 Original Article
Original Article

Affiliation:
1State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
ORCID: https://orcid.org/0009-0002-6154-6149
Affiliation:
2Biotech Drug Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
ORCID: https://orcid.org/0000-0002-9759-7856
Affiliation:
1State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
Affiliation:
1State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
Email: hliu@simm.ac.cn
ORCID: https://orcid.org/0000-0003-3685-6268
Affiliation:
2Biotech Drug Research Center, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, China
Email: chenshiyu@simm.ac.cn
ORCID: https://orcid.org/0000-0001-6578-561X
Explor Drug Sci. 2024;2:719–733 DOI: https://doi.org/10.37349/eds.2024.00071
Received: July 17, 2024 Accepted: September 12, 2024 Published: October 17, 2024
Academic Editor: Xuechen Li, The University of Hong Kong, China
The article belongs to the special issue Bioactive Peptides discovery and development
Aim: Develop technology to apply bicyclic peptides for discovering covalent inhibitors of proteases and use this technology to create bicyclic peptide—warhead conjugates for targeting the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) 3C-like (3CL) protease. Enhance the potency of the discovered bicyclic peptides for potential development into anti-SARS-CoV-2 drugs.
Methods: Rational design was employed to discover the initial bicyclic peptide—warhead conjugates. Medicinal chemistry optimization was conducted to improve the potency of these peptides. Enzymatic assays and mass spectrometry characterization were performed to validate the covalent inhibition of the target protease.
Results: The need for peptide display selection in discovering hit bicyclic peptides was overcome. Active bicyclic peptide—vinyl sulfone inhibitors with nanomolar potency were discovered. Optimization through medicinal chemistry strategies not only improved the potency of the peptides but also revealed residue preferences at individual positions of the bicyclic peptide inhibitors. The most potent bicyclic peptide can inhibit the target with a half-maximal inhibitory concentration (IC50) of 40.46 ± 6.35 nM. Mass spectrometry tests confirmed the covalent inhibition of the target protease by the developed peptides.
Conclusions: Bicyclic peptide and vinyl sulfone conjugates are a form of covalent and potent inhibitors for targeting proteases. The rational design of bicyclic peptide ligands is feasible when structural and amino acid preference information is available. Structural information is also crucial for optimizing the potency of bicyclic peptide ligands.
As COVID-19 evolves into a seasonal, flu-like virus that spreads similarly to the common cold, it increasingly resembles other circulating coronaviruses [1]. The pandemic underscored the critical importance of having technologies ready to rapidly generate effective therapeutic methods. Vaccines against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) were promptly approved for clinical use and administered globally 13.07 billion times, saving a large number of patients [2]. Additionally, over 20 drugs have been approved or granted emergency authorization for treating COVID-19 infections, including Remdesivir, Molnupiravir, Paxlovid, Azvudine, Ensitrelvir, VV116, Favipiravir, Baricitinib, Proxalutamide, SIM0417, and RAY1216 [3, 4]. However, due to escalating virus mutations and persistently high infection rates worldwide, the effectiveness and safety of these treatments may diminish over time [5, 6]. This highlights the ongoing need to develop clinically valuable anti-SARS-CoV-2 drugs targeting key components of the virus’s lifecycle.
3C-like (3CL) protease, also known as the main protease (Mpro), is a cysteine protease crucial for the replication and transcription of SARS-CoV-2 (Figure 1A) [7–9]. This homodimer consists of two nearly vertical monomers, each with three domains, totaling 306 residues. A long and narrow cleft between domain I (residues 8–101) and domain II (residues 102–184) forms the substrate-binding pocket, surrounded by multiple hydrophobic residues. The catalytic triad (Cys145, His41, and Asp187) is also in a generally hydrophobic environment and is the target for multiple covalent inhibitor warheads [10, 11]. Aldehydes, nitriles, ketones, and α,β-unsaturated carbonyl compounds have been used to target 3CL protease for generating covalent inhibitors [12–18]. Compounds containing vinyl sulfone (VS) exhibit good stability and selectivity in vivo and have been widely used to develop covalent inhibitors of various cysteine proteases [19–21]. The discovery of covalent inhibitors of proteases has traditionally relied on constructing chemical libraries, where each compound must be evaluated individually. In contrast, constructing and screening combinatorial libraries not only reduces the effort involved in library creation but also improves the efficiency of the screening process.
