 Review
Review

Affiliation:
1Hainan Institute, East China Normal University, Shanghai 200062, China
2State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
3Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
Affiliation:
1Hainan Institute, East China Normal University, Shanghai 200062, China
2State Key Laboratory of Petroleum Molecular & Process Engineering, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
3Shanghai Key Laboratory of Green Chemistry and Chemical Processes, School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200062, China
4School of Chemistry and Chemical Engineering, Henan Normal University, Xinxiang 453007, Henan, China
5State Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, Shanghai 200032, China
Email: xfjiang@chem.ecnu.edu.cn
ORCID: https://orcid.org/0000-0002-1849-6572
Explor Drug Sci. 2024;2:540–554 DOI: https://doi.org/10.37349/eds.2024.00060
Received: June 30, 2024 Accepted: August 11, 2024 Published: September 06, 2024
Academic Editor: Xuechen Li, The University of Hong Kong, China
The article belongs to the special issue Bioactive Peptides discovery and development
Chemo-select modification of peptides, targeting a handful of the most reactive proteinogenic amino acids (AAs), is gradually utilized to address the medical needs of peptide drugs and biopharmaceuticals. Cysteine (Cys), one of the less abundant AAs in many biological proteins, plays a vital role in the catalysis, signal transduction, and redox regulation of gene expression. In natural AAs (α-AAs) residues, Cys exhibits high nucleophilicity and low redox-active potential, making it a primary target for site-selective conjugation. This review summarizes several representative Cys-peptide/protein conjugation strategies developed in recent years, including polar reactions, radical coupling reactions, and stapling techniques.
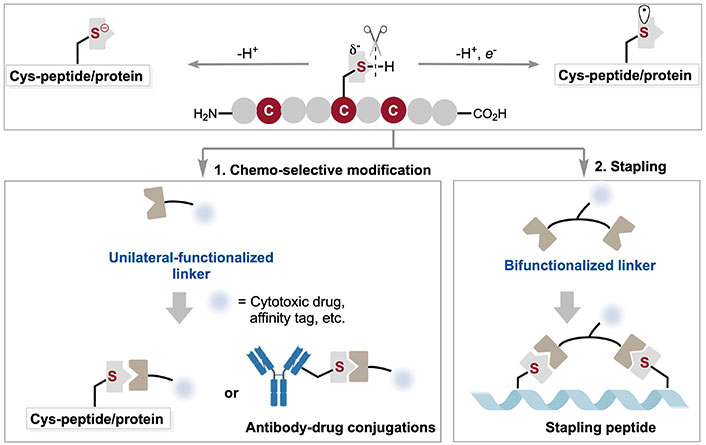
Chemo-selective modification of Cys residue: synthesis and application in the discovery of potential drug candidates. Cys: cysteine
Chemo-selective modification of peptides and proteins has been extensively utilized in molecular bioengineering, biorthogonality, and drug discovery, with applications in developing peptide drugs, antibody drugs, and molecular imaging probes [1]. Given the inherent structural complexity of peptides and proteins, chemo-selective modification [2] must not only proceed under physiological conditions (including aqueous phases and near-neutral pH) but also often require low reactant concentrations and high reaction rates to achieve high chemo-selectivity and site-specific bioconjugation. Although numerous strategies have been developed to directly target endogenous natural amino acids (AAs), achieving site-selective bioconjugation remains challenging due to the lack of significant differences in the activity of AAs at different sites within the same protein.
Considering that high-abundance AA residues on the surface of proteins often lead to a modified coupling mixture at various protein positions, a common approach is to select natural AAs with relatively lower abundance (Figure 1a), such as cysteine (Cys), tryptophan [3], methionine [4], histidine [5], and tyrosine [6], for modification. This strategy maintains the structural and functional integrity of the protein while ensuring the selectivity and specificity of the reaction. Among these methods, Cys peptide and protein bioconjugation protocols [7] have been extensively reported for their demonstrated chemo-selective reactivity under the requisite aqueous conditions, owing to its strong nucleophilicity and low redox potential (Figure 1b and 1c). Consequently, Cys is usually the primary target for site-selective coupling under physiological conditions [8]. Achieving site-selective chemical coupling is challenging due to the lack of significant differences in the activity of Cys at various sites within the same protein. Only a few specific reagents are utilized to modify N-terminal Cys [9]; most of chemical reagents used to modify this AA target the reactivity of the sulfhydryl side chain (-SH).
