antiparallel coiled-coil homodimerization domain (SAH-FOXP3), were aimed at disrupting the crucial FOXP3 PPI (Figure 1A). They demonstrated that the SAH-FOXP3s penetrate cells and effectively block the FOXP3 PPI, which elicits alterations in Treg cell gene expression in vivo and hence hinders the immune suppression functionality of Treg cells. For another example, the human angiotensin-converting enzyme 2 (hACE2)/spike PPI plays a pivotal role in the internalization process of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). A structural element on hACE2 named α1-helix involved most of the hACE2/spike PPI relative residues, also called the minimal fragment angiotensin-converting enzyme 2 (ACE2; 24–42). Due to the relatively short sequence length and defined helical conformation, the minimal fragment ACE2 (24–42) becomes a perfect choice of peptide sequence for developing inhibitors to disturb the hACE2/spike PPI and stop SARS-CoV-2 invading normal cells based on peptide stapling techniques. Quagliata and co-workers [71] introduced one or two triazole staples at different positions in the peptide based on copper(I)-catalyzed azide–alkyne cycloaddition (CuAAC), resulting in a competitive binder of the minimal ACE2 fragment with enhanced helicity in the bound state.
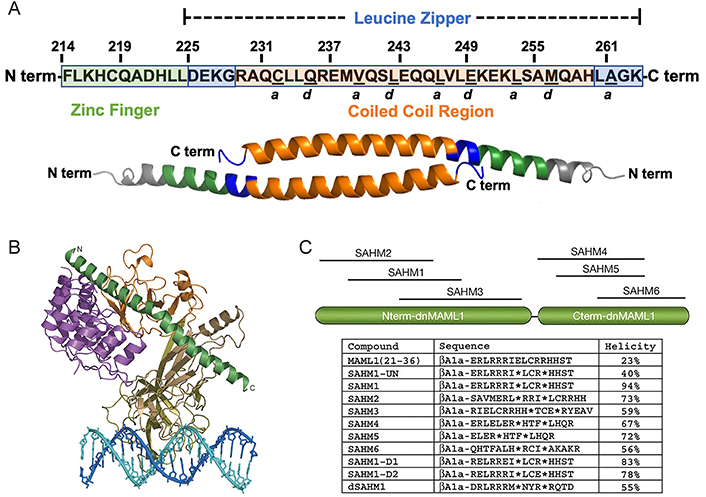
Stapled peptide inhibitors derived from natural PPI interfaces. (A) Human FOXP3 sequence. The segment from F214 to K263 comprises the complete helix encompassing the zinc finger (F214–L224; depicted in green) and leucine zipper (D225–K263; depicted in blue) regions. Within the leucine zipper, the coiled-coil (R229–H259; depicted in orange) corresponds to an area highly conserved between human and murine FOXP3. Residues underlined denote the “a” and “d” core residues, crucial for binding each other on an identical antiparallel helix and serving as key contact points for homodimerization; (B) overall structure of the mastermind-like transcriptional coactivator 1 (MAML1):ankyrin (ANK):CSL:DNA complex. The ANK domain is colored purple; the MAML1 polypeptide is colored dark green; and the RHR-N, b-trefoil, and RHR-C domains of CSL are colored light brown, gold, and orange, respectively. The two DNA strands are colored blue and cyan; (C) schematic, sequences, and helicity characteristics of MAML1-derived SAHM peptides. Locations of the incorporated hydrocarbon staple in all SAHM peptides except modified but unstapled peptide (SAHM1-UN), are indicated by asterisks. The βAla signifies a β-alanine spacer
Note. Figure 1A was adapted from “Inhibition of FOXP3 by stapled alpha-helical peptides dampens regulatory T cell function,” by Hawley KM, Eclov RJ, Schnorenberg MR, Tian Y, Shah RN, Thomas-Toth AT, et al. Proc Natl Acad Sci U S A. 2022;119:e2209044119 (https://doi.org/10.1073/pnas.2209044119). CC BY-NC-ND. Figure 1B was adapted with permission from “Structural basis for cooperativity in recruitment of MAML coactivators to Notch transcription complexes,” by Nam Y, Sliz P, Song L, Aster JC, Blacklow SC. Cell. 2006;124:973–83 (https://doi.org/10.1016/j.cell.2005.12.037). © 2006 Elsevier Inc. Figure 1C was adapted with permission from “Direct inhibition of the NOTCH transcription factor complex,” by Moellering RE, Cornejo M, Davis TN, Del Bianco C, Aster JC, Blacklow SC, et al. Nature. 2009;462:182–8 (https://doi.org/10.1038/nature08543) ©2009 Macmillan Publishers Limited.
A viable strategy was reported which involves the creation of peptide libraries with varying sequence lengths and strategic position selection in the truncation from the PPI interface. It has been proven to be a feasible approach to establish peptide libraries with different lengths when uncertain about how to excise the amino acid sequence from the natural PPI interface to achieve optimal receptor affinity. A notable example was reported by Moellering and co-workers [72], in which they discussed a series of HCS peptides targeting a critical PPI in an oncogenic transcription factor (TF), NOTCH1’s transactivation complex. Inappropriate NOTCH activation is directly linked to the pathogenesis of T-cell acute lymphoblastic leukemia (T-ALL) [73]. A fragment of MAML1 forms a continuous α-helix at the assembly interface of the NOTCH transactivation complex (Figure 1B) [74]. Through truncating and modifying the sequence of MAML1, a library of stapled peptides called SAHM was created by using the ring-closing metathesis (RCM) method (Figure 1C). Compared to unmodified and modified but unstapled counterparts, the stapled peptides demonstrated a remarkable increase in helical character, resulting in robust cellular uptake with enhanced binding affinity [72].
The rational design of amino acid sequences for stapled peptides emerges as a paramount approach when scientists aim to acquire stapled peptides with enhanced binding affinity, stability, and specificity towards target receptors beyond the natural ligands, or when the target receptor has undergone mutations that preclude the derivation of valuable design cues from natural ligands. This process entails the elucidation of the spatial conformation and distribution of amino acid residues within the interaction site on the receptor, along with a comprehensive understanding of the underlying mechanisms and principles governing PPIs. The first stage usually involves analyzing the conformation of the binding site on the target receptors through X-ray diffraction and molecular graphics software like PyMOL [75]. Next, the process of optimization encompasses two fundamental aspects, with the initial one focusing on the incorporation of amino acid residues capable of more efficiently occupying the binding pocket of receptors within a three-dimensional (3D) context. Meantime, it encompasses the fortification of influential interactions between key interacting residues on the receptor, including but not limited to π-π packing, cation-π interactions, electrostatic attraction, and salt bonding. Additionally, computational studies, such as molecular dynamics simulations and molecular docking, provide valuable information regarding the interaction of stapled peptides with their target proteins, which can predict the binding free energy and favorable binding orientation, and identify essential residues involved in interactions [76], leading to more insights into critical interaction motifs. Based on the above, it becomes feasible to enhance the affinity of a stapled peptide with its receptor by experimental validations, such as alanine scanning mutagenesis, isothermal titration calorimetry, and surface plasmon resonance, which can be employed to evaluate the binding affinity of the designed peptide and determine whether the predicted interactions are consistent with experimental observations. More importantly, iterative approaches to peptide sequence optimization are urgently needed to be developed, due to the introduction of amino acid substitutions and additional stabilizing modifications to further enhance the binding affinity and specificity. Interestingly, the incorporation of non-canonical amino acids, backbone N-methylation, or alternative staple designs can further modulate the physicochemical properties of peptides and extend the therapeutic potential.
In recent years, artificial intelligence (AI) has emerged as a prominent tool in the field of drug design [77]. It combines computer science, machine learning, and bioinformatics, and is widely applied throughout various stages of drug discovery and design [78]. AlphaFold, as a leading representative in this field, is a protein structure prediction system that utilizes deep learning and neural network technologies to accurately predict the 3D structures of proteins by learning from extensive data on protein sequences and structures [79]. Modi et al. [14] designed a stapled peptide named N1S based on predictions of the interactions between nuclear respiratory factor 2 (Nrf2) and small musculoaponeurotic fibrosarcoma (sMAF) homolog G (MafG) via AlphaFold. Since there are no crystal structures of the Nrf2/MafG interaction available to guide the design of the Nrf2 peptides, they utilized AlphaFold to predict the structure of the Nrf2/MafG heterodimer, then used the AlphaFold model to determine the best region to target for inhibition and designed three 16-amino-acid peptides that cover the predicted binding domain, they also used the model to determine the best locations for staples, specifically focusing on locations unlikely to interrupt the binding of the N1S to MafG. A subsequent cell-based reporter assay combined with in vitro biophysical assays demonstrated that N1S directly inhibits Nrf2/MafG heterodimerization. Overall, the introduction of AI technologies such as AlphaFold has brought about more efficient, accurate, and innovative approaches to drug design, holding the potential to accelerate the discovery and development of new drugs and offering tremendous opportunities in the pharmaceutical field.
Subsequent modifications focus on two aspects after obtaining the original peptide sequence bearing critical residues for PPIs. Firstly, introduce more non-covalent bonds to reinforce the α-helix conformation and enhance metabolic stability. Secondly, modify critical residues to increase the affinity of the peptide for the target. Free energy perturbations (FEPs)-based approaches such as virtual alanine scans have been successfully adapted to the design of stapled peptides [80], similar to their application in the development of small molecule binders and predicting binding pockets [75]. For instance, to disrupt the binding of Wiskott-Aldrich syndrome protein family (WASF) regulatory complex (WRC) to the WASF3 (Figure 2A), Limaye et al. [81] performed an in-silico alanine scan on the native sequence within an online platform for peptide-based PPI inhibitors design called Rosetta to identify individual residues that were predicted to energetically contribute to the binding interface (Figure 2B). Combining with the structural information, this technique could be used to identify positions where the olefinic amino acids could be introduced along the nonbinding interface to ultimately form the hydrocarbon staple (Figure 2C). In conclusion, the in silico alanine scan approach is a valuable tool in understanding protein structure-function relationships and guiding stapled peptide-based PPI inhibitor design by identifying potential hotspots for drug targeting within a protein.