In this work, we aimed to develop a potent covalent inhibitor targeting 3CL protease while minimizing unspecific off-target effects caused by the basal electrophilicity of the warhead. We selected a bicyclic peptide-based format for the inhibitors, conjugating them with a VS group. Due to the lack of technologies for screening bicyclic peptide- VS conjugation inhibitors, we explored the pathway of rational design. This approach successfully led to the discovery of several potent 3CL inhibitors. The discovered inhibitors demonstrated a half-maximal inhibitory concentration (IC50) inhibition in the 40.46 ± 6.35 nM range with 10 minutes of preincubation, and the covalent linkage between the peptide inhibitor and the target protease was also confirmed.
To a solution of N-Boc-L-glutamic acid dimethyl ester (1) (10.0 g, 36.3 mmol) in THF (100 mL) was added in portions a solution of lithium bis(trimethylsilyl)amide (LiHMDS) (80 mL, 1 M in THF) at −78°C under argon; then, the mixture was stirred at −78℃ for 1 h. Subsequently, bromoacetonitrile (2.70 mL, 38.8 mmol) was added dropwise to the dianion solution over 30 min while maintaining the temperature of −78℃, and the mixture was stirred at −78℃ for an additional 4 h. After the consumption of the reactant was consumed, the reaction was quenched with saturated aqueous NH4Cl (40 mL). After stirring for 30 min, the reaction mixture was warmed up to room temperature and extracted with EtOAc (50 mL × 3). The combined organic layers were concentrated under vacuum and purified by flash column chromatography [petroleum ether (PE)/EtOAc = 4:1] to afford 2 as a colorless oil (6.5 g, 58%). 1H-nuclear magnetic resonance [1H NMR (600 MHz, CDCl3)]: δ 5.20 (d, J = 8.8 Hz, 1H), 4.36–4.29 (m, 1H), 3.70 (s, 3H), 3.69 (s, 3H), 2.85–2.77 (m, 1H), 2.76–2.68 (m, 2H), 2.17–2.05 (m, 2H), 1.38 (s, 9H). Electrospray ionization mass spectrometry (ESI-MS) m/z 215.1[M–Boc+H]+ (see 1H NMR spectra at Figure S1).
A 250 mL round-bottomed flask was charged with compound 2 (6.5 g, 20.3 mmol) and CoCl2·6H2O (2.8 g, 12.1 mmol) in anhydrous MeOH (100 mL) at 0°C. Then, NaBH4 (4.7 g, 124.0 mmol) was added portion-wise at 0°C and the resulting solution was stirred at room temperature for 12 h until the reaction was completed. The mixture was quenched by saturated aqueous NH4Cl (30 mL). The solvent was removed under vacuum, and the residual mixture was diluted with water, and extracted with EtOAc (50 mL × 3). The combined organic layers were washed by saturated NH4Cl solution (100 mL × 3) and brine (100 mL × 3), dried over anhydrous Na2SO4, and concentrated to get the residue, which was purified by flash column chromatography (PE/EtOAc = 2:1) to obtain the product 3 as a white solid (3.0 g, 75%). 1H NMR (600 MHz, CDCl3): δ 6.57 (s, 1H), 5.55 (s, 1H), 4.29 (d, J = 10.7 Hz, 1H), 3.72 (s, 3H), 3.38–3.29 (m, 2H), 2.50–2.39 (m, 2H), 2.16–2.06 (m, 1H), 1.89–1.74 (m, 2H), 1.42 (s, 9H). ESI-MS m/z 187.7[M–Boc+H]+ (see 1H NMR spectra at Figure S2).