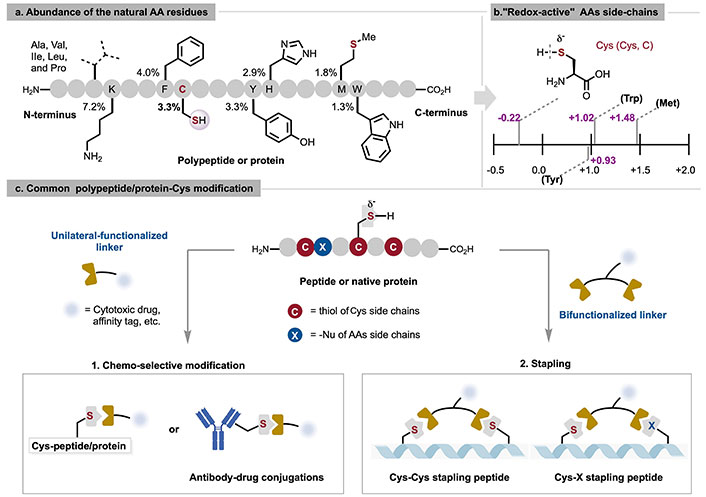
Graphical description of abundance of the natural AA residues in vertebrates (a), redox potentials of key proteinogenic AAs vs. Na+/H+ exchanger (NHE), at pH 7 [13] (b), and common polypeptide/protein-Cys modification (c). AAs: amino acids; Ala: alanine; Cys: cysteine; Ile: isoleucine; Leu: leucine; Met: methionine; Pro: proline; Trp: tryptophan; Tyr: tyrosine; Val: valine
Early strategies for the chemo-selective modification of Cys residues leveraged their high nucleophilicity to directly react with electrophilic reagents (e.g., disulfides [10], α-halogenated carbonyls [11], Michael acceptors, etc.) in nucleophilic substitution reactions (Figure 2). These methods have been widely used due to their rapid reaction kinetics and robustness. However, these coupling reagents can be easily interfered with by other nucleophilic AAs, resulting in poor chemical selectivity. Given the challenges associated with these bioconjugation strategies for nucleophilic substitution, some innovative methods have also been reported and applied, including rapid and effective Cys-arylation modifications with organometallic reagents, high-valent iodine reagents [12], or perfluorocarbons. In recent years, methods exploiting differences in the inherent reduction potential [13] of AA residues have been developed to achieve chemically selective modification of peptides and proteins, such as S-arylation, thio-ene/-yne reactions [14], and desulfurization reactions. Each strategy has unique advantages and disadvantages, which will be classified and analyzed in detail in the following sections.
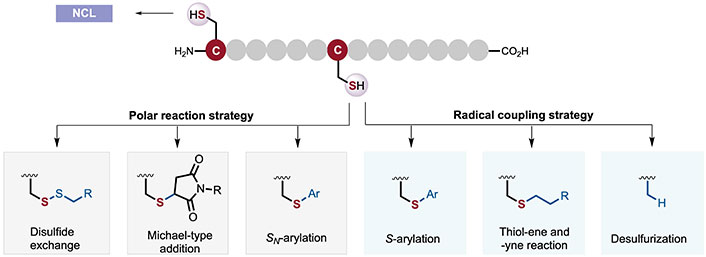
Methods for chemo-selective polypeptides/protein-cysteine (Cys) bioconjugation. NCL: native chemical ligation
As a promising drug for treating tumors, antibody-drug conjugates (ADCs) [15] covalently bind cytotoxic chemicals (known as warheads) to antibodies through linkers using a conjugation strategy. Eight of the thirteen ADCs approved by the FDA employ a Cys conjugation strategy, including Adcetris™, and Trodelvy™, Zynlonta™, among others [16] (Figure 3a). These marketed Cys-conjugated ADCs are primarily obtained by reacting thiol groups with various maleimide-based linkers [17]. The maleimide-thiol strategy has certain limitations, such as the need for a slightly acidic pH to avoid cross-reaction with lysine (Lys) residues and the retro-Michael reaction between the adduct and biological thiols in plasma, leading to premature release. To inhibit the retro-Michael reaction, although hydrolyzing the maleimide in the adduct can enhance conjugate stability and produce a more potent ADC, incomplete hydrolysis limits this strategy. Therefore, based on the maleimide-thiol strategy, an increasing number of Michael acceptors are being used as new linkers for Cys coupling [18–26], including 3-bromo-5-methylene pyrrolones (3Br-5MPs), α-unsaturated compounds, and vinyl/alkyne-phosphonamides, among others (Figure 3b).
