Review
Review

Affiliation:
Department of Clinical Pharmacy, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
Email: 3821010008@stu.cpu.edu.cn
ORCID: https://orcid.org/0000-0002-9071-4212
Affiliation:
Department of Clinical Pharmacy, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
ORCID: https://orcid.org/0009-0003-5199-2118
Affiliation:
Department of Clinical Pharmacy, School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, Nanjing 210009, Jiangsu, China
Email: mingxu@cpu.edu.cn
ORCID: https://orcid.org/0000-0002-0655-2610
Explor Drug Sci. 2024;2:449–472 DOI: https://doi.org/10.37349/eds.2024.00056
Received: March 15, 2024 Accepted: May 31, 2024 Published: August 02, 2024
Academic Editor: Santiago J. Ballaz, Yachay Tech University, Ecuador
Age-related pathologies, particularly cardiovascular disorders, pose a significant global health concern. The World Health Organization (WHO) predicts an increase in advanced mortality by 2030 unless critical interventions are implemented. Atherosclerosis remains the major cause of various cardiovascular diseases. Hence, this review focused on the interaction between known mechanisms of vascular aging, disease manifestation, and progression during atherosclerosis. In the review, we highlighted five altered vascular mechanisms in cardiovascular models: genomic instability, neurohormonal deregulation, epigenetics, protein regulation, and the gut microbiome. The articles were selected from various indexed scientific databases. It is important to note that the mechanisms are equally interrelated with other aging pathways, such as inflammation and senescence. In conclusion, atherosclerosis is multifaceted and cholesterol-lowering therapy has been widely used. However, more than one specific action line is required to eradicate or slow down its manifestation. Equally, establishing a balance between aging stressors resulting in vascular injuries and stress buffering mechanisms during aging is critical to the treatment of atherosclerosis. The promising therapeutic targets reviewed include the angiotensin (1–7)/MAS axis, the gut microbiome, histone deacetylases, DNA repair systems, noncoding RNAs, β3/dopamine adrenoceptors, senescence and inflammation checkpoints.
Cardiovascular diseases (CVDs) pose a significant global health problem, accounting for 29.6% of deaths in 2010, reaching 17.9 million in 2019, with the World Health Organization predicting an increase by 2030. Atherosclerosis remains the major cause of various CVDs [1, 2]. Despite links to fatty degeneration and vessel stiffening, with the low-density lipoprotein (LDL)/high-density lipoprotein (HDL) ratio being significant risk factors, atherosclerosis is a chronic inflammatory condition that primarily causes CVDs, as demonstrated by studies such as canakinumab anti-inflammatory thrombosis outcome study (CANTOS), colchicine cardiovascular outcome and rescue trials [2–5]. Morphological, cellular, genetic, and consequently, functional alterations occur during atherosclerosis and aging [6]. The arterial walls harden, accompanied by a reduced elasticity [7]. Atherosclerosis involves the formation of plaques comprising necrotic cores, calcified regions, reorganized lipids, inflamed vascular smooth muscle cells (VSMCs), endothelial cells (ECs), and inflammatory factors [8]. Aged vessels show similar modifications as observed in atherosclerosis [9]. However, even in the absence of atherosclerosis, vessels can undergo vascular remodeling, leading to decreased elasticity [10]. These mechanisms that lead to the manifestation of the disease are currently englobed as hallmarks of aging. Various authors have defined aging mechanisms and their significant role in scientific research, particularly in disease and drug development [11–13]. In this review, we focus on five vascular alterations (genomic instability, neurohormonal regulation, epigenetics, protein regulation, and gut microbiome) leading to atherosclerosis during aging. In addition, we also discuss how these mechanisms also induce other aging phenomena, such as inflammation and senescence, because these mechanisms are significantly interrelated [13, 14]. Furthermore, we highlight current and potential therapeutic interventions.
Preliminary atherogenesis involves the trapping of lipoproteins at the injury site, allowing LDL cholesterol to enter ECs through endocytosis, increasing the concentration of LDL in the intima. Passive movement of oxidized LDLs (ox-LDLs) in ECs is facilitated by compromised endothelial barriers, activin-like kinase receptor 1 (ALK1), and scavenger receptors (SRs) [15]. The next process is endothelial activation, which occurs due to glycocalyx dysfunction and reduced cell contact, leading to weak junctions, promoting the absorption, uptake, and oxidation of triglyceride-rich lipoproteins in subendothelial cells [16]. CD36 and lectin-like oxidized LDL receptor 1 (LOX-1) regulate the effects of ox-LDLs on ECs. CD36 is the main binding receptor. ox-LDL through CD36 inhibits macrophage migration, potentially causing atherosclerotic lesions. The C-reactive protein-CD36 interaction reinforces macrophage uptake of ox-LDL [17, 18]. ECs are activated by ox-LDL in subendothelial spaces, causing monocyte adhesion and rolling through the expression of chemoattractant proteins [vascular adhesion molecule 1 (VCAM1), intercellular adhesion molecule 1 (ICAM1), P-selectin, monocyte chemoattractant protein 1 (MCP-1), and CC-chemokine receptor 2 (CCR2) and CCR5] [19, 20]. The CCR pairs involved in macrophage transmigration also include; CCR2-CCL2, CX3C CCR1, and CCR5-CCL5. In addition, T cell helper 1 (Th1) cells express the T-box transcription factor TBX21 (T-bet) and CCRs such as the CXCR3, CCR5, and IFNγ. In atherosclerotic ApoE-/- mice, Th1 cells express the atherosclerotic plaque-homing receptor CCR5 in lymph nodes [21]. Blocking of these axes in ApoE-/- mice reduces atherosclerosis progression by 90% [22]. CD4+ T cells in the atherosclerotic plaque express other pro-inflammatory cytokines associated with Th1 in addition to IFNγ, such as IL-2, IL-3, tumor necrosis factor (TNF), and lymphotoxin, which can all activate macrophages and aggravate inflammatory responses [21, 23, 24]. On the other hand, Th2 cells activate the signal transducer and transcription 6 (STAT6) through IL-4, resulting in up-regulation of GATA-binding protein 3 (GATA3), the key regulator of Th2 cell development. Mouse atherosclerotic plaques include cytokines associated with Th2 cells such as IL-4, IL-5, IL-10 (secreted by regulatory T cells), and IL-13. However, whether Th2 cells possess a pro-atherogenic or atheroprotective effect is still a subject of debate [25]. Interestingly, individuals with elevated Th2 cells in the peripheral blood mononuclear cells (PBMC) fraction have less preclinical atherosclerosis risk, as indicated by a lower thickness of the common carotid intimal media, compared to people with reduced levels of Th2 cells. Furthermore, the release of IL-4 from human activated mononuclear leukocytes in vitro is inversely associated with clinical atherosclerosis in ApoE-/- and LDLR-/- mice [23, 24]. In addition to counteracting the effects of Th1 and Th2 cells, IL-4 also affects other cells, which complicates the understanding of its unique role in atherosclerosis. Mouse models have demonstrated that IL-4 production results in increased expression of CD36, SR-A1 on macrophages, VCAM1, matrix metalloproteinase 1, and MCP-1. These findings suggest a potential link between IL-4 production and the development of atherosclerosis [24]. It is also estimated that during aging, T cell subsets become more proinflammatory [12, 21]. To progress, through macrophage colony stimulating factor (M-CSF), monocytes in the tunica intima differentiate into inflammatory macrophages (M1) and anti-inflammatory macrophages (M2) [26]. Mice lacking M-CSF show resistance to atherosclerosis [27]. Atherosclerotic lipoproteins (ox-LDL) are then taken in by monocyte-differentiated macrophages through the SR-A1 and CD36 [28, 29]. SR-B1 targets macrophages to minimize cholesterol trafficking and foam cell formation in atherosclerosis [30]. Foam cell production increases the expression of chemoattractant factors, allowing VSMCs to penetrate the intima. Soluble N-cadherin inhibits VSMC proliferation and macrophage foam cell apoptosis in vitro [31, 32]. Intima monocyte-differentiated macrophages via IL-1 promote the proliferation of derived growth factors. This stimulates VSMC proliferation and formation of extracellular matrix components that form a fibrous cap. The fibrous cap, necrotic core thickening, and arterial remodeling index can cause plaque rupture and thrombosis [33]. Please see Figure 1 and Figure 2 below.