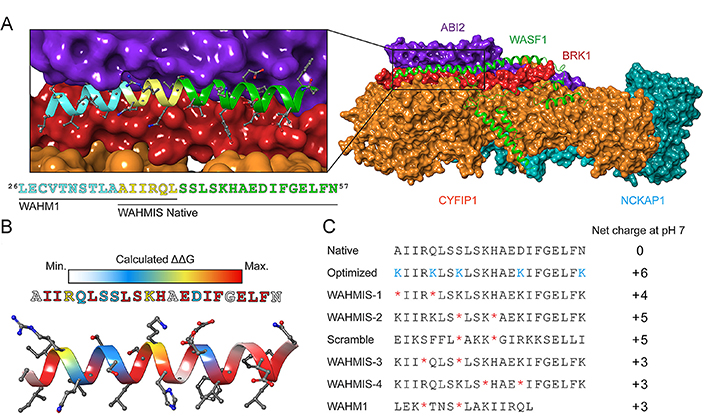
Design principle of stapled peptides derived from WASF. (A) Structure of the WRC. BRICK1 subunit of SCAR/WAVE actin nucleating complex (BRK1, shown in red), cytoplasmic FMR1 interacting protein 1 (CYFIP1, shown in orange), Nck associated protein 1 (NCKAP1, shown in cyan), Abelson interactor 2 (ABI2, shown in purple), and WASF1 (shown in green); (B) heatmap showing the binding energy contribution of each residue in the WASF3-derived peptide, which was calculated by in silico alanine scanning using Rosetta; (C) a library of stapled peptides by placing olefinic amino acid (*) at i, i + 4 positions and lysine (Lys) replacement (K) to achieve positive net charge (NC) at positions that were predicted to have almost no influence on protein-binding interfaces. WHM1/2: WASF helix mimics 1/2; WAHMIS: WAHM in silico
Note. Reprinted with permission from “In silico optimized stapled peptides targeting WASF3 in breast cancer,” by Limaye AJ, Bendzunas GN, Whittaker MK, LeClair TJ, Helton LG, Kennedy EJ. ACS Med Chem Lett. 2022;13:570–6 (https://doi.org/10.1021/acsmedchemlett.1c00627). © 2022 American Chemical Society.
As the core module of stapled peptides, the staple is focused on the design process, in which properties such as structural characteristics, physicochemical properties, and biological effects play a pivotal role. While scientists have identified some general principles for the incorporation of staples, it is necessary to tailor the approach to specific peptide-protein interfaces, given the unique context of each interaction. Firstly, the position of the staple is the primary consideration, since the correct introduction of the staple can confer enhanced structural stability, which serves as the foundation for the functional stability of stapled peptides. Besides, careful selection of the reaction type that forms the staple is another critical consideration, for different reactions necessitate varying reaction conditions to impose specific requirements on the amino acid residues within stapled peptides. Furthermore, it can also be divided into one-component and two-component strategies based on the composition of staples. One-component strategies offer the advantage of simplicity and rapidity, while two-component strategies enable the introduction of different types of staples with diverse properties and functionalities like plugins, and these multifunctional peptide staples greatly expand the potential applications of stapled peptides. Therefore, this chapter will primarily focus on three key aspects: position of the staple, reaction type for forming staples, and composition of the staple.
The position of staples plays a critical role in stabilizing the secondary structure and enhancing the bioactivity of stapled peptides. For efficient cross-linking, the residues involved in forming the covalent staples must be located on the same side of the helix [82]. Given that α-helix has 3.6 residues per turn, the most common patterns of inducing covalent cross-links between the side chains of amino acid residues in stapled peptides are separation by approximately one (i and i + 4 positions), two (i, i + 7), or three turns (i, i + 11) of the helix (Figure 3A) [83]. This approach has been proven to significantly enhance α-helicity, structure stability, binding affinity, protease resistance, and cell permeability of stapled peptides [45].
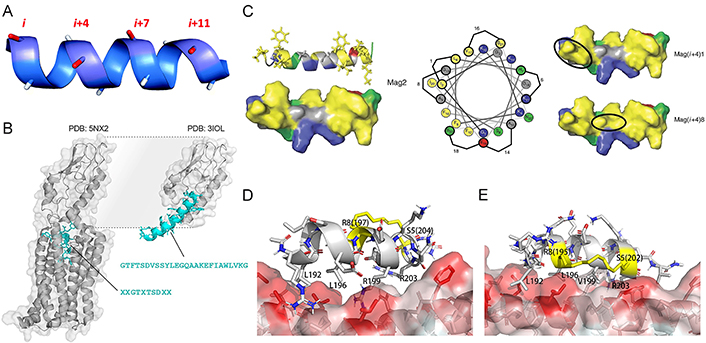
The position of staples plays a critical role in the properties of stapled peptides. (A) The spatial position of i, i + 4/7/11 locating amino acids on a helical peptide; (B) the structure of glucagon-like peptide 1 (GLP-1)’s N-terminus is buried in a complex with GLP-1 receptor (GLP-1R; PDB: 5NX2; left) and its interaction with the extracellular domain of GLP-1R (PDB: 3IOL; right); (C) the hydrophobic face of the amphipathic magainin II (Mag2) α-helix (PDB ID: 2MAG; left), which contains two patches of highly hydrophobic residues (yellow) separated by a small area of relative hydrophily (gray). Cationic residues are shown in blue, hydrophilic residues in green, and anionic residues in red on the structures and the helical wheel. The hydrocarbon staple (shown in a black oval) is located in superposition with a patch of high hydrophobicity such as in Mag(i + 4)1 (right up). The staple connects the highly hydrophobic patches into a continuous region of high hydrophobicity such as in Mag(i + 4)8 (right down). Staple locations for each stapled antimicrobial peptide (StAMP) are indicated on the helical wheel (middle); (D) binding mode of the SP4 to beclin 1 coiled-coil domain obtained in the previous study; (E) the newly designed i7-01s scaffold which the hydrocarbon staple is colored in yellow position close to the hydrophobic coiled-coil surface
Note. Figure 3A was reprinted with permission from “Peptide stapling techniques based on different macrocyclisation chemistries,” by Lau YH, de Andrade P, Wu Y, Spring DR. Chem Soc Rev. 2015;44:91–102 (https://doi.org/10.1039/c4cs00246f). © The Royal Society of Chemistry 2015. Figure 3B was adapted with permission from “Hydrocarbon-stitched peptide agonists of glucagon-like peptide-1 receptor,” by Bird GH, Fu A, Escudero S, Godes M, Opoku-Nsiah K, Wales TE, et al. ACS Chem Biol. 2020;15:1340–8 (https://doi.org/10.1021/acschembio.0c00308). © American Chemical Society. Figure 3C was adapted with permission from “Design of stapled antimicrobial peptides that are stable, nontoxic and kill antibiotic-resistant bacteria in mice,” by Mourtada R, Herce HD, Yin DJ, Moroco JA, Wales TE, Engen JR, et al. Nat Biotechnol. 2019;37:1186–97 (https://doi.org/10.1038/s41587-019-0222-z). © The Author(s), under exclusive licence to Springer Nature America, Inc. Figure 3D and E was adapted with permission from “Optimization of beclin 1-targeting stapled peptides by staple scanning leads to enhanced antiproliferative potency in cancer cells,” by Yang Q, Qiu X, Zhang X, Yu Y, Li N, Wei X, et al. J Med Chem. 2021;64:13475–86 (https://doi.org/10.1021/acs.jmedchem.1c00870). © American Chemical Society.
It is crucial to prevent the replacement or blockage of key interaction residues in the staple. A straightforward strategy that involves situating the staple-forming residues on the solvent-exposed side of peptides in protein-peptide interactions was conducted to achieve this goal, which ensures the preservation of PPI interfaces without disruption [84]. For instance, in the quest to develop alternative GLP-1R agonists for glycemic control, Bird and co-workers created an i, i + 7 hydrocarbon staple scanning library of GLP-1 to study its full functional GLP-1R binding activity [85]. However, they found that ninety percent of N-terminal staple positions were not compatible with GLP-1R binding and internalization. The N-terminus of GLP-1 must remain buried within the receptor to facilitate the interaction, which is the reason for the incompatibility (Figure 3B). Staples placed near the N-terminus of GLP-1 will cause inadequate steric hindrance, preventing the N-terminus of GLP-1 from burying within the receptor, thereby disrupting the agonistic function of GLP-1.
It is a good idea to choose suitable positions along the helical peptide for the placement of staples. Placing the hydrophobic hydrocarbon staples on the native hydrophobic regions of helix peptides, without violating the preceding rule, can preserve the natural performance of peptides to a great extent. Hemolytic activity is a serious issue in cationic antimicrobial peptides (AMPs) and is related to their hydrophobicity [86]. In their efforts to develop a series of StAMPs with limited nonspecific membrane toxicity based on Mag2, Mourtada and co-workers [87] discovered that when the hydrocarbon staple was positioned within one of the high hydrophobicity patches on the AMPs helical, the hemolytic activity remained similar to the native AMP. However, when the low hydrophobicity region was replaced with the hydrocarbon staple moiety, the hydrophobic area was integrated and became bigger and more hydrophobic, leading to a significant increase in hemolytic activity (Figure 3C). Furthermore, staples placed at the binding interface can enhance binding affinity by forming a noncovalent bond between the partially exposed staple and the protein [69]. For coiled-coil domains in the PPI interface that lack conventional binding sites, a promising approach to enhance the binding affinity of stapled peptides is to position the hydrocarbon staple closer to the coiled-coil surface and incorporate hydrophobic residues near the staple, effectively expanding the hydrophobic binding interface. In the case of a series of stapled peptides with the hydrocarbon staple positioned closer to the beclin 1-peptide interface (Figure 3D and E), their binding affinity was significantly enhanced by approximately 10- to 30-fold [88].
Schafmeister and co-workers [37] introduced the pioneering all-hydrocarbon peptide stapling strategy using olefin RCM (Figure 4A), which was inspired by a similar approach developed by Blackwell and Grubbs [89]. This strategy involves incorporating unnatural α-methyl-substituted amino acids with alkene residues of varying lengths on the same side of the helix. Subsequently, ruthenium-mediated RCM is applied to the resin-bound material to form macrocyclic hydrocarbon cross-links across either one or two turns (i and i + 3/4, or i + 7 positions, respectively) of the helix. The efficiency of RCM is highly influenced by the ring size of the macrocyclic cross-linking. Generally, a larger ring size typically results in a higher RCM conversion rate, and in certain cases, the addition of one carbon unit can increase the RCM conversion rate by nearly 50% [37, 90]. Regarding stereochemistry, the most preferred cross-link in i, i + 4 stapling is formed by two units of S-configurated α-methyl,α-pentenylglycine residues, which is referred to as the S(i, i + 4)S(8) staple. RCM of two R-configurated amino acids forming an R(i, i + 4)R(8) staple also proceeds efficiently, but this particular staple neither stabilizes helices well nor provides peptides with high cell-penetrating ability, and RCM reactions using one R- and one S-configurated amino acid are found to be very inefficient [83]. All-hydrocarbon peptide stapling strategy based on RCM is still the most popular approach in peptide stapling nowadays, however, there are limitations to the method. For instance, the yield of the RCM product would be low if the side-chain orientation of the alkene pair is not in close proximity, and the separation of Grubb’s catalyst from the product by high-performance liquid chromatography (HPLC) is difficult, it is also challenging to control E/Z isomerism and distinguish between the isomers by solution nuclear magnetic resonance (NMR) spectroscopy [91, 92].