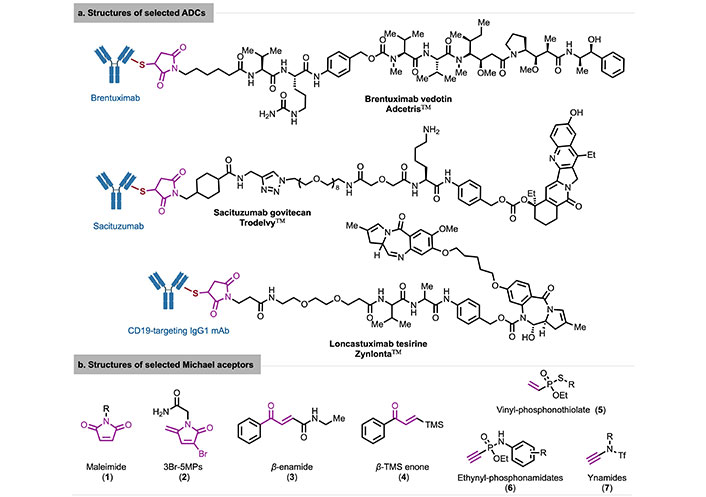
Structures of selected maleimide-containing ADCs (a) and selected Michael receptors (b). 3Br-5MPs: 3-bromo-5-methylene pyrrolones; ADCs: antibody-drug conjugates; TMS: trimethylsilyl
In 2019, Bernardim et al. [18] utilized carbonyl acrylic acid reagents as Michael acceptors to achieve chemo-selective and irreversible bioconjugation of Cys residues under biocompatible conditions (Figure 4a). The adduct conjugates formed by this strategy not only prevent the retro-Michael reaction with biological thiols in plasma but also remain unaffected by competing nucleophilic AAs (e.g., Lys) at slightly basic pH. Interestingly, modification of Thiomab LV-V205C with carbonyl acrylic acid reagent 3 resulted in a single modification in the light chain while leaving the heavy chain unaffected. The modified bioconjugate retained the antibody’s natural binding affinity to the HER2 antigen.
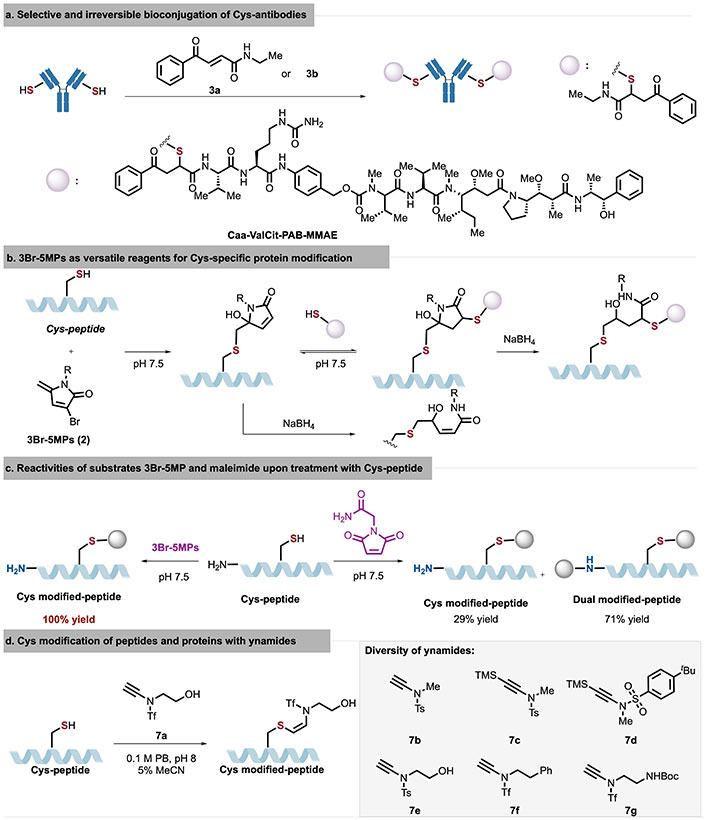
Graphical description of selective and irreversible bioconjugation of Cys-antibodies (a), 3Br-5MPs as versatile reagents for Cys-specific protein modification (b), reactivities of substrates 3Br-5MP and maleimide upon treatment with Cys-peptide (c), and Cys modification of peptides and proteins with ynamides (d). 3Br-5MPs: 3-bromo-5-methylene pyrrolones; Cys: cysteine; MeCN: methyl cyanide; NaBH4: sodium borohydride; PB: phosphate buffer
In 2020, Zhang et al. [21] employed 3Br-5MPs (2), an analog of maleimide, to achieve highly chemo-selective modification of Cys peptides/proteins, demonstrating labeling efficiency comparable to that of maleimide (Figure 4b). Following the initial addition of 3Br-5MPs to Cys peptides/proteins, a second addition can occur with thiol or Cys of another protein. Single or double adduct-products can be generated under sodium borohydride (NaBH4) reduction. Comparative analysis with different Michael acceptors (maleimide and 5MP) revealed that 3Br-5MPs exhibit higher Cys specificity than maleimide and greater reactivity than 5MP (Figure 4c).