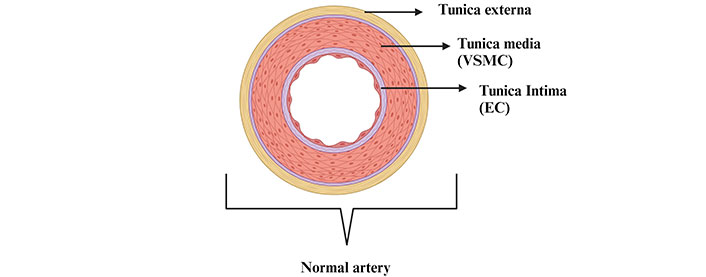
Anatomy of the normal artery [tunica externa is the outer layer; tunica media is the middle layer comprising mostly vascular smooth muscle cells (VSMCs); tunica intima is the inner most layer encompassing endothelial cells (ECs)]. Created with BioRender.com
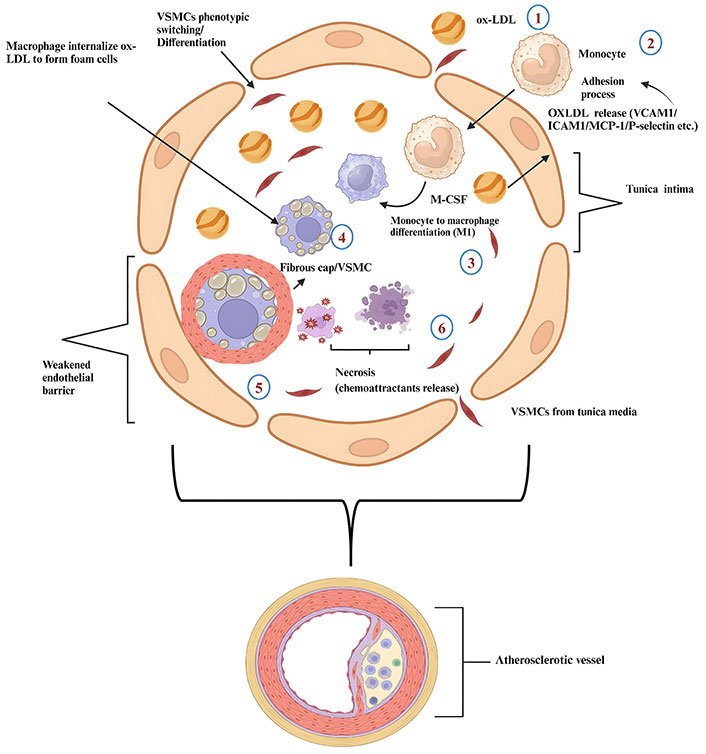
Pathophysiology of atherosclerosis 1: entrance of oxidized low-density lipoprotein (ox-LDL); 2: entrance of monocytes in response to ox-LDL secreted by ox-LDL; 3: differentiation of monocytes into M1 macrophages via macrophage colony stimulating factor (M-CSF); 4: foam cell formation; 5: vascular smooth muscle cells (VSMCs) and fibrous cap formation; 6: necrosis and liberation of pro-inflammatory factors to enhance the process of atherosclerosis]. ICAM1: intercellular adhesion molecule 1; MCP-1: monocyte chemoattractant protein 1; VCAM1: vascular adhesion molecule 1. Created with BioRender.com
Data from experimental aged animal models have portrayed the associations between DNA damage and atherosclerosis [34]. End-replication and oxidative DNA damage mechanisms alter DNA stability; therefore, failures in DNA damage response (DDR) sequences cause genome instability by activating DDR [35, 36]. The result of constant DDR is the activation of cyclin-dependent inhibitor pathways, including p53/p21Cip and p16INK4A/Rb [37]. With the successful repair of damaged DNA regions, standard cellular mechanisms are resumed [38]. However, unresolved DNA damage induces nuclear events that activate ataxia-telangiectasia mutated (ATM) and post-translational modifications of the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) essential modulator (NEMO), thus activating NF-κB, senescence (characterized by the expression of pro-inflammatory and immune-modulatory chemokines and cytokines, growth factors and matrix metalloproteinases) and, consequently, inflammatory circuits in atherosclerosis [39, 40]. This is evident in aged animal models that have been studied [41, 42]. Another evidence of an association between atherosclerosis, inflammation, and genetic instability involves the immune checkpoints CD80/CD86-CD28 and CTLA4. Breaks in DNA cause the expression of class I MHC and costimulatory molecules (CD80/CD86-CD28) in fibroblasts, together with adhesive molecules such as VCAM1 and ICAM1 [43]. Evidently, genetic deletion of CD80/86 vitiated the development of atherosclerosis in LDLR-/- mice by lowering the inflammatory feedback driven by T cells [44]. However, in the aortic regions, which comprise more advanced plaques, αCTLA4 antibody (negative regulator CD80/CD86-CD28) increased plaque T cell composition, indicating the importance of this pathway in interventional therapy for accelerated atherosclerosis [44, 45]. Patients with Hutchinson-Gilford progeria syndrome (HGPS) and Werner syndrome (WS) also show DNA detrition, cellular senescence, deregulated mTOR pathways (nutritional sensing pathway), and upregulated inflammatory responses and consequently succumb to complications from atherosclerosis [19, 46]. For instance, administering mTOR inhibitors in HGPS patients delays senescence [47]. Microsatellite instability (MSI) and loss of heterozygosity (LOH) are also commonly observed in atherosclerosis. For example, LOH at the hMSH2, hPMS1, and hMLH1 loci occurs in atherosclerosis. MSI and LOH have also been found on chromosomes 1, 2, 8, 9, and 17 in cerebral atherosclerotic plaques [48]. DNA repair has been shown to be less accurate in atherosclerotic tissues due to changes found in mismatch repair (MMR) genes [48]. The aortic samples from people with atherosclerosis had more damage to mitochondrial DNA (mtDNA) than the samples of healthy donors of identical age. These samples also had metabolic enzymes involved in fatty acid oxidation [49]. Manganese superoxide dismutase 2 (MnSOD2) deficiency and p66sch overexpression increase reactive oxygen species (ROS)-induced damage in mtDNA, accelerating atherosclerosis in ApoE-/- mice [50, 51]. In the same way, ApoE-/- mice models that were haploinsufficient for ATM/angiotensin (Ang) II receptor (ATR) repair mechanisms show worsened atherosclerotic lesions together with inflammatory markers in the mitochondria [52, 53]. The telomerase enzyme also ensures