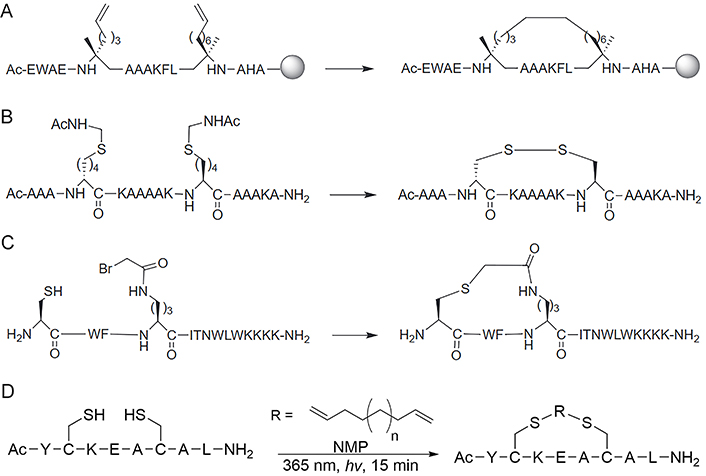
Different reaction strategies for the synthesis of staples. (A) RCM or RCM reaction for the synthesis of the all-HCS peptides; (B) disulfide staple formed between D and L amino acids bearing thiol-side chains; (C) thioether crosslink formed between cysteine (Cys) thiol and α-bromo amide groups; (D) the thiol-ene stapling system allows a diene of unspecified length to couple two Cys thiols in N-methylpyrrolidone (NMP) to cyclize through the tether
Note. Figure 4A, B, and C was reprinted from “Stapled peptides inhibitors: a new window for target drug discovery,” by Ali AM, Atmaj J, Van Oosterwijk N, Groves MR, Dömling A. Comput Struct Biotechnol J. 2019;17:263–81 (https://doi.org/10.1016/j.csbj.2019.01.012). CC BY-NC-ND. Figure 4D was adapted with permission from “Macrocyclic control in helix mimetics,” by Guarracino DA, Riordan JA, Barreto GM, Oldfield AL, Kouba CM, Agrinsoni D. Chem Rev. 2019;119:9915–49 (https://doi.org/10.1021/acs.chemrev.8b00623). © 2019 American Chemical Society.
Disulfide bonds have exerted a significant influence on the folding and structural stability of native proteins, typically originating from cysteine residues in oxidizing environments. These bonds were deliberately selected by researchers during the initial investigations to modulate the conformation of proteins (Figure 4B) [15, 93, 94]. Cysteine can undergo a variety of chemical modifications through nucleophilic substitution and other chemical reactions with various electrophilic reagents due to the strong electron-rich nature of thiols (Figure 4C) [95, 96]. Cysteine residues used in stapling can be inherent or easily incorporated at the right position in standard solid-phase peptide synthesis (SPPS). After purification, the peptides need to co-incubate with reducing reagents, such as tris(2-carboxyethyl)phosphine (TCEP), DL-dithiothreitol (DTT), and glutathione reduced (GSH) to ensure the reduction of any disulfide [41], followed by cyclization with appropriately functionalized cross-linkers in solution. Cysteine-mediated two-component peptide stapling is a strong competitor to the all-hydrocarbon stapling strategy now (Figure 4D) since it can introduce staples with diverse functionalities, such as aromatic, perfluoroaromatic, aliphatic, ketone, maleimide, and tetrazine linkages [55, 70, 97–100].
Forming a lactam crosslink between endogenous residues Lys and aspartic acid (Asp)/glutamic acid (Glu) by amidation reaction is the first stapling strategy of peptide (Figure 5A) [101], which has been widely used nowadays since there is no need for expensive unnatural amino acids or specialized catalytic conditions. Although Lys, Asp, and Glu appear with great frequency in natural protein sequences [41], the lactamization between specific residues can be achieved by incorporating amino acids with different protecting groups in SPPS, the deprotection of these amino acids can programmatically proceed under different chemical conditions [41, 102]. For instance, Wegener et al. [103] incorporated covalent i, i + 4 lactam linkages to constrain the peptides derived from the p21 into the 310-helical structure required for proliferating cell nuclear antigen (PCNA) inhibiting. The stapled peptide is designed based on p21, named ACR1 and ACR2. The side chains of Glu and Lys for lactamization were protected by hydrazine-labile groups namely Fmoc-Glu(Dmab)-OH for position 149, Fmoc-Dab(ivDde)-OH (ACR1) and Fmoc-Lys(ivDde)-OH (ACR2) for position 145 respectively. After the completion of peptide synthesis, the peptide resin was treated with 2% hydrazine/N,N-dimethylformamide (DMF; 2 × 5 min) to remove the Dmab and the ivDde protecting groups simultaneously, which was fully orthogonal to chemical conditions used in the deprotection of other residues. The lactam bond between positions 145 and 149 was then formed by activation with PyBOP, and the peptide was finally deprotected and cleaved from the resin by treatment with trifluoroacetic acid (TFA)/triisopropylsilane/H2O = 95/2.5/2.5 (v/v/v). Lactam stapling can enhance both the helicity of peptides and the stability of the helix, and the induction of helix by lactam crosslinks depends on the length of the lactam linker, the position of the linking residues in a peptide sequence, and the placement and orientation of the amide moiety along the cross-link [104].
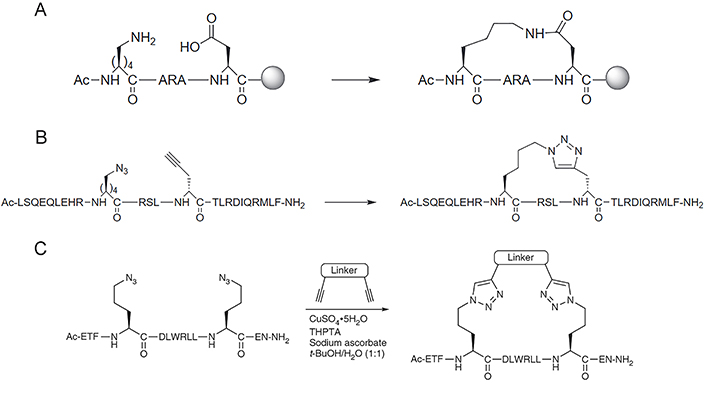
Synthesis of staples by lactamization and CuAAC “click” reaction. (A) Lactamisation stapling between Lys and Asp residues; (B) the CuAAC stapling strategy is achieved by generating a 1,4-substituted 1,2,3-triazole linker formed between azido and alkynyl group; (C) the two-component CuAAC “click” reaction stapling strategy allows incorporating various crosslinks with different chemical properties and functionalized modifications through double “click” reactions under copper catalyzation. THPTA: tris(3-hydroxypropyltriazolylmethyl)amine
Note. Figure 5A and B was reprinted from “Stapled peptides inhibitors: a new window for target drug discovery,” by Ali AM, Atmaj J, Van Oosterwijk N, Groves MR, Dömling A. Comput Struct Biotechnol J. 2019;17:263–81 (https://doi.org/10.1016/j.csbj.2019.01.012). CC BY-NC-ND. Figure 5C was reprinted with permission from “A two-component ‘double-click’ approach to peptide stapling,” by Lau YH, Wu Y, de Andrade P, Galloway WR, Spring DR. Nat Protoc. 2015;10:585–94 (https://doi.org/10.1038/nprot.2015.033). © 2015 Nature America, Inc.
The CuAAC, known as Huisgen cycloaddition, is the classic click reaction that has also been widely used in the field of stapled peptides [105]. The reaction is achieved by anchoring non-natural amino acids with azido and alkynyl groups at appropriate positions and generating a 1,4-substituted 1,2,3-triazole linker through copper catalyzation (Figure 5B). The driving force of the reaction is the formation of the triazole ring, which displays great stability due to the aromatic properties of the ring (∆G° = –61 kcal mol–1) [106]. Compared with other stapling strategies, triazole stapling is better in terms of biocompatibility since the functional groups involved in the reaction are orthogonal to other functionalities present in the natural cellular environment, and the copper-catalyzed mild chemical conditions for the reaction [107]. Quagliata et al. [71] reported that the triazole stapling of a short peptide derived from the α1-ACE2 helix can stabilize its helical conformation, and improve its antiviral activity against SARS-CoV-2 relative to the native peptide. The triazole stapling strategy was extended to be used in the construction of two-component stapled peptides (Figure 5C). Due to the high reaction specificity and orthogonality of the CuAAC “click” reaction, this two-component stapling strategy can incorporate crosslinks with different chemical properties and functionalized modifications through double-click reactions involving two side chains with azido functional groups at the i, i + 4, or i + 7 positions and a dialkynyl linker. Lau et al. [108] made a series of representative works in the field of triazole-based two-component stapled peptides. Firstly, they used the double-click chemistry strategy to staple α-helical peptides derived from the tumor suppressor p53 for the inhibition of p53-murine double minute 2 (MDM2) interaction and introduced cationic arginine (Arg) residues on the staple linkage to improve the cell permeability and activate capability of p53. They also attached cell-penetrating peptide (CPP) sequences onto the crosslink of a non-helical conformational peptide targeting the tankyrase (TNKS) proteins to further enhance the cell-penetrating capability while maintaining TNKS binding affinities [109]. Furthermore, they developed a toolbox of diverse dialkynyl crosslinks to the staple of MDM2-binding peptides via the double-click approach and conducted a study about the effect of these crosslinks on cellular uptake and activation of p53 [110].