Recent progresses in employing diverse Michael acceptors with electron-deficient alkynes, including phosphonamide [20] and ynamides [26], have been explored. Expanding their studies on ynamide chemistry, Hu and Zhao [27] developed a chemo- and regio-selective Cys modification protocol by taking advantage of the less studied β-addition of ynamides under the basic reaction conditions (Figure 4d). Ynamides showed unique reactivities owing to three modifiable sites, including the alkynyl moiety, the flexible modulation of electron-withdrawing group (EWG), and substituent on the nitrogen atom. Through a rational adjustment of three modifiable sites of the ynamide reagent 7, ynamide 7a possessed a stronger electron-withdrawing trifluoromethylsulfonyl group [triflyl (Tf)] on the nitrogen atom, which could enhance its activity as a Michael acceptor for nucleophiles. The conjugation reaction of ynamide 7a proceeded smoothly with an excellent efficiency (up to 95% yield) and stereoselectivity (Z:E > 99:1). Compared to those modified by maleimides and other related strategies, ynamide-modified peptide conjugate displayed a remarkable stability under a series of conditions in stability studies, such as acidic, basic, and oxidative environments. Interestingly, the Z-conjugated peptide could be further functionalized via an additional C-C triple bond to undergo click reaction, introducing a biotin tag or other useful functional groups.
Considering that Cys bioconjugation reagents need to operate under biomolecule-compatible conditions, they must modify the target with high selectivity and exhibit effective reaction kinetics. Reaction kinetics is typically an easily adjustable parameter, promoting the development of rapid Cys bioconjugation reagents. In recent years, various novel bioconjugation reagents have been reported, including hypervalent iodine reagents (8–10) [28, 29], organometallic reagents (11) [30–33], and perfluorocarbons (13) [34] with effective loads (Figure 5a).
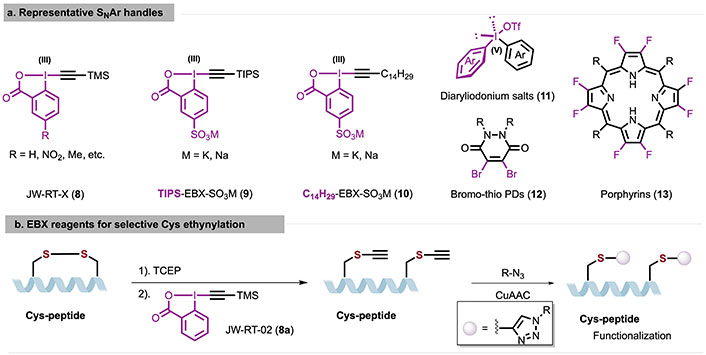
Graphical description of representative SNAr handles (a) and EBX reagents for selective Cys ethynylation (b). CuAAC: copper-catalyzed alkyne-azide cycloaddition; Cys: cysteine; EBX: ethynylbenziodoxolone; PDs: pyridazinediones; TCEP: tris(2-carboxyethyl)phosphine; TIPS: triisopropylsilyl; TMS: trimethylsilyl
In recent years, the hypervalent iodine reagents have been used as electrophilic group transfer reagents in peptides and proteins modification, as they exhibit low toxicity, high functional group tolerance, and stability in biocompatible media. Tessier et al. [29] have developed a class of electron-deficient high-valent iodine alkynyl reagents [ethynylbenziodoxolone (EBX)] to achieve thiol-alkynyl modification of Cys under near-physiological conditions. In 2020, Tessier et al. [29] used unstable trimethylsilyl (TMS) to design the EBX reagent into a finely tunable TMS-EBX reagent, allowing adjustment of the reaction rate and physical properties of the reagent for constructing bioconjugates (Figure 5b). The alkyne-modified Cys can be further functionalized by copper-catalyzed alkyne-azide cycloaddition (CuAAC). Compared to the labeling reaction rates of classic iodoacetamide (IAA) or maleimide for trastuzumab antibodies, EBX reagents exhibit similar or higher reaction rates and chemo-selectivity.