stable telomere lengths (TLs) and stability through the telomerase RNA component (TERC), telomerase reverse transcriptase (TERT), and dyskerin [54]. Their activities are reduced with age progression [54]. Despite contradictory results, associations have been observed between shorter TLs and CVDs [55]. For example, data from the West of Scotland Coronary Prevention Study Group (WOSCOPS) trial and other studies indicate a shorter TL as a predictor of cardiovascular risk factors such as atherosclerosis. Fibroblasts derived from patients with HGPS show premature senescence and telomere shortening [56]. VSMC, EC, and fibrous cap leukocytes also show shorter telomeres than usual during aging and plaque progression [57, 58]. The decreased expression of SRY-box transcription factor 4 (SOX4) genes in peripheral blood cells during aging also causes a shortening of TL, which contributes to senescence and atherosclerosis [59]. This is achieved by activating the tumor suppressor p53, reduction in levels of the coactivator 1- and the transcriptional regulator peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1-α (PGC1-α) and PGC1-β leading to mitochondrial dysfunction, deficient apoptosis, or EC senescence [60]. In short, DNA damage involves genomic alterations, adducts, and shortening of the telomere. In the same line, blood cells undergo somatic genetic mutations as we age, leading to expanded leukocyte clones and clonal hematopoiesis of indeterminate potential (CHIP) (another aging mechanism) [61]. Individuals with CHIP have increased cardiovascular related mortality. Data shows that mutations in DNA methyltransferase 3A (DNMT3A), tet methylcytosine dioxygenase 2 (TET2), and ASXL1 facilitate the clonal expansion of leukocytes. Furthermore, Macrophages from TET2-knockout hyperlipidemic mice result in abnormal activation of the NACHT, LRR, and PYD domain-containing protein 3 (NLRP3) and are absent in melanoma 2 (AIM2) inflammasome and contribute to enhanced atherosclerosis [60, 62, 63].
The renin-Ang-aldosterone system is crucial in the pathobiology of vascular disease, with Ang II acting through receptors ATR1 and ATR2. ATR1 promotes vasoconstriction, endothelial and cardiac dysfunction, aldosterone release, ROS, inflammation, and prothrombin activity [28]. During atherogenesis, Ang II stimulates Nicotinamide Adenine Dinucleotide Phosphate (NADPH) oxidase, causing the generation of superoxides in arterial wall cells. Superoxide is converted to hydrogen peroxide (H2O2), which promotes degradation of the IκB kinase complex, nuclear translocation of NF-κB, and transcriptional induction of pro-inflammatory genes such as VCAM1, MCP-1, or IL-6 [29, 64]. NF-κB inhibition reduces the expression of endothelial lipase, an enzyme that plays a role in ApoE and leukocyte and EC uptake during atherosclerosis when Ang II is present [65]. Furthermore, Ang II can potentially induce the polarization of RAW264.7 macrophages to the M1 type through the connexin 43/NF-κB pathway which may play a role in the development of atherosclerosis [66]. ATR2 and Ang-1–7/renin-Ang system (RAS) axis ensure cardioprotection by increasing resistance to cardiac remodeling mechanisms by modulating the calcineurin nuclear factor of activated T cells (NFAT) pathway (calcineurin/NFAT) [67]. In a cecal ligation and puncture (CLP)-induced inflammatory model, Ang converting enzyme 2 (ACE2)-Ang-(1–7)-Mas axis modulated M1/M2 macrophage polarization to reduce inflammation via toll-like receptor 4 (TLR4)-Mediated NF-κB and mitogen-activated protein kinase (MAPK) pathways [68]. Ang II, renin, and ACE activities physiologically decline with age, but local tissues show a marked increase in expression [69]. Ang II, ACE, and ATR1 levels have been found to be higher in the hearts of older mice and the thoracic aorta of C57/BL6 mice [70]. Ang II also induces premature senescence of VSMC and accelerates atheroma progression through the p53/p21 pathway. Inhibition of this pathway decreases pro-inflammatory signals and arrests premature senescence, preventing age-induced atherosclerosis in mouse models [65]. Collectively, Ang II facilitates aging by increasing free radical production, mitochondrial dysfunction, and telomere attrition equally driving atherosclerosis [29].