Using nature or non-nature amino acid-bearing side-chain functional groups that can be directly coupled and forming covalent bonds to constrain the conformation of peptides is termed as one-component stapling strategy [45, 111]. The length, structure, and chemical functionality of staple linkages are dictated by the selection of non-native amino acids during SPPS. One-component stapling techniques have been extensively explored and applied in early stapled peptide research. Their primary advantage lies in structural simplicity, particularly the appeal of the “all-hydrocarbon” linkage, which represents a straightforward and minimal motif of staple design [112–120]. However, the all-hydrocarbon strategy faces challenges. It is still difficult to introduce the required α,α-disubstituted unnatural amino acids with olefinic side-chains through SPPS, while the routes of HCS peptides with i, i + 3/4/7 spacings have been established and optimized. Although lactam stapling is more accessible, the disadvantage of this strategy is the need for introducing additional orthogonal protective groups to selectively deprotect the amine and acid functional groups before lactamization [57]. Additionally, emerging one-component stapling strategies have shown promising prospects as cycloaddition chemistry of triazole stapling. Triazole stapling amino acids are readily available, and conditions for intramolecular CuAAC are established. In a word, classic one-component stapling strategies, among the earliest and most extensively studied, are widely applied due to their structural simplicity and convenient synthesis. Meanwhile, the ongoing development of new one-component stapling strategies is enriching the toolkit of scientists [121, 122].
Two-component peptide stapling strategies entail the utilization of a linear peptide in conjunction with a separate staple crosslink [45], which means the feasibility of incorporating staples of different features simultaneously. Compared to flexible linear staples such as alkanes, alkenes, and ethers, aromatic staples (Figure 6A) exhibit stronger hydrophobicity and structural rigidity, the latter will promote the cell permeability of peptides, and biphenyl staples will further enhance these two properties [123]. However, the ability of these staples to induce peptide helicity still depends on the specific circumstances. A comparative study on the physiochemical properties of SAH peptides with different types of crosslinks conducted by Tian and co-workers [124] showed that when stapled at the same position of a peptide sequence targeting the estrogen receptor (ER) coactivator, the lactam and all-HCS peptides exhibited the highest helical contents, followed by the triazole and the m-xylene crosslink, the helicity of vinyl sulfide stapled peptides is slightly higher than the m-xylene crosslink. Perfluoroaryl and decafluorobiphenyl stapling techniques positioned in i, i + 4/7 fashion brought by Spokoyny et al. [125] are featured in the combination of rigidity (Figure 6B), lipophilicity, and easy synthesis. Introducing hydrophilic staples that have carbonyls, such as lactone, might improve the solubility and biodegradation properties [126].
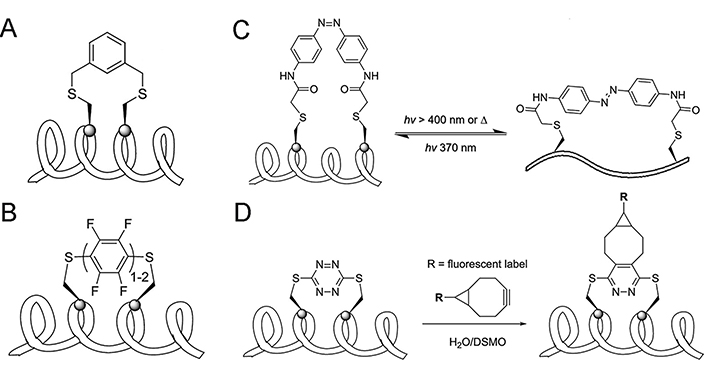
One-component and two-component peptide stapling strategies. (A) Aromatic stapling strategy; (B) perfluoroaryl and decafluoro-biphenyl stapling strategy; (C) azobenzene-mediated photoswitchable i, i + 7 Cys-Cys stapling strategy; (D) incorporation of s-tetrazine staple serving as the socket of other functional groups. DSMO: dimethyl sulfoxide
Note. Adapted with permission from “Stapled helical peptides bearing different anchoring residues,” by Li X, Chen S, Zhang WD, Hu HG. Chem Rev. 2020;120:10079–144 (https://doi.org/10.1021/acs.chemrev.0c00532). © 2020 American Chemical Society.
Since the binding affinity between the helical ligand and receptor is highly related to the helical content of the peptide ligand, the ability to reversibly and intentionally control the helicity of peptides can be useful in peptide-based targeted therapy. Kumita et al. [127] reported the first photo-switchable stapled peptide using azobenzene-based i, i + 7 crosslink, which can change the distance between two sides of the crosslink through a light-driven isomerization (Figure 6C). Begin with an ideal non-helical trans-azobenzene stapled peptide, the helical cis-azobenzene stapled peptide could be obtained after irradiation at 370 nm for 5 min and returned into the trans-form by keeping the peptide in dark conditions for several hours. Staples can also be the socket of other functional groups, Brown and Smith [99] developed facile protocols for the incorporation of s-tetrazine between unprotected adjacent cysteine sulfhydryls of peptides. A significant advantage of the S,S-tetrazine staple is the ability to introduce probes exploiting inverse electron demand Diels–Alder reactions, which make tetrazine staples hold the promise of dual roles: confining peptide conformation and introducing photophysical or other potential probes (Figure 6D) [128].
The characterization of biochemical functions is an essential and necessary factor in the transition from synthetic experiments of peptides to cellular and in vivo characterizations, as the inevitability of failures is higher when conducted directly without pre-verification of the biochemical functions. The advocated approach follows a sequential path, starting with the optimization of biochemical functional characterization for peptides, followed by cellular uptake, and then, gradually advancing to in vivo experiments. An overview is presented of the biochemical performance of designed stapled peptides engaged in various PPIs, including high specific targeting, increased protein binding affinity, and superior resistance to proteolysis.
Therapeutic stapled peptides generally show high targeting specificity activities, which represents possessing the necessary qualification to be involved in the interface of PPIs [87, 114, 129, 130]. The structural constraints brought by the insertion of stapled bonds can maintain the bioactivity of the peptide by stabilizing the secondary structure, and ensuring that it can specifically combine with the target protein to play an accurate biological function. Recently, Speltz et al. [131] developed a class of synthetic transcriptional repressors (STRs) derived from the basic helix-loop-helix (bHLH) domain of the core DNA-binding domain (DBD) bHLH-TFs (such as MYC and MAX1) protein that was equipped with the minimal structural properties necessary for high specific DNA recognition and can compete with full-length MYC/MAX to bind with DNA binding. They found that certain stapled STRs exhibit increased specificity compared to the non-stapled progenitor B–Z, which can be attributed to considerably more helical and thermal stability. They additionally demonstrated that the STR scaffold as a modular platform can be reprogrammed to develop highly specific mimics of the bHLH-TF family targeting other diverse DNA sequences.
The primary interface between synaptotagmin-1 (Syt1) and the neuronal soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complex is vital for the rapid synchronization of neurotransmitter release triggered by Ca2+. Lai et al. [119] designed an HCS peptide SP9 that can specifically interrupt membrane fusion triggered by Ca2+ via disturbing the primary interface between SNARE and the Ca2+-binding C2B domain of Syt1 (Figure 7). Introducing hydrocarbons as staples stabled the α-helical conformation of SP9 and tremendously strengthened the interaction with Syt1 (Figure 7B–E). In the reconstruction system comprised of the neuronal synaptic proteins or airway homologs [including synaptosomal-associated protein of 23 kDa (SNAP-23), syntaxin-3 (Stx3), Syt2, vesicle-associated membrane protein 8 (VAMP-8), mammalian homolog of Caenorhabditis elegans uncoordinated gene 13 (Munc13-2), and Munc18-2], the stapled peptide SP9 has exhibited strong suppression to fusion triggered by Ca2+ at physiological Ca2+ concentrations, whereas the non-stapled peptide penetratin (PEN)-P9-sulfo cyanine3 (Cy3) did not show any inhibitory effects at the same concentration, suggesting the specificity of the stapled peptide SP9 (Figure 7F). Additionally, SP9 could be efficiently uptake into mouse airway epithelium and cultured human epithelial cells when conjugated with CPPs, reducing mucus occlusion of mouse airways and secretion of stimulated mucin specifically and significantly.
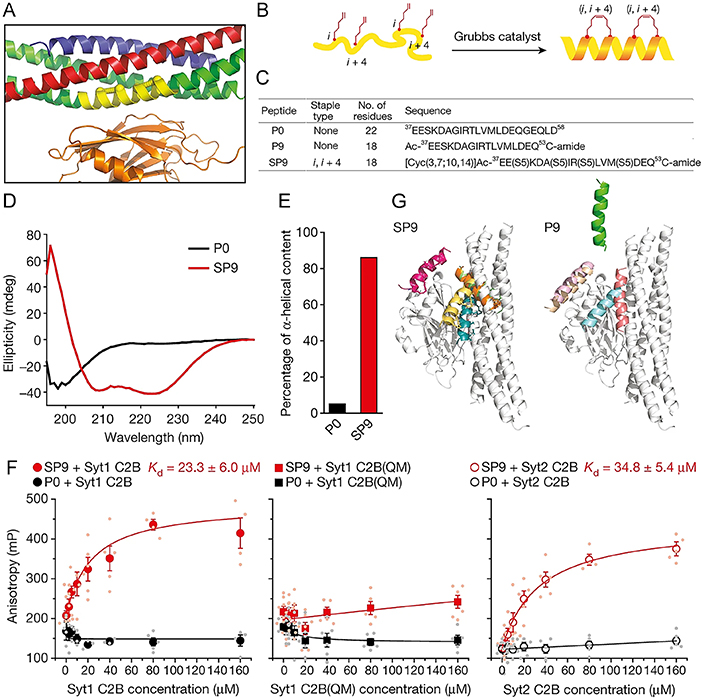
Characterization of designed stapled peptide SP9. (A) Magnified view of the primary interface [(VAMP-2; blue), SNAP-25A (green), Stx1 (red), and the C2B domain of Syt1 (orange)]; (B) schematic of SP9; (C–E) sequences (C), circular dichroism spectra (D), and percentage of α-helical content (E) of peptides used in the research; (F) interactions between Cy3-labelled SP9/P0 and Syt1 C2B, Syt1 C2B(QM) mutant and Syt2 C2B; (G) peptide conformations of SP9-Syt1 C2B (left) and P9-Syt1 C2B (right) superimposed onto the primary interface by molecular dynamics simulations
Note. Reprinted from “Inhibition of calcium-triggered secretion by hydrocarbon-stapled peptides,” by Lai Y, Fois G, Flores JR, Tuvim MJ, Zhou Q, Yang K, et al. Nature. 2022;603:949–56 (https://doi.org/10.1038/s41586-022-04543-1). CC BY.