Visible light-induced [35–39] or electrochemically driven [40] Cys residues effectively generate reactive thiyl radicals without interference from other nucleophilic residues. This allows thiyl radicals [41] to participate in unique bond formations, creating more stable bioconjugates and providing a variety of tools for bioorthogonal applications (Figure 6). In addition to utilizing its nucleophilicity, the photocatalytic thiol-ene reaction has also been applied to Cys-peptides and proteins in recent years. The thiol-ene reaction, typically initiated by a free radical initiator or UV light, is considered a new type of click reaction. In 2020, Choi et al. [35] employed a water-compatible fluorescent photosensitizer, QPEG, and QCAT itself as photosensitizers to undergo hydrogen atom transfer (HAT) with blue LEDs, thereby highly chemo-selectively activating Cys residues to form thiyl radicals for thiol-ene reactions (Figure 6a). The constructed QPEG-Cys-conjugate possesses dual functionality: generating photoluminescence and singlet molecular oxygen (1O2), offering potential applications in image-guided photodynamic therapy. Slightly modified QCAT can introduce various important biologically relevant molecules (such as affinity tags like biotin, bioorthogonal handles like azides, and crizotinib, an ALK inhibitor) into Cys-peptide/proteins, thereby achieving efficient photocatalytic Cys-specific bioconjugation.
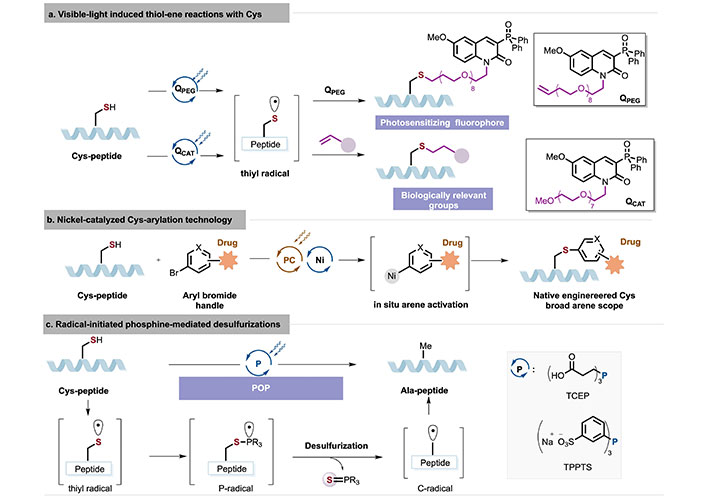
Graphical description of visible-light induced thiol–ene reactions with Cys (a), nickel-catalyzed Cys-arylation technology (b), and radical-initiated phosphine-mediated desulfurizations (c). Ala: alanine; Cys: cysteine; POP: phosphine-only photodesulfurization; TCEP: tris(2-carboxyethyl)phosphine; TPPTS: tris(3-sulfonatophenyl)phosphane trisodium salt
In 2023, Bacauanu et al. [39] reported a nickel/photoredox-mediated bioconjugation of Cys peptides/antibodies with aromatic bromides under the aqueous conditions. In the aqueous phase, photocatalysis facilitated the formation of low-valent nickel species and aryl-halide oxidative addition complexes in situ, while avoiding the oxidation of highly reactive AA residues (Figure 6b). Both native and engineered Cys-antibodies could be rapidly and highly chemo-selectively conjugated to a series of aromatic scaffolds (such as small molecules, probes, and cytotoxic payloads). All conjugates exhibited potent cytotoxic activity proportional to the drug-antibody ratio (DAR) in cell-killing assays, indicating that the target-mediated in vitro cytotoxic activity correlates with the stability of the S-arylation in bioconjugation.