Transforming growth factor-β (TGF-β) exhibits anti-aging properties. Some recent studies have described its involvement in cellular senescence and atherosclerosis via various mechanisms [71]. Older mice portray increased TGF-β expression [72]. At higher vascular levels, TGF-β with Ang II in the presence of T cells increases the collagen/elastin ratio in favor of collagen, drives inflammation, senescence, and consequently, EC dysfunction, a common characteristic of atherosclerosis [72–75]. TGF-β has both anti- and pro-inflammatory properties. It lowers the expression of pro-inflammatory cytokines MCP-1 and inducible nitric oxide synthase (iNOS), while simultaneously increasing the expression of IL-10 via the SMAD/p21/MAPK/Rho/ROCK pathways, as reviewed by Chen et al. [76]. On the other hand, defective TGF-β expression of TGF-β could be a driver of cardiac aging and atherosclerosis at higher concentrations (observed in aged mice) by stimulating the expression of chemoattractants such as IL-1 and IL-6, negatively regulating noncoding atheroprotective mRNAs, collagen synthesis, and upregulating some adhesion molecules and pathways dependent on plasminogen activator inhibitor (PAI) [72, 76].
Insulin-like growth factor-1 (IGF-1) also has cytoprotective properties through antioxidant and anti-inflammatory effects on the vascular systems, contributing to atherosclerotic stability and ensuring longevity at lower levels [77]. Generally, these observations are made in the early stages of an organism’s lifespan [47, 78]. Its involvement in cardiovascular pathologies and cell senescence remains vague [79]. Some models show that the over secretion of IGF-1 and growth hormone (GH) is associated with atherosclerosis, longevity, and inflammation [80]. Interestingly, GH receptor (GHR) knockout mice with decreased serum IGF-1 levels lived 38–55% longer than wild-type mice [81]. Increased non-conjugated IGF-1 in the circulation stimulates atherosclerosis and senescence [82]. To support these results, the models of Harrington et al. [83] showed a connection between the genetic deletion of pregnancy-associated plasma protein A (PAPP-A), a metalloproteinase in the IGF system that increases the bioactivity of IGF, and resistance to the development of atherosclerotic lesions in high-fat mice lacking ApoE, which was accompanied by an extended life span. Mechanistically, PAPP-A impairs liver X receptor expression through the IGF-1/PI3K/Akt signaling channel, hence reducing ATP binding cassette A1 (ABCA1), ABCG1, and SR-B1 expression and cholesterol efflux in macrophage-derived foam cells thereby promoting plaque formation [84, 85]. However, Mediterranean diet supplementation promotes IGF-1 inhibition downregulates mTORC1 which activates the fork head box O 3A (FOXO3A), resulting in anti-inflammatory and gene homeostasis effects, thereby promoting longevity [47]. Equally, FOXO3 protects vascular ECs and VSMCs against oxidative stress injury by up-regulating the expression of antioxidant enzymes such as catalase and MnSOD, enzymes implicated in atherosclerosis [86]. Therefore, in both cases, the results remain contradictory because vascular concentrations leading to the occurrence of lesions have not been universally established.
Catecholamine levels increase with age, leading to increased tissue spillover and reduced plasma clearance [87]. Studies in adults with prevalent CVDs show increased white blood cell count, reduced baroreflex sensitivity, and advanced carotid atherosclerosis compared to those without a history of CVD [20]. Age-related increases in catecholamine can lead to β-adrenergic desensitization, as seen in the infusion of isoproterenol and dobutamine models [88]. In atherosclerosis, excessive sympathetic activation leads to cardiac hypertrophy, necrosis, and apoptosis and promotes inflammatory signaling [88]. Non selective inhibition of β1 and β2 reduces M1 content in atherosclerotic lesions [89]. Opposingly, β3-adrenoceptor and dopamine D1-like activation improve atherosclerotic plaques in aged ApoE-/- mice by attenuating lipid profiles (free cholesterol, cholesteryl ester, and non-HDL triglycerides) and suppressing macrophage proliferation induced by ox-LDL through the PI3/AKT and MAPK/ERK signaling pathways [90, 91]. The implication of α adrenergic receptors in atherosclerosis is not well elucidated. However, impeding α1 receptors improves lipid profiles in favor of atherosclerosis prevention while the latter is observed in α2 activation [81, 90, 92]. Furthermore, catecholamines autoxidize into highly reactive pro-oxidant species, including amino chromes, o-semiquinones, OH-, and O2-, promoting lipid peroxidation and cellular necrosis. The activation of catecholamine monoamine oxidase (MAO) in the plasma membrane also generates H2O2 and aldehydes, which opens the mitochondrial permeability transition pore (MPTP) to promote mtDNA damage induced by oxidative stress, cardiomyocyte death, and heart failure [87]. Both enzymes (MAO-A/B) are expressed on the mitochondrial surface. MAO-A inhibits mitophagy and induces senescence. However, MAO-B impairment retards atherosclerosis and the progression of nonalcoholic fatty liver disease [93]. To further study outcomes of sympathetic and atherosclerosis association, Li et al. [94] designed a model in which they observed the effects of renal denervation (RDN) and atherosclerosis progression in ApoE-/- mice. RDN reduces atherosclerosis, EC mitochondrial oxidative stress and inflammation by impairing MAO-A activities [94]. Acetylcholine is a protective neurotransmitter in atherosclerosis due to its role in impeding NF-κB, IL-6, TNF-α, and C-reactive protein expression [95], modulating senescence, proliferation of VSMC and antioxidant pathways via the α7 nicotinic acetylcholine receptor (α7nAchR) [96]. Therefore, its decrease in aged models leads to a change in the immune reflex axis towards NF-κB, hence driving senescence and the inflammatory pathways that contribute to atherosclerosis [97].
Epigenetics and aging mechanisms are linked to age-related diseases [98]. Individuals with advanced epigenetic caducity have an increased cardiovascular age and a higher risk of subclinical atherosclerosis progression [99]. Hypomethylation and DNA hypermethylation have been observed in the atheroma of ApoE-/- mice and monocyte/macrophage cell lines before and after the onset of atherosclerosis [100–103]. Patients with atherogenesis and familial hypercholesterolemia (FH) often exhibit hypermethylation of promoters and protective genes such as ABCA1 and Krüppel-like factor 4 (KLF4) [104]. Atherogenic enzymes and receptors, including SOD, endothelial nitric oxide synthase (eNOS), and estrogen receptors, are hypomethylated [105, 106]. This overshadows the protective mechanisms. DNMT1 disrupts PPARγ signaling, leading to atherosclerosis. TET2 counteracts this, restoring autophagic activities in EC and VSMC [107]. The somatic mutation TET2, which leads to accelerated atherosclerosis, is associated with NLRP3-mediated IL-1β production in clonal hematopoiesis [108].