The compound 3 (3.0 g, 2.0 mmol) was dissolved in dry THF at 0°C, then NaBH4 (1.36 g, 37.0 mmol) was added slowly. Subsequently, the reaction mixture was warmed up to room temperature and stirred for 3 h. The completion of the reaction was monitored by thin-layer chromatography (TLC). To the reaction mixture was added water and concentrated to get a crude residue. The residue was dissolved in DCM and washed with saturated aqueous NH4Cl (50 mL × 3), NaHCO3 (50 mL × 3), and brine (50 mL × 3), dried over Na2SO4 and concentrated to afford a crude which was purified by column chromatography (DCM/CH3OH, 20:1 v/v) to afford the pure product 4 as a light solid (2.0 g, 75%). 1H NMR (600 MHz, CDCl3): δ 6.48 (s, 1H), 5.49 (s, 1H), 3.75 (s, 1H), 3.62–3.55 (m, 2H), 3.38–3.30 (m, 2H), 2.53–2.45 (m, 1H), 2.45–2.35 (m, 1H), 2.00–1.90 (m, 1H), 1.85–1.78 (m, 1H), 1.62–156 (m, 1H), 1.42 (s, 9H). ESI-MS m/z 259.2[M+H]+ (see 1H NMR spectra at Figure S3).
DMP (1.68 g, 4.0 mmol) was added slowly to the solution of the 4 (1.50 g, 3.3 mmol) in DCM (30 mL). The resulting mixture was stirred for 5 h at room temperature and monitored by TLC. The reaction mixture was concentrated and filtered through Celite. The solution was washed with saturated NaHCO3 solution (50 mL × 3) and brine (50 mL × 3), dried over Na2SO4, and concentrated to remove all the solvents to get the residue. The residue was purified by column chromatography (DCM/CH3OH, 20:1 v/v) to afford the pure product 5 as a light solid (0.79 g, 52%). 1H NMR (600 MHz, CDCl3): δ 9.56 (s, 1H), 6.09 (d, J = 7.9 Hz, 2H), 4.22–4.16 (m, 1H), 3.43–3.30 (m, 3H), 2.50–2.46 (m, 1H), 2.04–1.98 (m, 1H), 1.89–1.82 (m, 2H), 1.42 (s, 9H). ESI-MS m/z 257.3[M+H]+ (see 1H NMR spectra at Figure S4).
A dried flask under an argon atmosphere was charged with 4-hydroxy-thiophenol (2.52 g, 20.0 mmol) in THF (30 mL). The NaH (0.84 g, 21 mmol) was added to the solution slowly at 0°C and stirred for 30 min. Then, diethyl iodomethyl phosphonate (5.56 g, 20 mmol) was added as a solution in THF (10 mL). The reaction mixture was stirred for another 4 h at room temperature. The mixture was quenched with 20 mL of 5% potassium hydrogen sulfate and extracted with EtOAc (50 mL × 3). The combined organic layers were washed with brine (50 mL × 3), dried over anhydrous Na2SO4, and concentrated. The residue was purified by column chromatography (DCM/CH3OH, 20:1 v/v) to afford the pure product 8 as an oil (4.0 g, 58%). 1H NMR (600 MHz, CDCl3): δ 7.57 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 8.2 Hz, 1H), 4.19–4.2 (m, 2H), 4.12–4.05 (m, 2H), 3.49 (t, J = 15.0 Hz, 1H), 3.37 (t, J = 14.7 Hz, 1H), 1.33 (t, J = 7.0 Hz, 3H), 1.29 (t, J = 7.0 Hz, 3H). ESI-MS m/z 277.3[M+H]+ (see 1H NMR spectra at Figure S5).
To a solution of compound 8 (4.0 g, 14.4 mmol) in DCM (80 mL) was added m-chloroperoxybenzoic acid (m-CPBA, 5.0 g, 28.96 mmol), and then the resulting suspension was stirred for 2 h at room temperature. The completion of the reaction was confirmed by TLC and then quenched with aqueous saturated sodium thiosulfate. The resulting solution was diluted with water and concentrated by vacuum. The residue was extracted with EtOAc (50 mL × 3), and washed with H2O (50 mL × 2) and brine (50 mL × 2). The organic layer was dried with anhydrous Na2SO4 and concentrated in vacuo, and the residue was purified by column chromatography (DCM/CH3OH, 20:1 v/v) to afford the pure product 9 as a white solid (4.5 g, 73%). 1H NMR (600 MHz, CDCl3): δ 7.79 (d, J = 8.5 Hz, 1H), 6.75 (d, J = 8.6 Hz, 1H), 4.28–4.18 (m, 4H), 3.81 (d, J = 16.7 Hz, 2H), 3.77 (d, J = 11.9 Hz, 2H), 1.48–1.33 (t, J = 7.0 Hz, 6H). ESI-MS m/z 307.5[M–H]– (see 1H NMR spectra at Figure S6).