In 2023, Venneti et al. [37] reported phosphine-only photodesulfurization (POP) for Cys-peptides using near-UV light (Figure 6c). This strategy does not require the addition of thiols to maintain the free radical chain and utilizes Cys as a hydrogen atom donor. It is noteworthy that a novel mode of alkyl thiol initiation was represented, in which photochemical cleavage of the S-H bond of alkyl thiols was induced by various phosphines. Furthermore, under very dilute conditions, one-pot disulfide reduction and multiple desulfurizations of linaclotide, aprotinin, and wheat protein were achieved, as well as the desulfurization of cyclic peptides and glucagon-like peptide-1 [GLP-1, (GLP-1(7–36))] A12C.
Many of the aforementioned methods enable rapid and efficient Cys bioconjugation. Despite the great potential of these approaches, they are often limited to the modification of a single Cys residue and/or a single transformation. Compared with single-Cys residue modification, stapling techniques involving the specific modification of two-Cys residues are much more complex. These techniques require a bifunctional bioconjugation reagent to react with one Cys while retaining an electrophilic site [42–44], which can then rapidly and efficiently couple with a nucleophilic residue of another molecule (such as Lys or Cys) to form a stapled peptide. Cys-stapling is a powerful technique for developing protein-protein interactions (PPIs) inhibitors for drug discovery [45], as it involves inserting bifunctionalized bioconjugation agents onto Cys residues to enhance the helical conformation of bioactive peptides, improve binding to biological targets, increase proteolytic stability, and enhance bioactivity/membrane permeability. Therefore, numerous Cys two-component stapling [46] techniques utilize alkylation or arylation chemistry of Cys residues. These techniques will be analyzed and explained below in terms of the two major categories of symmetric and asymmetric bioconjugation agents.
The use of bifunctional linkers aims to convert conformationally flexible natural peptide precursors into constrained and stable cyclic peptides through the formation of chemical bonds. Early methods employed various symmetrical dielectrophilic halides (such as allyl or alkyl halides) [47] for efficient and highly selective stapling of Cys residues (Figure 7a). Ceballos et al. [48] and Luo et al. [46] further expanded the variability of coupling linkers by introducing hypervalent iodine reagents and 1,4-dinitroimidazoles (1,4-DNIms). In 2023, Yu et al. [49] designed a thioether chloride reagent 14 with adjustable length, angle, rigidity, and site to address issues related to α-helix stability and peptide drug delivery for sulfur-containing peptides (Figure 7b). This reagent can efficiently link with Cys residues of peptides/proteins via Cys-Cys-(Cys) double/triple-click stapling in just 1 min constructing a series of 17-membered cyclic peptides, 18- to 48-membered macrocyclic peptides, and stapled bicyclic peptides. The stapled Cys-Cys-peptides were then subjected to tris(2-carboxyethyl)phosphine (TCEP), regenerating the native peptides with superb efficiency. The circular dichroism (CD) spectrum of the Cys-Cys-stapled peptides and biological characterization of anti-HCT-116 cells in this stapling protocol indicated an enhancement of both the α-helicity and the biological activity of linear peptides.