Histone deacetylases (HDACs) affect the progression of atherosclerosis in various ways, from lipid metabolism, EC integrity, inflammatory pathways, and plaque disruption to modulation of oxidative pathways. The role of HDACs in atherosclerosis often leads to conflicting results. HDAC3 plays a role in EC integrity, but mice lacking it exhibit more stable plaques, decreased NADPH oxidase 4 (Nox4), reduced lipid content, and an anti-inflammatory phenotype during atherosclerosis. HDAC3 also inhibits PPAR and enhances foam cell formation [109]. HDAC9 interacts with the IκK/NF-κB p65 complex, leading to inflammatory phenotypes in macrophages and ECs linked to atherosclerosis. HDAC9-/- mice exhibit increased histone H3K9 acetylation at the ABCA1 and ABCG1 promoters, which affects macrophage cholesterol efflux and highlights its significant role in atherosclerosis [110]. HDAC6 causes EC injury and autophagy [111]. HDAC1/HDAC2 disrupts prolyl-4-hydroxylase alpha1 (P4Hα1) transcription, an enzyme in collagen formation, enhancing plaque destabilization. Furthermore, HDAC2 exhibits sexual dimorphic activity when inhibiting the expression of the antioxidant glutaredoxin-1 [111, 112]. HDAC7 suppresses VSMC proliferation and neointima formation by preventing β-catenin nuclear translocation [113, 114]. Recently, data suggest a more protective role for HDAC11 primarily through the expression of IL-10, IL-17, IL-10, IL-12, and TNF [115]. Members of the sirtuin (Sirt) family (Sirt1, Sirt3, and Sirt6) are studied for their antiaging and vasoprotective activities in atherosclerosis. Sirt1 modulates FOXO signaling, upregulates 5’-AMP-activated protein kinase (AMPK) (in low energy conditions), and restores normal autophagic activities (mTOR inhibition) [47], while Sirt6 targets inflammatory factors and loss of telomeres. SIRT3 protects against oxidative stress by upregulating PPAR‐α and eNOS and downregulating iNOS [116–118]. Sirt activities decline with age.
Non coding RNAs (ncRNAs) play a crucial role in atherogenesis, with their expression linked to EC dysfunction, inflammation, lipogenesis, and macrophage deregulation in atherosclerosis, with knockdown mice models demonstrating alleviation. Long ncRNAs (LncRNAs) and circular RNAs (circRNAs) modulate atherosclerotic progression via microRNAs (miRNAs) at the transcriptional and post-transcriptional levels. In cardiovascular development, three novel LncRNAs (TERMINATOR, ALIEN, and PUNISHER) have attracted great attention [119]. LncRNA H19 promotes the area of atherosclerotic plaque area in ApoE-/- by inhibiting cholesterol efflux mechanisms and upregulating NF-κB-mediated inflammatory responses in vascular ECs by miR-130b [120]. The lncRNA RP5–833A20.1 THP-1 was found to increase oxidized LDL uptake, lower cholesterol efflux, and increase the production of inflammatory mediators like TNF-α, IL-1β, and IL-6 in macrophages [121]. However, the expression of LncRNA-DYNLRB2-2 lowered the amount of cholesterol in macrophages by increasing the expression of G-protein-coupled receptor 119 (GPR119) and the cholesterol transporter ABCA1 [122]. LncRNA SMILR (smooth muscle-induced LncRNA) enhances replication and aggravates atherosclerosis by targeting the miR-10b-3p/KLF5 axis [123, 124]. LncRNA-p21 improves atherosclerosis by regulating the miR-221/SIRT1/Pcsk9 axis [125]. LncRNA-MALAT1 sponges miR-330-5p to activate the NF-κB signal pathway in THP-1 macrophage-derived foam cells in atherosclerosis pathogenesis [126]. Similarly, circRNAs are differentially expressed in atherosclerotic regions. Li et al. [127] found that 52 circRNAs were up-regulated more than twice and 47 circRNAs were down-regulated more than 1.5 times in atherosclerotic aortic vessels and of the circRNA examined, Mmu_circRNA_36781 (circRNA ABCA1) and 37699 (circRNA KHDRBS1) were significantly up-regulated in atherosclerotic aortic vessels and H2O2-treated mouse aortic ECs. They potentially regulated atherosclerosis and vascular endothelial injury by targeting the miR-30d-3p-TP53RK and miR-140-3p-MKK6 axes [127, 128].
Protein homeostasis declines with age, leading to abnormalities in the neurological and cardiovascular systems [129]. Numerous studies show that one of the main factors influencing atherogenesis is cell death [130, 131]. The signaling channels involved include the ubiquitin- proteasome, chaperons, and lysosome-dependent autophagy, with evidence found in human and animal cardiovascular models leading to atherosclerosis [132]. Madrigal-Matute et al. [129] found a decrease in activities and elevated inflammatory responses after chaperone-mediated autophagy (CMA) blockage in a fluorescent reporter mouse model, consistent with the results observed by Qiao et al. [133] (2021). Subclinical models demonstrate the essential involvement of the ubiquitin proteasome system (UPS) in NF-κB activation, where impaired UPS is associated with uncontrolled NF-κB pathways that drive inflammation in inflammatory-related disorders [131, 134]. Exploring the connection between atherosclerosis and UPS has revealed both pro-atherogenic and potentially preventive impacts. Increased inhibition of the proteasome accelerates the progression of atherosclerosis, while reduced inhibition enhances cholesterol efflux through ABCA1 and ABCG1 [135]. UPS is essential for the intracellular breakdown of proteins in all cells. Under stress, these proteasome activities are replaced by immunoproteasomes (IP), which mediate immune responses in various inflammatory diseases. Nevertheless, in LDLR-/-LMP7-/- and LDLR-/- mice fed on a Western-type diet for 6 weeks or 24 weeks to induce early and advanced stage atherosclerosis, respectively, depleted IP subunit β5i/LMP7 did not alter protein homeostasis and macrophage polarization, thus not aggravating atherogenesis in LDLR-/- mice [96]. The degradation of perilipin (protects lipid droplets from lipase activities) through the UPS pathway leads to the progression of atherosclerosis, while inhibition of adiponectin receptor 1 (ADR1) protects atherosclerotic models by altering the generation of foam cells [136]. Ubiquitin-specific peptidase 9 X-linked (USP9X), a deubiquitinating enzyme, inhibits macrophage lipid uptake by downregulating SR-A1, preventing foam cell formation [137]. Furthermore, ubiquitin C-terminal hydrolase L1 (UCHL1) and USP14 promote CD36 autophagic degradation, but their mechanical effects have opposite results: Inhibition of USP14 leads to the suppression of foam cells while deubiquitinating UCHL1 promotes the formation and stabilization of foam cells [138, 139]. Heat shock protein 60 (HSP 60) is the most recognized HSP 60 with known direct atherogenic potential. However, HSP 70, HSP 90, and HSP 47 have shown the potential to aggravate atherosclerosis in ApoE-/- mice and THP-1 derived macrophage models as reviewed by various authors [139–144]. In the same line, HSP 27 demonstrates atheroprotective activities in vitro [142].