To a solution of 9 (647 mg, 2.1 mmol) in dry THF (20 mL), NaH (92 mg, 2.3 mmol) was added at 0℃ under argon. After stirring for 15 min, compound 5 (538 mg, 2.1 mmol) was added into the solution and the reaction mixture was stirred for another 1 h at room temperature. The reaction mixture was then extracted with EtOAc (20 mL × 3), washed with saturated brine, dried over anhydrous Na2SO4, and concentrated in vacuo to obtain the residue, which was purified by flash column chromatography (DCM/CH3OH, 20:1 v/v) to afford the compound 10. 1H NMR (600 MHz, DMSO-d6): δ 10.63 (s, 1H), 7.65 (d, J = 8.6 Hz, 2H), 7.61 (s, 1H), 7.26–7.18 (m, 1H), 6.94 (d, J = 8.8 Hz, 2H), 6.77–6.67 (m, 1H), 6.68–6.54 (m, 1H), 4.37–4.21 (m, 1H), 3.22–3.01 (m, 2H), 2.21–2.06 (m, 2H), 1.85–1.78 (m, 1H), 1.68–1.58 (m, 1H), 1.35 (s, 9H). 13C NMR (151 MHz, DMSO-d6): δ 178.59, 162.57, 155.61, 145.87, 131.14, 130.29, 130.20, 116.48, 78.65, 55.39, 49.48, 38.21, 35.56, 28.56, 27.86. ESI-MS m/z 409.5[M–H]– (see 1H and 13C-1H NMR spectra at Figures S7–8).
Compound 10 (300 mg, 0.73 mmol) was dissolved in DCM (10 mL), then the HCl (9 mL, 4 M in dioxane) was added. The reaction mixture was stirred for 12 h at 20°C, and the mixture was concentrated in vacuo to get a white solid 11, which could be used for the following step without purification.
All peptides were synthesized using the MultiPep 2 parallel peptide synthesizer (CEM) with standard solid-phase peptide synthesis (SPPS) protocols and Fmoc-protected amino acids. Each peptide was synthesized on a 50 µmol scale. The first Fmoc amino acid was manually loaded onto 2-chlorotrityl chloride resin (50 mg, 1.31 mmol/g, in 3 mL anhydrous DCM) by adding Fmoc-AA-OH (1 equivalent) and DIPEA (35 μL, 4 equivalent). After being shaken at room temperature for 1 h, the resin was washed with DCM/MeOH/DIPEA (17:2:1 v/v/v, 5 times), DCM (3 times), and dried with MeOH (3 times). Fmoc groups were then removed using 300 μL of a 20% (v/v) solution of piperidine in dimethylformamide, and amino acid coupling was achieved using a 4:4:6 ratio of amino acid: HBTU/HOBt/DIPEA in DMF. Following the completion of peptide synthesis on the solid phase, the peptide-loaded resin was treated with a 20% (v/v) solution of hexafluoroisopropanol in dichloromethane at room temperature for 1 h, which cleaved the peptide from the resin with side chain-protecting groups intact. The fully protected peptides were dried and coupled with (3S)-γ-lactam alanine VS under the condition of using a 1:2:4 ratio of (3S)-γ-lactam alanine VS/HATU/HOBt in DMF. Following water precipitation and filtration, the crude peptides were treated with cleavage cocktail K [1 mL, 90:2.5:2.5:2.5:2.5 (v/v) mixture of TFA:thioanisole:water:phenol:1,2-ethanedithiol] for 2 h, which also removed all protecting groups.
The diethyl ether-precipitated peptides were washed and recovered by centrifugation. 10 mg of each crude peptide was cyclized with 3.5 mg of 1,3,5-triacryloylhexahydro-1,3,5-triazine (TATA) or 1,3,5-tris(bromomethyl)benzene (TBMB) in 10 mL of 50% acetonitrile with 100 mM NH4HCO3. After incubation at 30°C for 1 h, the cyclized peptides were purified on a 1260 Infinity high performance liquid chromatography (HPLC, Agilent) equipped with a semi-prep C18 column (5 µm C18, 100 Å, 21.2 × 50 mm preparative LC column; Agilent) and separated over a linear gradient from 95% solvent A (water and 0.1% trifluoroacetic acid) to 95% solvent B (acetonitrile and 0.1% trifluoroacetic acid) in 18 min. HPLC fractions with the correct mass were determined and lyophilized to obtain the desired peptides.