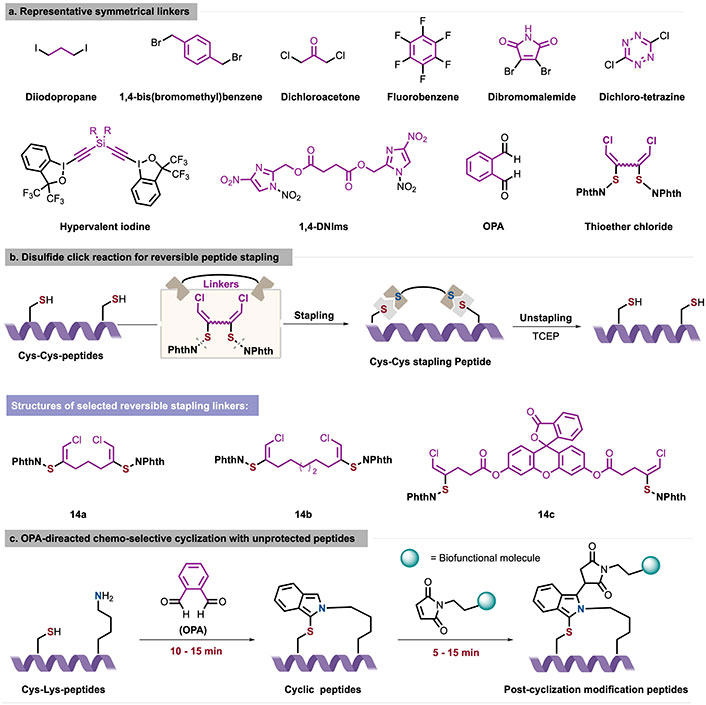
Graphical description of representative symmetrical linkers (a), disulfide click reaction for reversible peptide stapling (b), and OPA-directed chemo-selective cyclization with unprotected peptides (c). 1,4-DNIms: 1,4-dinitroimidazoles; Cys: cysteine; Lys: lysine; OPA: o-phthalaldehyde; TCEP: tris(2-carboxyethyl)phosphine
Although the symmetrical stapling strategy exhibited high chemical selectivity and orthogonality, these bifunctional linkers are limited to symmetrical stapling of two Cys residues. Notably, in 2019, Zhang et al. [50] and Cheung et al. [51] utilized o-phthalaldehyde (OPA) as a bifunctional symmetrical handle to achieve unsymmetric Cys-Lys stapling in native peptides. In aqueous PBS buffer (pH 7.4), the OPA-mediated cycling achieved full conversion within 10 min without any trace of the two-component reaction (2CR) product, indicating high chemo-selectivity under physiological conditions (Figure 7c). The OPA peptide cyclization product can further react with maleimide analogs in a one-pot manner to introduce additional functional motifs, such as fluorophore probes, glycans, and DNA. In the same year, Todorovic et al. [52] employed a similar intramolecular OPA-amine-thiol 3CR for kinase probe development.
The intramolecular OPA-amine-thiol reaction on peptides modification exhibits a simplicity, rapidity, and chemo-selectivity, hence, Cheung et al. [51] further achieved intermolecular OPA-mediated three-component conjugation in a single-step operation by suppressing the undesired rapid OPA-amine 2CR in 2023. This strategy avoided using an excess of thiol and utilized guanidine as an effective additive, which enabled the stoichiometric intermolecular OPA-amine-thiol 3CR by reversibly blocking the aldehyde groups in the key intermediates. Moreover, a set of peptide-peptide conjugates and peptide-drug conjugates (PDCs) have been constructed with excellent conversions and isolated yields in this method. In the same year, Li et al. [53] further applied OPA as a potential cross-linking anchor to primary amino groups of various AA residues.
Unlike the OPA-Lys-Cys stapling, various unsymmetric linkers [54–58] have been gradually reported for use in Lys-Cys stapling, including bischlorooxime (BC), N-hydroxysuccinimide (NHS)-activated acrylic ester, vinyl thianthrenium (VTT) salts, and vinyl tetrafluorothianthrenium (VTFT) salts, among others (Figure 8a). Here, we discuss two notable examples to demonstrate progress in this field. In 2023, Chen et al. [59] reported a Cys-directed proximity-driven strategy to achieve bicyclization of native peptides using a novel chlorooxime-based multifunctional cross-linker (Figure 8b). The bicyclization of peptides was achieved by kinetically controlling sequential residue modifications, which involved rapid Cys conjugation followed by proximity-driven intramolecular amide bond formation. This approach efficiently constructs a variety of peptide chain/side chain bicyclized peptides under biocompatible reaction conditions, such as N-terminal-Cys-Cys, Cys-Lys-Cys, and Lys-Cys-Lys cross-coupling. Additionally, site-specific bicyclization of peptide-protein [staphylococcal superantigen-like protein 11 (SSL11)] fusions can be achieved, reflecting the precise site selectivity of the Cys-directed strategy for site-specific Lys bicyclization in complex protein modifications. The model studies of N-terminus-Cys-Cys directed bicyclization on M13 bacteriophage indicate that a bicyclic AX3CX7C phage library could be readily constructed using this protocol, providing new structure diversity for peptide therapeutics.