The gut microbiome and the immune system are interdependent, and disruptions can increase vulnerability to disease and influence drug candidates’ pharmacokinetic or pharmacodynamic results. Studies show that as we age, the diversity of the gut microbiome decreases due to a loss in organisms’ physiological and symbiotic capacities, leading to reduced longevity and age-related anomalies [145]. Reprogramming of the gut microbiome induced by trimethylamine N-oxide (TMAO) is a typical example that serves as a prognostic index for CVDs and senescence [146, 147]. In aged animal models, TMAO is elevated and accelerates cell senescence by causing mitochondrial damage, superoxide formation (ROS), and promoting the generation of pro-inflammatory factors such as caspase-1 and IL-1β through the ROS-TXNIP-NLRP3 signaling pathway, thus aggravating atherosclerosis [148, 149]. It can also stop the SIRT3-SOD2-mitochondrial ROS signaling pathway, which damages ECs and leads to the development of atherosclerosis [148]. Zhu et al. [150] found that an enriched titanium dioxide diet (E71)/TMAO in ApoE-/- mice caused the appearance of lesions, mediated by E71, which transformed choline into pro-atherogenic TMAO. Consistently, Traughber et al. [151] reversed cadmium and high-fat-induced atherosclerosis by inhibiting TMAO production, NLRP3 machinery, and macrophage polarization in the gut microbiota of mice. TMAO also reduces bile acid formation, increases foam cell formation, and promotes oxidative stress-induced mitochondrial damage, leading to pro-inflammatory factors, senescence, and atherosclerosis during aging [148]. The alteration of the intestinal microbiome reduces butyrate-producing bacteria, increasing intestinal permeability and lipopolysaccharide levels, which activate inflammatory pathways, primarily in gram-negative species [152]. The intestinal microbiomes also produce hydrogen sulfide (H2S), which supports vascular tone, VSMC, cholesteryl ester (CE) activities, and energy production under normal conditions, but under strenuous conditions, H2S becomes pro-atherogenic [153–156]. Specifically, H2S is produced by three key enzymes; cystathionine β-lyase (CBE), cystathionine γ-lyase (CSE), and 3-mercaptopyruvate sulfurtransferase (MST)/cysteine aminotransferase (CAT) [157]. CSE is predominantly in the cardiovascular system [147]. These enzymes can be suppressed under conditions of high glucose, oxidative stress, and high fat diet (HFD) thus leading to a reduction in H2S production [156, 158]. For example, inhibition of MST by 2-[(4-hydroxy-6-methylpyrimidin-2-yl)sulfanyl]-1-(naphthalen-1-yl)ethan-1-one (HMPSNE) resulted in adipocyte differentiation and lipid accumulation with increased inflammation of adipose tissue [159]. CSE exhibits its antiatherosclerotic effect through 3 mechanisms, namely reduction of the chemoattractant factor, ICAM1, and CX3C CCR 1 (CX3CR1), inhibition of macrophage lipid uptake, and induction of VSMCs apoptosis through the MAPK pathway [157]. Although it has not been clinically tested, H2S has excellent potential as a treatment for this pathophysiological condition. Figure 3 below summarizes vascular mechanisms associated with aging and atherosclerosis materialization.
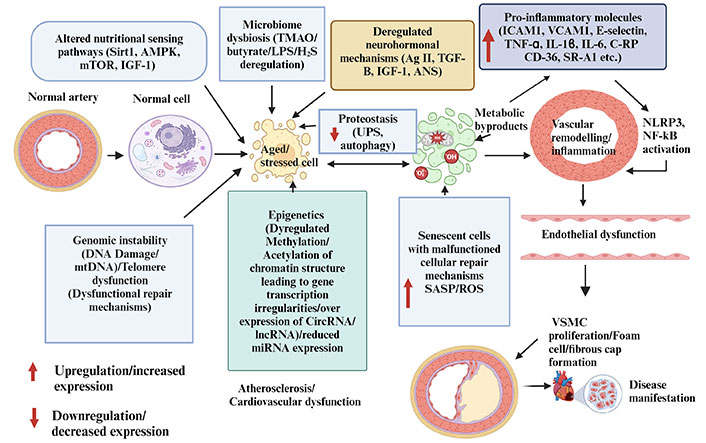
Vascular mechanisms (genomic instability, microbiome dysbiosis, neurohormonal deregulation, epigenetics, and proteostasis/protein deregulation) and atherosclerosis. These vascular mechanisms lead to increased senescent activities accompanied by elevated senescence associated secretory patterns which further aggravate inflammatory signals implicated in atherosclerosis. AMPK: 5’-AMP-activated protein kinase; ANS: autonomous nervous system; CircRNA: circular RNA; C-RP: C-reactive protein; H2S: hydrogen sulfide; ICAM1: intercellular adhesion molecule 1; IGF-1: insulin-like growth factor-1; LncRNA: long noncoding RNA; LPS: lipopolysaccharide; miRNA: micro RNA; mtDNA: mitochondrial DNA; NF-κB: nuclear factor kappa light chain enhancer of activated B cells; NLRP3: NACHT, LRR, and PYD domain-containing protein 3; ROS: reactive oxygen species; SASP: senescence-associated secretory phenotype; Sirt1: sirtuin 1; SR-A1: scavenger receptor A1; TGF-β: transforming growth factor-β; TMAO: trimethylamine N-oxide; TNF-α: tumor necrosis factor-α; UPS: ubiquitin-proteasome system; VCAM1: vascular adhesion molecule 1; VSMC: vascular smooth muscle cell. Created with BioRender.com
Statin therapy remains the gold standard in atherosclerosis due to its pleiotropic nature [160]. Given the complexity of atherosclerosis, it is essential to find more adjuvant compounds (Figure 4).