Quagliata et al. [71] designed the stapled analog P3 derived from the minimal fragment ACE2 (24–42), with the triazole-containing bridge in the positions at i to i + 4, to stabilize the α-helical secondary structure that may contribute to enhancing target specificity. Modi et al. [14] developed a designed stapled peptide N1S based on the prediction from AlphaFold to specifically block the heterodimerization of Nrf2 with MafG directly through binding the sMAF proteins immediately, and no binding between biotin-labeled N1S and bovine serum albumin (BSA) was observed, demonstrating that N1S could not bind to other proteins non-specifically. Cathcart et al. [115] employed the α1 helix of E2 in the ubiquitin-proteasome system (UPS) as the model for the design of stapled peptides binding to the groove of E1, inducing the change of consequential conformational and inhibiting the transfer of E1 ubiquitin. They demonstrated the binding specificity between SAH-ubiquitin-conjugating enzyme E2 A (SAH-UBE2A) and E1 through a fluorescence microscopy-based qualitative binding assay and examined the specificity for disrupting the transfer of UBE1 thioester in the reconstituted system.
Based on their previous research [132], Rezaei Araghi et al. [49] synthesized new variants of HCS peptide MS1, a BIM-based peptide, which was previously engineered to have high affinity and specificity for myeloid cell leukemia-1 (Mcl-1), performing the optimization by iteration steps, and found that the conversion of staple positions is closely related to the specificity of the stapled peptide, while the unmodified template peptide (BIM SAHBA1) peptide possessed higher Mcl-1-specificity when the staple was located at the 2e–3b position. Wu et al. [133] rationally designed HCS peptides that could specifically target the coiled-coil domain of beclin 1, which led to the reduction of self-association for beclin 1 and an increase in the formation of beclin 1-ultraviolet (UV) radiation resistance-associated gene protein (UVRAG) complexes.
In general, the side chains and the backbone of stapled peptides are usually structurally constrained, stabilizing the peptide to a predetermined conformation, thereby reducing the entropy penalty associated with binding to molecular targets and significantly improving the binding affinity of the peptides, which is known as the prepayment of entropy costs bound up with the binding process. A study showed that the high affinity and selectivity of stapled peptides were governed by the chemical structures of the staples by analyzing the obtained crystal structure, even a small change, which can trigger different binding modes with high affinity [134]. Subsequent studies indicated that the two peptides (P11 and P12) with the stapling to i, i + 7 position derived from tryptophan 13 (Trp13) to Glu33 of autophagy related 16 like 1 gene (ATG16L1) showed high binding affinity to ATG5 (Kd = 3–6 nmol/L) compared to the control linear peptide (P13) with the Kd of 0.072 μmol/L, which could be partially given the credit to the stabilization of the helical by stapling [135]. As CD studies showed, the helix content of the stapled peptide solution conformation ranges from 17% to 22%, whereas the control peptide (P13) mainly exhibited a random coil conformation with a helix content of 5%. Mayer et al. [136] developed a chimeric stapled peptide inhibitor named c‑ML 9GS, incorporating an additional LinkTer peptide derived from disordered p53 into the N-terminal domain of MDM2. Compared with the two components separately (Kd c‑MDM2 = 2.49 μmol/L ± 0.04 μmol/L and Kd p53 LinkTer = 22 μmol/L ± 1 μmol/L), c‑ML 9GS targeted inhibitor of apoptosis-stimulating protein of p53 (ASPP; iASPP) with an increased binding affinity (Kd c‑ML 9GS = 1.59 μmol/L ± 0.02 μmol/L) which can be interpreted for the increased binding enthalpy with larger binding interface (Figure 8A).
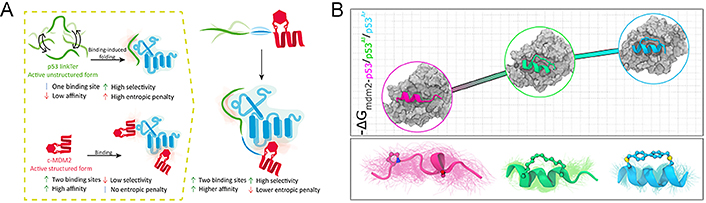
Stapled peptides can improve the binding affinity of the peptide. (A) Several binding mechanisms of chimeric stapled peptide inhibitors; (B) effect of stapling on the thermodynamics of MDM2-p53 binding
Note. Figure 8A was reprinted with permission from “Targeting protein interaction hotspots using structured and disordered chimeric peptide inhibitors,” by Mayer G, Shpilt Z, Kowalski H, Tshuva EY, Friedler A. ACS Chem Biol. 2022;17:1811–23 (https://doi.org/10.1021/acschembio.2c00177). © 2022 American Chemical Society. Figure 8B was reprinted with permission from “Effect of stapling on the thermodynamics of mdm2–p53 binding,” by Maity A, Choudhury AR, Chakrabarti R. J Chem Inf Model. 2021;61:1989–2000 (https://doi.org/10.1021/acs.jcim.1c00219). © 2021 American Chemical Society.
The binding inhibition of p53 to MDM2 is an efficient approach for restoring p53 activity and promoting apoptosis in tumor cells. Peptide stapling is recognized as a practical strategy for generating α-helical peptides to mimic the p53 transactivation domain. By targeting dual inhibition of the interaction between p53-MDM2 and MDMX as the validation target, Ricardo et al. [137] implemented a diversity-driven stapling method that constrained peptides to p53-like bioactive conformations while regulating the helical properties, hydrophobicity, and flexibility of the staple. All the designed peptides bind to the deep grove of MDM2 with high affinity and a similar pattern to the wild type (WT)-p53. Maity et al. [112] used all-atom molecular dynamics simulations to systematically investigate the interaction between MDM2 and three p53 variants (including WT, p53 with a biaryl aromatic staple, and p53 with an aliphatic hydrocarbon staple) (Figure 8B), demonstrating the stapled peptides of p53 were more thermodynamically favorable than the WT, and proposed that the introduction of restriction to the peptide conformation can effectively increase the binding affinity and the rigidity of the staple is more beneficial for the binding [112]. They justified the idea that the introduction of a staple to peptides decreases the enthalpic stabilization through the lowering of the entropic penalty and enhances the overall binding affinity, although it increases the hydrophobicity of peptides to a certain extent. Ye et al. [138] developed an unlabeled combinatorial alanine affinity selection platform to promote the optimization of PPI modulators based on peptides, in which the i, i + 4 perfluoroaryl stapling strategy was applied to three highest frequency alanine-substituted sites of peptides and contributed to the formation of three highest affinity stapled peptides monitored by biolayer interferometry (BLI) competition assays.
A study indicated that the HCS peptide with the ability to disrupt the RNA polymerase sigma-54 factor (RpoN)-DNA binding via binding the promoter sequence of RpoN-DNA and inhibiting expression mediated by RpoN in Escherichia coli has improved the binding affinity constants for DNA in the high nanomolar range [139]. Learte-Aymamí et al. [140] demonstrated that a novel coordination stapled peptide αH-histidine-2 (His2)[Pd] consisting of a bis-His peptide based on the αH-helix of the cofactor son of sevenless homolog 1 (SOS1) and coordination with Pd(II) can bind to Kirsten rat sarcoma viral oncogene homolog (KRAS) with high binding affinity, which showed a marked increase in the anisotropy when titrated with the preformed coordination stapled peptide to KRAS, while the control peptide αH-His2 exhibited lower binding affinity to KRAS. Jeganathan et al. [84] reported the first structurally based design of a stapled peptide for blocking the assembly of NF-Y, by changing the α-methylation mode of the non-natural amino acids involved and enabling fine-tuning of peptide flexibility, leading to enhanced affinity for TF subunits, thereby inhibiting their functional assembly and other biological functions. By optimizing the staple and sequence for the beclin 1-targeting peptides, Yang et al. [88] placed the hydrocarbon staple closer to the interface of the beclin 1-peptide to increase the binding affinity by approximately 10- to 30-fold. Based on the factors mentioned above, it is evident that the position, property, conformation, number, and other factors of the staple all have a significant impact on the binding affinity to target PPIs.
The high susceptibility to proteases is one of the chief defects of peptides as therapeutic drugs, and a main property that is generally addressed by stapled peptides. Constraining the structure of the stapled peptide actually renders a more compacted framework, resulting in reduced exposure of vulnerable groups, thus preventing the attack of the protease [58, 61, 134, 141–145]. Since proteases require the peptide to adopt an extended conformation to facilitate hydrolysis of the amide bond, resistance towards protease is usually associated with the degree of α-helix stability and the number of staples, thus enhancing the helical structures of peptides may render them protease-resistant.
Indeed, generous studies indicated that stabilizing α-helix conformation through peptide stapling can improve protease resistance. Zheng et al. [117] synthesized a stapled peptide library by introducing an all-hydrocarbon stapling strategy and screened out two optimized peptides with enhanced helicity and proteolytic stability that showed better inhibitory activities towards pseudo and authentic SARS-CoV-2 compared with the linear CoVHR2-0 peptide. Subsequent studies showed that proper placement of all-hydrocarbon staples endowed peptides with enhanced α-helical structure and reduced exposure to proteases, giving rise to striking resistance to proteolysis (Figure 9A) [135, 136, 146, 147]. Significantly, the increased half-life owing to the insertion of the staple is not only limited to the extracellular environment where most proteases are located but also recorded in the intracellular environment (Figure 9B). Kuster et al. [148] attempted to target the “dimer-of-dimers” interface of the carboxy-terminal binding protein (CtBP) interacting protein (CtIP) via an innovative stapled peptide-based approach, in which they found that the helical content upon hydrocarbon stapling showed a sharp increase and a higher ability to resist protease hydrolysis as only a slight effect on the steady state level of CtIP was observed in the cycloheximide (CHX) chase experiment. Mourtada et al. [87] discovered Mag(i + 4)1,15(A9K) as a selective candidate drug for StAMP via the design algorithm and proved that the stapled peptide is an attractive strategy for AMP-based antibiotic development due to the enhancement of the α-helical structure with biological activity can twist the amide bonds and make stapled peptide becoming a poor substrate for proteases.