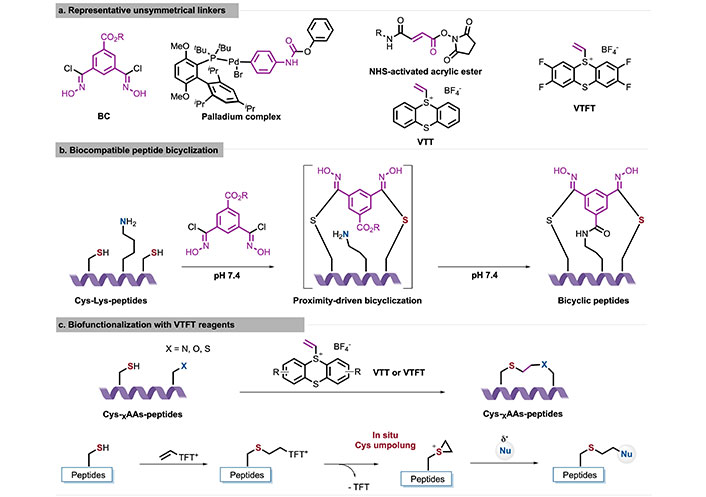
Graphical description of representative unsymmetrical linkers (a), biocompatible peptide bicyclization (b), and biofunctionalization with VTT reagents (c). AAs: amino acids; BC: bischlorooxime; Cys: cysteine; Lys: lysine; NHS: N-hydroxysuccinimide; VTFT: vinyl tetrafluorothianthrenium; VTT: vinyl thianthrenium
In 2024, Hartmann et al. [60] reported a Cys bioconjugation method using VTT/VTFT to convert Cys in situ into highly reactive electrophilic episulfonium intermediates, enabling bioorthogonal conjugation with various nucleophiles in a single step (Figure 8c). The site-specific bioconjugation of Cys residues with VTT exploits the cationic charge of thianthrene which discourages interaction in hydrophobic regions or environments with a high concentration of positively charged AAs. In the absence of exogenous nucleophiles, intramolecular nucleophilic AA residues can react with the episulfonium intermediate to achieve stapled peptides and protein-protein linkages. It is worth that a short two-carbon linker was introduced by VTT, which could recognize PPIs that were undetectable with other reagents making it an ideal choice for introducing infrared labels, post-translational modifications, or nuclear magnetic resonance probes.
Due to the limited abundance of Cys and the unique reactivity of its side chain functional group (-SH), chemo-selective bioconjugation of Cys residues plays a crucial role in peptides/protein modification. When the linker handle is equipped with a fluorescent group or a drug, it can be used for fluorescent labeling of proteins in living cells, aiding in the analysis of protein function, structure, dynamics, and intracellular trafficking pathways. Additionally, it can be used to create potential anticancer drugs with enhanced efficacy and reduced side effects by coupling potent cytotoxic substances with antibodies. Many FDA-approved ADCs in recent years are based on Cys site functionalization. Furthermore, various bifunctional linker handles are used to construct stapled helical peptides with more stable α-helical conformations, improving the pharmacokinetics and therapeutic properties of peptides, and thereby providing a broad platform for the development of PPI inhibitors for drug discovery. However, these new strategies still require further investigation. For example: 1) an unknown chiral center generated via thio-ene reaction results in two configurations of ADC drugs, raising concerns about toxic side effects; 2) site-selective coupling of Cys-peptides/proteins; and 3) the development of novel bifunctional linkers that not only enhance helicity or binding affinity but also improve cell membrane permeability. The ongoing development and advancement of peptide drugs and basic biological research will promote the diversified advancement of strategies for bioorthogonal and chemo-selective peptides/protein modification.
2CR: two-component reaction
3Br-5MPs: 3-bromo-5-methylene pyrrolones
AAs: amino acids
ADCs: antibody-drug conjugates
Cys: cysteine
EBX: ethynylbenziodoxolone
Lys: lysine
OPA: o-phthalaldehyde
PPIs: protein-protein interactions
VTT: vinyl thianthrenium
YL: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. XJ: Conceptualization, Writing—original draft, Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was supported by NSFC [22125103], STCSM [22JC1401000], and the Fundamental Research Funds for the Central Universities. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Qiushi Chen ... Xuechen Li
Ping Zeng ... Lanhua Yi
Shoko Tanaka ... Kohei Sato
Feliciana Real-Fernandez ... Paolo Rovero
Chloé O. Sebilleau, Steven J. Sucheck
Qian Zhang ... Chunqiu Zhang
Suze Ma ... Qi Zhang
Qian Wang ... Shiyu Chen
Jarais Fontaine, Jianfeng Cai
Oscar Noya ... Sandra Losada