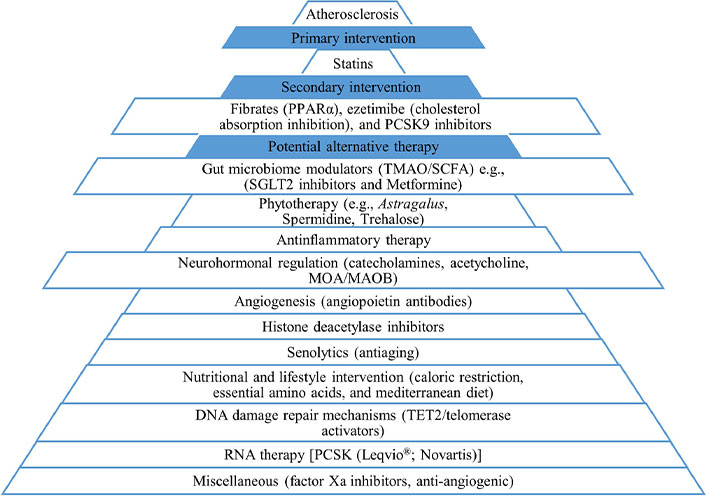
Various pharmacological interests in the treatment of atherosclerosis. PCSK9: proprotein convertase subtilisin/kexin type 9; PPARα: peroxisome proliferator-activated receptor α; SCFAs: short-chain fatty acids; SGLT2: sodium glucose transporter 2; TET2: tet methylcytosine dioxygenase 2; TMAO: trimethylamine N-oxide
Potentially beneficial HDAC inhibitors in the treatment of atherosclerosis mainly target class I and II HDACs. These include TMP195, RGFP966, MC1568, and sodium valproate (antiepileptic). Other nonspecific HDAC inhibitors are trichostatin and vorinostat. HDAC inhibitors that have entered clinical trial phases primarily target neurological and oncological disorders [161, 162]. Please see Table 1.
Histone deacetylases (HDACs) inhibitors
| Drug | Model and mechanisms | Structure | Author | Reference |
|---|---|---|---|---|
| TMP195 | HDAC9 inhibition (hyperlipidemic mice) | 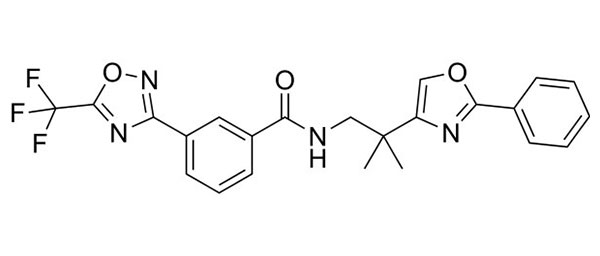 | Asare et al. (2020) | [163] |
| RGFP966 | HDAC3 inhibition (ApoE-/- mice) | 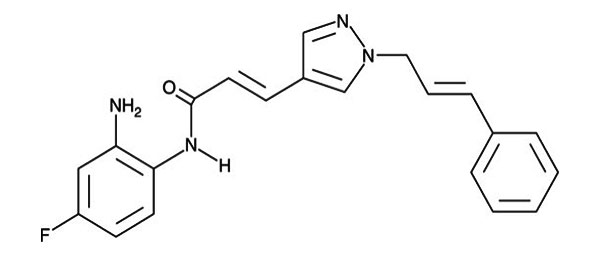 | Chen et al. (2021) | [164] |
| MC1568 | HDAC9 inhibition (ex vivo genetic knockout HDAC9 endothelial cells) | 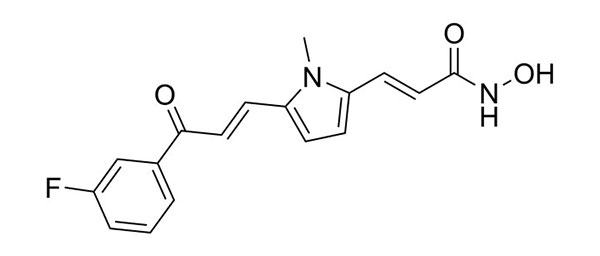 | Lecce et al. | [165] |
| Sodium valproate | HDAC9 inhibition (human patients) | 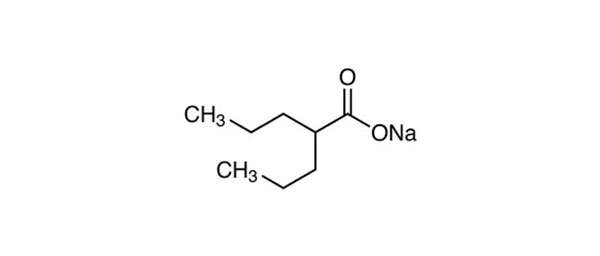 | Brookes et al. (2018) | [166] |
Reconstitution of TERC or TERT expression in TERC- or TERT-deficient mice with critically short telomeres resulted in telomere elongation, less DNA damage, decreases in aging biomarkers, and delay in age-related pathologies. For example, in preclinical models, gene therapy involving the administration of TERT adeno-associated virus (AAV), lentivirus, anellovirus, and lipid nanoparticles to young and old mice resulted in elongated telomeres that can prolong life expectancy and delay age-associated pathologies [61, 167]. Partially, the delivery of modified mRNA encoding TERT to human fibroblasts and myoblasts increased telomere proliferation in a dose dependent manner by 1012 fold. However, after 4 days of treatment, all treated cell populations eventually stopped increasing in number and expressed senescence markers to the same extent as untreated cells. This method is rapid, shows minimal risks of mutagenesis, and could have wide utility in regenerative medicine, drug development, and disease comprehension [60]. Furthermore, studies demonstrate that the use of key regulatory enzymes 8-oxoguanine DNA glycosylase (OGG1) can recruit TET1 into damaged DNA regions, thus preventing the activation of the inflammasome [168]. TA-65 telomerase activator (Astragalus, an antiaging agent), resveratrol, pterostilbene, PPAR agonists (e.g., pioglitazone), and statins, alleviate inflammation, senescence, increase leukocyte TL, promote stabilization of the Nijmegen breakage syndrome-1 protein, upregulate telomere repeat binding factor 2 (TRF2) and improve endothelial function with dietary supplementation, caloric restriction, and inclusion of polyunsaturated fatty acid [61, 169–171]. Clustered regularly interspaced short palindromic repeats-associated protein 9 (CRISPR/CAS9) technology has shown significant success in telomere-targeted therapy. However, little effort has been directed towards cardiovascular treatment [172]. A recent study by Xu et al. [173] (2024) used a CRISPR/CAS9 method to target the proprotein convertase subtilisin/kexin type 9 (Psck9) gene and successfully prevented the occurrence of atherosclerosis in hyperlipidemic mice. However, telomerase-targeting therapies in CVD face carcinogenic concerns.