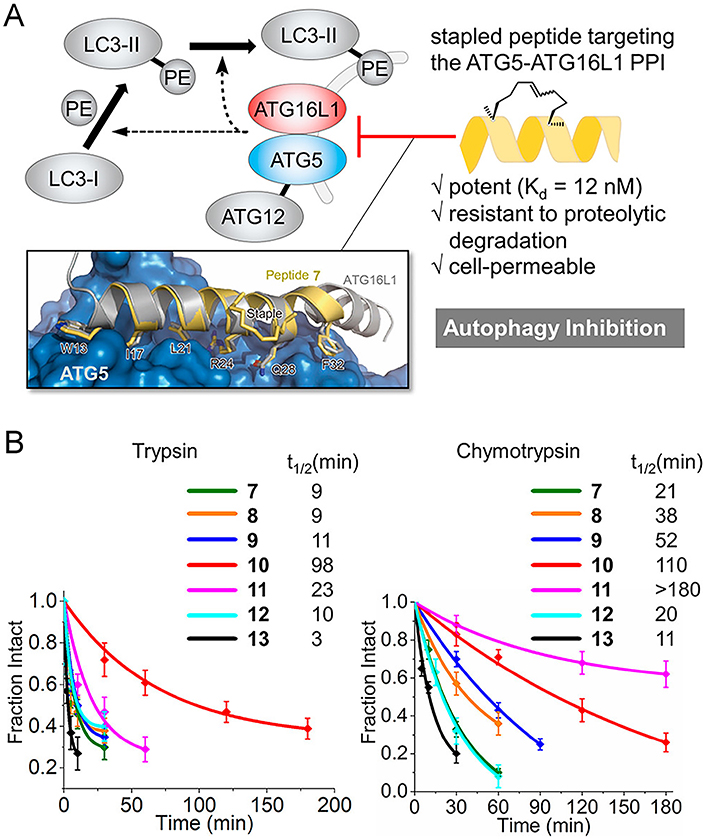
Stapled peptides can enhance the protease resistance of the peptide. (A) Targeting the ATG5-ATG16L1 PPI with a stapled peptide derived from ATG16L1 for autophagy inhibition; (B) tolerance of staple peptides toward trypsin and chymotrypsin. PE: phosphatidylethanolamine
Note. Reprinted with permission from “Targeting the ATG5-ATG16L1 protein–protein interaction with a hydrocarbon-stapled peptide derived from ATG16L1 for autophagy inhibition,” by Cui J, Ogasawara Y, Kurata I, Matoba K, Fujioka Y, Noda NN, et al. J Am Chem Soc. 2022;144:17671–9 (https://doi.org/10.1021/jacs.2c07648). © 2022 American Chemical Society.
The susceptibility to proteolytic cleavage and lack of oral bioavailability of the linear peptide-based inhibitors led to its being downgraded to the final option of treatment. Yang et al. [149] optimized the GLP-1R/glucose-dependent insulinotropic polypeptide (GIP) receptor (GIPR) dual inhibitors by introducing biaryl-based stapling to the chimera of GLP-1 and GIP peptides RG7697, which displayed a more stable α-helix structure, markedly improved proteolytic stability and excellent pharmacokinetics exposure upon subcutaneous injection. Speltz et al. [131] found that stapled peptides STRs derived from MAX protein displayed enhanced thermal and proteolytic stability, for both STR116 and STR118 were observed intact in the cells after 12 h for visualization by gel electrophoresis, showing longer half-time (> 10-fold) compared to linear peptide B–Z. In the previous study, Li et al. [63] explored the influence of the stapled strategy on the bioactivity and proteolytic stability of hymenochirin-1B and found that certain analogs have increased protease resistance relative to the liner peptide. In the subsequent research, they further optimized the peptide, adopting a new strategy of glycosylation combined with stapling for the first time, and successfully acquired the optimal peptide H-58 with high proteolytic stability and anti-tumor selectivity [150]. Similarly, based on the previous proof that introducing double-stapling at i, i + 4 to exendin-4 (Byetta) provides enhanced proteolytic stability [151], Bird et al. [85] attempted to determine whether stapling can generate alternative GLP-1 therapeutic candidate drugs through the use of simplified validation workflows and stapling design. It has been proven that staple-scanning and stitching methods can generate GLP-1R agonists with stable structure and protease resistance in mouse models, accompanied by good therapeutic activity.
As mentioned previously in “Introduction”, one of the main limitations of peptide therapeutic drugs is that they are difficult to penetrate the cellular membranes, which is vital to the modulation of PPIs as they only begin working after binding to the target protein intracellular. The explicit mechanism of cell uptake is still an active field of research, but it seems to relate to energy-dependent pinocytosis and may directly penetrate under certain circumstances. The design of peptides for cell delivery is one of the most breathtaking yet challenging frontiers in the field of therapeutic drugs. Based on the summary and analysis of a series of research works on stapled peptides, it has been proposed that the stapled peptide strategy is an effective technique for improving membrane permeability, and the capacity of cell penetrability has a direct correlation to the characteristics of peptides, such as helicity, hydrophobicity, and charge, among other factors.
Due to the direct dependence of cell permeability on the hydrophobicity and helicity of peptides, it is evident that stapled peptides can promote cellular uptake of peptides (Figure 10A). Indeed, the insertion of staples forces the peptide into α-helical conformation, which enhances the helicity of the peptide and also increases its hydrophobicity by exposing non-polar side chains to the environment outside. Many studies have proven that α-helical stapled peptides show tremendous potential for improving cell permeability, providing a pathway for intracellular proteins as drug targets [148, 152–158].
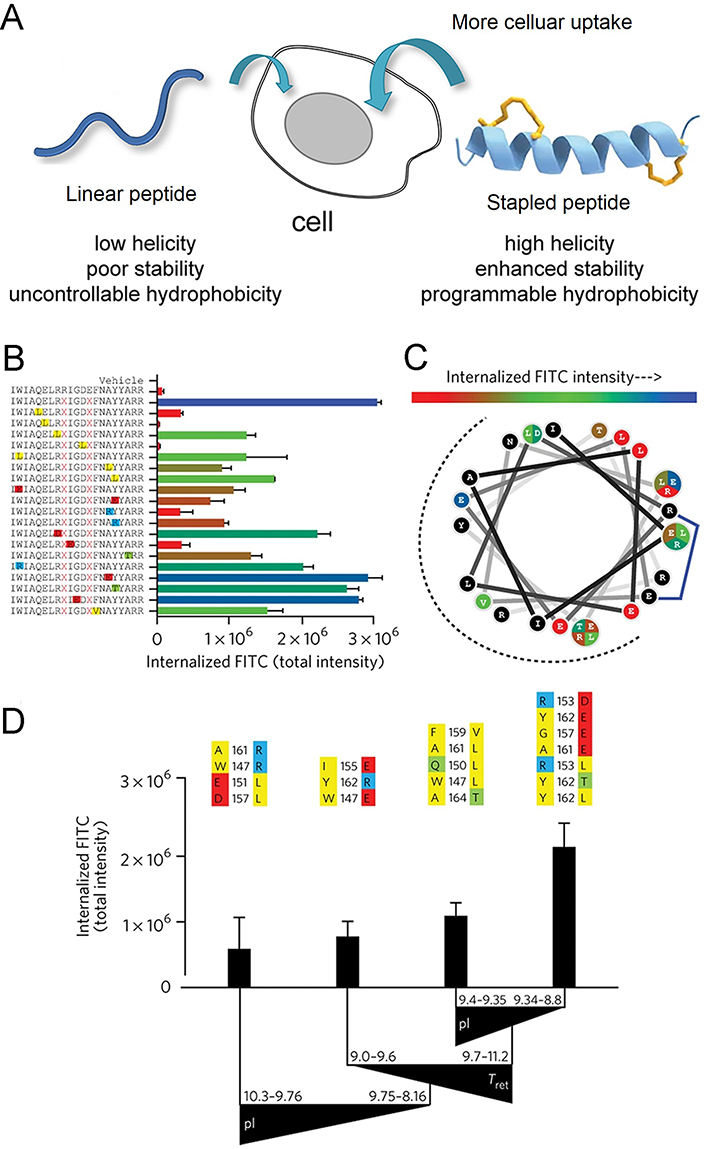
Stapled peptide strategy is an effective technique for improving membrane permeability. (A) Scheme of the mechanism for stapled peptides alter the cell permeability; (B–D) impact of mutagenesis on cellular uptake of the stapled analog, corresponding to the BIM SAHBA1. (B) the library of BIM SAHBA1 mutant peptides and corresponding total internalized fluorescein 5-isothiocyanate (FITC) intensity (TIFI) values; (C) wheel depiction of the α-helical BIM SAHBA1; (D) the influence of principal components on TIFI outcome [isoelectric point (pI), hydrophobicity/HPLC retention time (Tret), and α-helicity]
Note. Figure 10A was adapted with permission from “Importance of net hydrophobicity in the cellular uptake of all-hydrocarbon stapled peptides,” by Sakagami K, Masuda T, Kawano K, Futaki S. Mol Pharm. 2018;15:1332–40 (https://doi.org/10.1021/acs.molpharmaceut.7b01130). © 2018 American Chemical Society. Figure 10B, C, and D was adapted with permission from “Biophysical determinants for cellular uptake of hydrocarbon-stapled peptide helices,” by Bird GH, Mazzola E, Opoku-Nsiah K, Lammert MA, Godes M, Neuberg DS, et al. Nat Chem Biol. 2016;12:845–52 (https://doi.org/10.1038/nchembio.2153). © 2016 Nature America, Inc.
Based on the importance of eukaryotic initiation factor 4E (eIF4E) in cancer, many groups have made significant efforts in the development of inhibitors of eIF4E PPI [50, 52, 159–161]. Gallagher et al. [50] and Song et al. [52] first synthesized an HCS peptide HCS-4E-eIF4E binding protein 1 (BP1) with increased stability of i, i + 4 helix, nanomoles binding to eIF4E, and suppression of eIF4E PPIs in vitro. However, the poor solubility because of strong hydrophobicity limits its further development as eIF4E PPI inhibitor. Then they further optimized the design and synthesized two kinds of lactam-stapled peptides to study their properties on helicity, bioactivity, and cell uptake, in which the lactam-stapled peptide galactose-6-phosphate isomerase subunit (LacB)-4E-BP1 exhibits extremely high cellular uptake efficiency (84% ± 20%) that is sufficient to bind to the cell membrane, trigger endocytosis and subsequently lead to endosome escape [162]. Paterson et al. [97] reported a series of novel stapled peptides SIGKAFKILGYPDYD (SIGK) that target the hotspot of Gβγ proteins PPI, in which the double thiolene stapling consists of a divinyl diester and two cysteine residues, with improved α-helicity and enhanced cell permeability relative to the unstapled peptide. Fluorescein-labeled stapled peptides with perfect cell penetrability can also serve as cell-based chemical probes.
Bird et al. [163] found that the position of the stapling at the amphipathic boundary has the ability to adjust the hydrophobicity of the overall interface, besides optimal hydrophobic and helical content, stapled positions are also the key driving force for cell uptake (Figure 10B–D). This observation was further supported in their subsequent work, which suggested that the hydrocarbon staple motif can affect the cellular permeability and protease resistance of peptides [49]. They reconstructed Mcl-1-binding peptides into cell-penetrating SAH peptides SAH-MS1-14 and SAH-MS1-18, both of which can mediate the changes of conformation for the α3–α4 region of Mcl-1, leading to an increase in the hydrophobicity of the interaction surface, revealing the correlation between hydrophobicity and cell permeability. Inhibiting the abnormal elevation of Parkinson’s disease (PD)-related leucine-rich repeat kinase 2 (LRRK2) can act on neuroprotection, Helton et al. [130] designed stapled peptides containing all hydrocarbons as PPI disruptors to inhibit the dimer interface of interaction for investigating the role of LRRK2 dimerization in kinase regulation. The stapled peptide they used involved olefinic amino acid Fmoc-(S)-2-(4’entenyl)-alanine bound at suitable i, i + 4 positions at the non-binding interface as the staple, and it was found that both selected stapling peptides LCIP1 and LRIP4 exhibited improved cell permeability as compared to the native one in a dose-dependent manner monitored by flow cytometry [130].
When referring to the charge of stapled peptides, several studies have proven that the positive charge is significant for the first step of cell internalization, as it provides electrostatic interactions with the negatively charged cell membrane, while peptides with negative charges rarely penetrate the membrane components, and in the presence of peptides with an NC ranging from 0 to +2, the permeation is more advantageous [98, 108, 134, 164–166]. Generally, introducing positively charged residues at the N- or C-terminus of peptides that do not affect the interactions is the commonly used method to enhance cell permeability. Substituting neutral amino acids [glutamine (Gln) and asparagine (Asn)] for anionic counterparts (Glu and Asp) on the backbone of peptides that are not involved in interactions is also a solution [26]. Additionally, strategies including the introduction of guanidine have also become a promising method for promoting endocytic uptake [167].
Li et al. [156] designed a series of stapled peptides derived from the trans-acting activator of transcription (Tat) peptide, Tat47–57, to investigate the impact on cellular uptake of the number and location of cationic motifs on stapled helical peptide backbones (Figure 11A–C). Peptides A6–A8 carrying charges of +5, +4, and +3 respectively, which are obtained by the substitution of Lys4, Lys4Arg11, or Lys4Arg6Arg11 of A4 with L-alanine, showed gradually decreasing cell penetration ability compared with A4 with +6 positive charges (Figure 11C). In addition, Limaye et al. [81] employed the positively charged Lys (K) residues to replace the solvent-exposed residues at different positions of peptides, resulting in peptide libraries with different net positive charges, among which WAHMIS-2 with the highest positive charge exhibits higher cell penetration ability compared to others. Kuster et al. [148] found that the overall positive charge of SP18–28, which is more positively charged by the removal of E29, combined with the extended hydrophobic interface, resulted in a higher intracellular uptake rate of the peptide compared to its longer corresponding peptide SP18–31, indicating that positive NC seems to have the ability to enhance cell uptake and biological activity. Wu et al. [110] reported a peptide library comprising a series of functionalized dialkynyl linkers equipped with double CuAAC staple. The results showed that different linkers did not have a remarkable influence on the in vitro binding affinity of MDM2, while positively charged residues produced a significant enhancement in cell uptake and targeted capability in cellular reporter assay, indicating that positive charge is crucial for achieving cell viability using WT-p53 sequence-based stapled peptides.
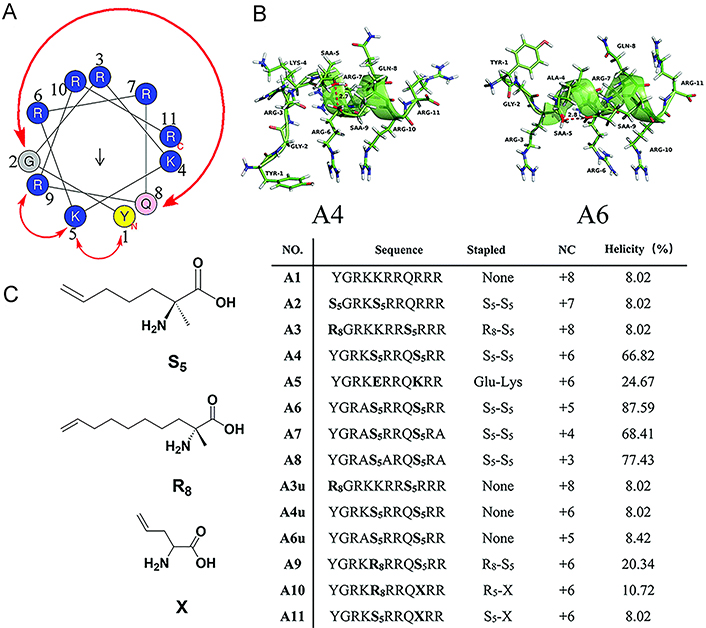
NC is an important consideration for the cell internalization of the stapled peptide. (A) Wheel of the α-helical Tat transduction domain sequence; (B) 3D structure molecular models of peptide A4 and A6; (C) sequences, NCs, and helicities of Tat peptide (A1) and its stapled derivatives (A2–A11). TYR-1: tyrosine-1; GLY-2: glycine-2; SAA-9: (S)-2-(4’pentenyl)-alanine-9
Note. Reprinted with permission from “Hydrocarbon staple constructing highly efficient α-helix cell-penetrating peptides for intracellular cargo delivery,” by Li S, Zhang X, Guo C, Peng Y, Liu X, Wang B, et al. Chem Commun (Camb). 2020;56:15655–8 (https://doi.org/10.1039/d0cc06312f). © The Royal Society of Chemistry 2020.
Yet it’s worth noting that a high positive charge does not necessarily indicate a stronger cell-penetrating ability, that is, the two are not in a simple linear relationship, as researchers have shown that peptides with an NC higher than +7 rapidly reduce cellular pathways [168]. It means that excess positive charge may trigger membrane lysis at increased peptide dosing. In fact, the NC for the first clinical candidate of stapled peptide for intracellular targeting is negative. An equally important issue to be aware of is that some peptides (such as cationic AMPs) [169–173] can disrupt the membrane as its amino acid composition, thus, it owns significant meaning to perform maximum tolerance dose titration before treating cells with stapled peptides to ensure the maximum dose, which will cause damage or destruction to the membrane once the excess is exceeded. That is to say, stapled peptides can only be used at tolerable doses, and if necessary, strategies need to be optimized to eliminate undesirable biophysical properties, for achieving specific targeting and other biological activities in cellular and other environments.
Besides functioning as PPI inhibitors, scientists have been diligently exploring ways to expand the capabilities of stapled peptides, such as intracellular tracking, subcellular localization, and theranostics. While certain functionalities can be achieved by directly modifying functional groups on the peptide sequence, this alteration may inevitably impact the primary role of the stapled peptide as a ligand in PPIs, mainly due to steric hindrance and other factors. Consequently, introducing functional groups onto the staple seems to be a promising strategy, as modularized staple functionalization can be achieved through the development of two-component stapling strategies, which have become a tangible reality in recent years [39, 42, 174].
Theranostics is a cutting-edge approach in modern cancer therapy, representing the seamless integration of targeting, imaging, and therapeutic capabilities [175]. The stapled peptide ligand is an excellent platform to realize this comprehensive and advanced treatment approach. Fluorescence is frequently employed to track the uptake of peptides in cells and tissues, and various techniques have been devised to attach fluorescent probes to peptides [174, 176, 177]. Assem et al. [100] demonstrated the utilization of dichloroacetone (DCA) as a powerful tool for synthesizing stapled cyclic and helical peptides through chemoselective oxime ligation within the tether (Figure 12A). They successfully attached various aminooxy tags to the acetone linker, including fluorophores like Alexa Fluor 488 and Alexa Fluor 647. Grison et al. [178] synthesized an alkyne-dibromomaleimide stapled peptide by introducing an alkyne group onto the dibromomaleimide through Mitsunobu alkylation. They then performed a subsequent “click” reaction with azide-fluorescein azides to convert them into fluorescein-conjugated stapled peptides (Figure 12B). Afterwards, they demonstrated that the introduction of a fluorophore at this position did not have any detrimental consequences, as confirmed through a direct fluorescence anisotropy titration.
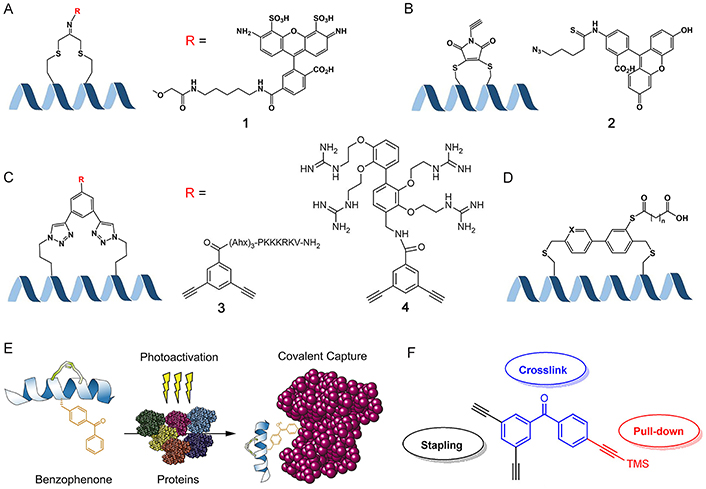
Biomedical application potentialities of stapled peptides. (A) Introducing functional groups on the staple by forming the oxime ligation with the DCA linker; (B) introducing functional groups on the staple by a “click” reaction between alkyne-dibromomaleimide stapled and azide-functionalized groups; (C) introducing functional groups through double “click” reactions; (D) two different lengths of fatty diacids to the biaryl staple via labile ester bonds; (E) to generate photoreactive stabilized α-helices of BCL-2 domains for protein capture, a process involves the substitution of native aromatic residues with 4-benzoyl-phenyl-alanine (Bpa) and subsequently performing RCM on olefinic non-natural amino acids located at i and i + 4 positions; (F) multifunctional crosslink with two alkynes for double “click” stapling (black), a benzophenone group for photo-crosslinking (blue), and the protected alkyne handle for pull-down assays (red). TMS: tetramethylsilane; Ahx: 6-aminohexanoic acid. R = X, X = 1, Alexa Fluor 488; X = 2, azide-FITC; X = 3, nuclear localization signal (NLS) sequence PKKKRKV; X = 4, small molecule carriers (SMoCs) on the dialkynyl aromatic staple
Note. Figure 12D was adapted with permission from “Design of potent and proteolytically stable biaryl-stapled GLP-1R/GIPR peptide dual agonists,” by Yang Y, Lee C, Reddy RR, Huang DJ, Zhong W, Nguyen-Tran VTB, et al. ACS Chem Biol. 2022;17:1249–58 (https://doi.org/10.1021/acschembio.2c00175). © 2022 American Chemical Society. Figure 12E was adapted with permission from “Photoreactive stapled BH3 peptides to dissect the BCL-2 family interactome,” by Braun CR, Mintseris J, Gavathiotis E, Bird GH, Gygi SP, Walensky LD. Chem Biol. 2010;17:1325–33 (https://doi.org/10.1016/j.chembiol.2010.09.015). © 2010 Elsevier Ltd. Figure 12F was reprinted from “Development of a multifunctional benzophenone linker for peptide stapling and photoaffinity labelling,” by Wu Y, Olsen LB, Lau YH, Jensen CH, Rossmann M, Baker YR, et al. Chembiochem. 2016;17:689–92 (https://doi.org/10.1002/cbic.201500648). CC BY.