Butyrate, a short-chain fatty acid (SCFA), has anti-inflammatory effects in animal models by creating barriers between lipopolysaccharides and the intestinal microbiome [174]. Research shows that reduction in choline substrate intake, along with acetylsalicylic acid and metformin therapy, can effectively lower TMAO in humans and ApoE-/- mice [175].
It should be noted that cellular senescence is an aging hallmark characterized by the presence of senescence associated secretory phenotypes (SASPs) [11–13, 176]. There is evidence atherosclerotic plaques encompass SASP components such as matrix metalloproteinases and multiple inflammatory factors [176]. SASPs drive atherosclerosis by upregulating inflammatory cascades involving the NF-κB, IL-1α, TGF-β, IL-8, and IL-6 [50]. Hence, senescence collectively alters DNA repair mechanisms and impairs antioxidant defenses, causing vascular inflammation and atherosclerosis in the presence of defective apoptotic pathways [176]. Clearing senescent cells in atherosclerosis is a therapeutic achievement. However, approved senolytic drugs, such as navitoclax (ABT-263) have been shown to make plaque less stable and increase mortality in atherosclerotic models [177]. Resveratrol indirectly prevents and attenuates cellular senescence by positively modulating energy and nutritional sensing pathways including AMPK and FOXO3A via Sirt1 [47]. Other drug candidates with antisenescent and anti-aging effects useful in atherosclerosis management have been summarized in the review by Sun et al. [176] (2022).
Targeting non-coding RNAs produces both intended and unintended effects. Mechanisms of drug delivery to atherosclerotic regions remain challenging in atherosclerosis. Inclisiran (Leqvio®; Novartis) is a first-in-class, cholesterol-lowering small interfering RNA (siRNA) approved by the FDA/EMA to target PCSK9 and has a favorable administration regimen compared to normal PSCK9 antibodies [178, 179]. It is used in patients who cannot achieve LDL cholesterol goals with the maximum tolerated doses of statins [180].
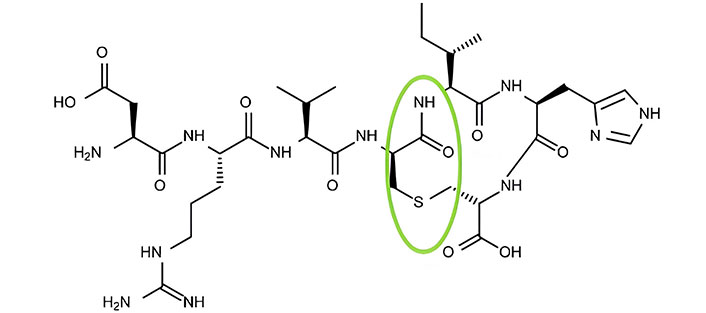
The multifaceted role of Ag II makes targeting RAAS equally crucial in the treatment of atherosclerosis. ACE inhibitors attenuate pro-inflammatory and oxidative pathways [70]. Ang (1–7)/RAS/ACE2 is atheroprotective, but its half-life and instability hinder drug delivery methods. Hydroxypropyl β-cyclodextrin-incorporated (HPβCD/Ang-1–7) formulations (HPβCD/Ang-1–7) address these problems by incorporating a thioether bridge, stabilizing the peptide against hydrolysis and increasing its half-life by eliminating the affinity of the AT1 receptor [181, 182] (Figure 5).
The development and progression of atherosclerosis are also related to various modalities of cell death (autophagy, apoptosis, necroptosis, pyroptosis, and ferroptosis) [183, 184]. Sodium glucose transporter 2 (SGLT2) and factor Xa inhibitors are additional main drugs that show beneficial effects when targeting these cell death pathways in atherosclerosis and require more extensive research [185, 186].
Recent investigations also evoke inflammation-induced intraplaque angiogenesis as a potential therapeutic target, as reviewed by Perrotta et al. [187]. Documented angiopoietin antibodies decreased intimal fatty streak formation in some mouse models of hypercholesterolemia-induced atherosclerosis, but did not affect preexisting plaques [188–190].
Although not fully covered, the review also highlights the importance of phytoextracts (e.g., Astragalus polysaccharides) in the management of atherosclerosis. Other natural pharmacological interventions that stimulate autophagy and Sirt activities (e.g., flavonoids, trehalose, or spermidine treatment) have been reported to reverse aspects of arterial aging and atherosclerosis [11, 191, 192]. Above all, contextualizing atherosclerosis to one particular pathway is impossible; therefore, more extensive research is paramount.
ABCA1: ATP binding cassette A1
ACE2: angiotensin converting enzyme 2
Ang: angiotensin
ATR: angiotensin II receptor
CCR2: CC-chemokine receptor 2
circRNAs: circular RNAs
CSE: cystathionine γ-lyase
CVDs: cardiovascular diseases
DDR: DNA damage response
ECs: endothelial cells
FOXO3A: fork head box O 3A
H2O2: hydrogen peroxide
H2S: hydrogen sulfide
HDACs: histone deacetylases
HGPS: Hutchinson-Gilford progeria syndrome
HSP 60: heat shock protein 60
ICAM1: intercellular adhesion molecule 1
IGF-1: insulin-like growth factor-1
LDL: low-density lipoprotein
LncRNA: long noncoding RNA
LOH: loss of heterozygosity
MAO: monoamine oxidase
MAPK: mitogen-activated protein kinase
MCP-1: monocyte chemoattractant protein 1
mtDNA: mitochondrial DNA
NF-κB: nuclear factor kappa light chain enhancer of activated B cells
NLRP3: NACHT, LRR, and PYD domain-containing protein 3
ox-LDLs: oxidized low-density lipoproteins
PPARγ: peroxisome proliferator-activated receptor γ
RAS: renin-angiotensin system
ROS: reactive oxygen species
SASPs: senescence associated secretory phenotypes
Sirt: sirtuin
SRs: scavenger receptors
TERC: telomerase RNA component
TERT: telomerase reverse transcriptase
TET2: tet methylcytosine dioxygenase 2
TGF-β: transforming growth factor-β
Th1: T cell helper 1
TLs: telomere lengths
TMAO: trimethylamine N-oxide
TNF: tumor necrosis factor
UPS: ubiquitin proteasome system
VCAM1: vascular adhesion molecule 1
VSMCs: vascular smooth muscle cells
We thank all members of the Department of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University, and the National Natural Sciences Foundation of China for their support.
SCW: Writing—original draft. XMJ: Conceptualization, Investigation, Writing—review & editing. MX: Conceptualization, Investigation, Writing—review & editing, Funding acquisition.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work is supported by the National Natural Science Foundation of China [82373870]. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.