The second challenge in stapled peptide-based theranostics is to target the delivery of peptide ligands to specific subcellular compartments. To promote peptide delivery into the nucleus, Wu et al. [110] explored staples functionalized with an NLS sequence PKKKRKV derived from the polyomavirus simian virus 40 (SV40) large T-antigen (Figure 12C) [179]. This sequence is known to bind to the major site on importin α, which facilitates the transport of NLS-containing proteins into the nucleus [180]. While a variable number of Ahx spacers was inserted to minimize steric clash between the NLS and the MDM2-binding interface, the NLS containing stapled peptides showed comparable binding values for MDM2, and were found to have sub-micromolar affinity for importin α in a direct fluorescence polarization (FP) assay. However, the result of confocal imaging showed that the majority of the administered stapled peptide remains trapped in the lysosomal trafficking pathways, despite the NLS sequence promoting cellular uptake to a certain degree, indicating that endosomal escape continues to be the primary limiting factor affecting proper targeting and the technology of stapled peptides subcellular delivery remain further developed.
In addition to its primary role in inhibiting disease-relevant PPIs, the integration of stapled peptide ligands with small molecule drugs can enhance the effectiveness of treatments and improve the targeting specificity. This combination approach leads to an overall increase in the efficiency and utilization of small-molecule drugs, offering promising prospects for more effective and targeted therapies. For example, the melanocortin 1 receptor (MC1R) is a G-protein-coupled receptor belonging to a family of five MCR subtypes [181], which plays a significant role in melanoma, and the overexpression of it can serve as a molecular marker for these types of tumors [182, 183]. White et al. [128] synthesized a series of stapled peptides that target MC1R derived from the backbone cyclic and disulfide bridged sunflower trypsin inhibitor-1 (SFTI-1) framework bearing various crosslinkers. They selected cobimetinib as a payload for peptide-drug conjugate since it is an approved treatment for inoperable or metastatic melanomas [184]. More importantly, it provided a convenient secondary amine as a suitable site for the installation of a functionalization handle. An azide-functionalized cobimetinib was linked to the tetrazine-stapled peptide precursor through a bifunctionalized bicyclononyne polyethylene glycol (PEG)-based linker to generate the peptide-drug conjugate. The receptor binding activity test of the peptide-cobimetinib drug conjugate demonstrated that this molecule exhibited strong binding activity across all receptors, confirming the efficacy of the stapling/conjugation approach in creating peptide conjugates that target MC1R as a molecular marker-drug combination. Furthermore, the introduction of certain small molecule moieties can modulate the interaction between stapled peptide ligands and receptors. Wu et al. [110] incorporated a group of SMoCs developed by Gooding et al. [185] on the dialkynyl aromatic staple (Figure 12C), resulting in an enhanced p53 response in comparison to analog with no SMoC groups. Optimizing the pharmacokinetics of peptide ligands is of great significance for their practical application [186, 187]. Xiao et al. [188] used a one-component strategy to study the influence of PEGylation on the conformational stability of the β-sheet WW domain. They incorporated amino acids containing both PEG motifs and an alkene function and utilized RCM to cross-link the staple. Besides PEG, there are other reagents used in improving the pharmacokinetics of peptides. Yang et al. [149] designed and synthesized a series of biaryl-stapled peptides, which displayed potent GLP-1R/GIPR dual agonist activity. To prolong their serum half-life, they chose to link two different lengths of fatty diacids to the biaryl staple via labile ester bonds (Figure 12D). Through incubation in fresh mouse serum, the results showed that the peptide modified with a C18 fatty diacid chain has a three-fold longer half-life in the serum compared to the peptide modified with a C10 fatty diacid chain. This is likely due to the longer chain fatty diacid promoting a tighter binding with serum albumin, thereby protecting the lipid-modified stapled peptide from esterase degradation.
Expanding the range of functionalities needed in the laboratory is also of great importance for the application of peptide ligands. Photoaffinity labeling is valuable for investigating the interactions between proteins and ligands or biomolecules [189]. It involves the use of a photoactivatable functional group that, upon exposure to UV light, can form a covalent bond with nearby biomolecules [190]. This irreversible process allows for subsequent analysis of the protein-ligand interaction [191]. In a study, Braun et al. [192] utilized photoaffinity probes to capture proteins in the BCL-2 homology domain 3 (BH3)/BCL-2 complex covalently by introducing unnatural amino acids with benzophenone tags into HCS BH3 peptides (Figure 12E). Similarly, Wu and his team [193] combined double-click stapling groups with photoaffinity labeling within a single linker, and then added a protected alkyne handle to incorporate a biotin moiety for pull-down assays (Figure 12F). The binding affinity of the stapled probe was comparable to that of previously studied p53 stapled peptides. After UV irradiation, the probe effectively crosslinked with MDM2 and displayed specificity towards MDM2 over competing BSA. Currently, this methodology is limited to labeling purified proteins and known PPIs. However, this linker strategy shows promise for future application in studying other PPIs and their corresponding peptidic inhibitors.
In recent years, stapled peptides have shown significant application promise in specific inhibition of PPIs, continuously attracting the attention of drug developers as they have the potential ability to combine optimal properties of both small molecules and biologics. Stapled peptides can better target PPIs compared to small molecule drugs, partly due to their larger size involved in the interaction, in addition, they have enhanced cell permeability and proteolytic stability compared to biological biologics. Hence, this work gives an overview of the continuous efforts of researchers in this field mainly in the past five years, aimed at providing a broad reference for accelerating the development of stapled peptides as the next-generation therapeutic peptide drugs targeting various human diseases. This review focuses on several significant considerations demanded for the design of therapeutic stapled peptides by extracting information from multitudinous recent literature. It starts with the design principles of peptide sequences, after that the screening for different stapled positions, reaction types, and properties of staples, and then the characterization of in vitro bioactivity including targeting specificity, binding affinity of target proteins, and ability to resist protein hydrolysis, and next the cell permeability required for targeting intracellular PPIs, finally the optimization of stapling such as the introduction of stapling with different properties or functions. Taken together, stapled peptides allow for the high selectivity and efficiency in targeting PPIs within cells, and have the potential to achieve precise adjustments to physical and chemical properties.
Despite the various advantages mentioned above, stapled peptides still confront great challenges, particularly high synthetic costs. In addition, ALRN-6924, as the only stapled peptide drug that has entered clinical trials so far, has a serious drawback of not being able to produce long-term inhibition of related PPI in vivo. The increasing awareness of the utility of stapled peptides prompts a higher demand for the development and optimization of performance, which means that it is imperative to develop AI approaches as powerful assistants to achieve a higher success rate of therapeutic peptides in a much-shortened timeframe. The approach used for drug development using AI is not significantly different from the analysis or prediction of proteins. Based on a solid database formed by massive previous research, once the target protein is determined, the incorporation of these features and the existing library can be used as parameters for AI to achieve accurate design of drug molecules (Figure 13). The recent surge of the application of AI methods in biomedical research showcases the potential as a promising designer candidate for in-depth investigations of therapeutic peptides [194–196], Bhardwaj’s [194] and Ma’s group [196] have demonstrated the feasibility of using machine learning methods to achieve de novo design of cell-penetrating macrocycles and accurate design of AMPs. The evolution of AI in the design of therapeutic peptides is undoubtedly with epoch-making significance, which perhaps will bring about the transformation of the new drug development model or even more new industrial revolutions in the near future, at when how to better transform biological problems into computing problems and then solve them with digital means will be an important challenge.
3D: three-dimensional
ACE2: angiotensin-converting enzyme 2
AI: artificial intelligence
AMPs: antimicrobial peptides
Arg: arginine
Asp: aspartic acid
ATG16L1: autophagy related 16 like 1 gene
BCL-2: B-cell lymphoma-2
CuAAC: copper(I)-catalyzed azide–alkyne cycloaddition
eIF4E: eukaryotic initiation factor 4E
Fmoc: 9-fluorenylmethyloxycarbonyl
FOXP3: forkhead box protein P3
GLP-1: glucagon-like peptide 1
GLP-1R: glucagon-like peptide 1 receptor
Glu: glutamic acid
hACE2: human angiotensin-converting enzyme 2
HCS: hydrocarbon-stapled
His: histidine
KRAS: Kirsten rat sarcoma viral oncogene homolog
Lys: lysine
MafG: musculoaponeurotic fibrosarcoma homolog G
Mag2: magainin II
MAML1: mastermind-like transcriptional coactivator 1
MC1R: melanocortin 1 receptor
Mcl-1: myeloid cell leukemia-1
MDM2: murine double minute 2
NC: net charge
NLS: nuclear localization signal
Nrf2: nuclear respiratory factor
PDB: Protein Data Bank
PEG: polyethylene glycol
PPIs: protein-protein interactions
RCM: ring-closing metathesis
RpoN: RNA polymerase sigma-54 factor
SAH: stapled α-helical
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
SMoCs: small molecule carriers
SPPS: solid-phase peptide synthesis
StAMP: stapled antimicrobial peptide
STRs: synthetic transcriptional repressors
Syt1: synaptotagmin-1
Tat: trans-acting activator of transcription
TF: transcription factor
Treg: regulatory T
UV: ultraviolet
WASF: Wiskott-Aldrich syndrome protein family
WT: wild type
QZ and ZW equally contributed to: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. XM: Investigation, Writing—review & editing. QC: Validation, Writing—review & editing, Supervision. CZ: Conceptualization, Writing—original draft, Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Qiushi Chen ... Xuechen Li
Ping Zeng ... Lanhua Yi
Shoko Tanaka ... Kohei Sato
Feliciana Real-Fernandez ... Paolo Rovero
Chloé O. Sebilleau, Steven J. Sucheck
Suze Ma ... Qi Zhang
Yanyan Liao, Xuefeng Jiang
Qian Wang ... Shiyu Chen
Jarais Fontaine, Jianfeng Cai
Oscar Noya ... Sandra Losada