The 3CL protease substrate peptide (Dacbyl-KTSAVLQSGFRKME-Edans) was synthesized by standard Fmoc-based SPPS on Rink Amide resin (0.458 mmol/g load). The resin was suspended in dry DMF (5 mL) and allowed to swell for 30 min. The solution was drained from the resin and the Fmoc group was removed with 20% (v/v) piperidine in DMF for 20 min at room temperature. The solution was drained and washed with DMF (5 mL × 5). Fmoc-Glu(EDANS)-OH (2 equivalent), HATU (2 equivalent), and DIEA (4 equivalent) in dry DMF (8 mL) were then added to the resin and mixed for 30 min. Fmoc-Glu(EDANS)-Rink amide AM resin was subjected to cycles of peptide coupling with Fmoc-protected amino acid building blocks as described above. After deprotecting the Fmoc from the N-terminus of the synthesized peptide, Dabcyl acid (2 equivalent), HATU (2 equivalent), and N,N-diisopropylethylamine (4 equivalent) were added to the washed resin and incubated for 30 min at room temperature. After TFA releasing and sidechain deprotection, the peptide was purified by HPLC and lyophilized to dryness.
Peptide stock solutions were injected into an Arc HPLC system (Waters) equipped with an XbridgeTM Peptide BEH C18 2.5 μm analytical column and run with a linear gradient of a mobile phase composed of eluent A [99.9% (v/v) water and 0.1% (v/v) formic acid] and eluent B [99.9% (v/v) acetonitrile and 0.1% (v/v) formic acid] from 5% to 95% over 5 min at a flow rate of 0.6 mL/min. The absorbance at the wavelength of 220 nm and 280 nm was used to generate plots of peptide purity.
A gene encoding the SARS-CoV-2 3CL protease was optimized and custom-synthesized. Following sequence confirmation, the gene was cloned into the vector pET28b between the NcoI and XhoI sites with a 3CL protease cleavage site before the C-terminal HisTag. The sequence-verified expression vector was then transformed into BL21(DE3) and induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) for expression in 500 mL of Luria-Bertani (LB) medium supplemented with 50 μg/mL kanamycin at 16°C for 16 h. The expressing cells were harvested by centrifugation and lysed in 50 mL of buffer R (20 mM Tris, 150 mM NaCl, pH 7.8) for releasing the expressed 3CL protease by sonication on ice. The centrifuged supernatant was loaded onto a HisTrap column for immobilization, and the His-tagged 3CL protease was eluted with 5 mL of buffer B (buffer R with 500 mM imidazole). HiPrep Sephacryl S-100 HR gel-filtration chromatography (GE Healthcare) was employed to further purify the fractions under the running condition of buffer R. PreScission protease (P2302, Beyotime) was used for hydrolyzing the C-terminal His-tag, and impurities were removed by flowing through glutathione S-transferase (GST) and nickel resin. The final 3CL protease was pure in gel and possessed authentic N- and C-terminal. The purified protein concentration was determined via bicinchoninic acid (BCA) protein assay and was > 90% pure, as estimated by a 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
The inhibitory activity of bicyclic peptides was determined by incubating SARS-CoV-2 3CL protease (50 nM) with different concentrations of inhibitors and quantifying residual activity using a fluorogenic substrate (100 μM, Dabcyl-KTSAVLQSGFRKME-Edans). Peptide inhibitors and SARS-CoV-2 3CL were incubated at 37°C for 10 min in buffer containing 50 mM Tris-Cl, 1 mM EDTA, 1 mM DTT, and 0.01% Triton X-100 pH 7.4. The residual activities of the target protease were monitored by adding the substrate and measured by monitoring the change in fluorescence intensity over 1 h using a SpectraMax M5e Microplate Reader (excitation, 340 nm; emission, 490 nm; Molecular Devices). For IC50 determination, 100% enzymatic activity was defined as the initial velocity of control triplicates containing no inhibitor, and the percentage of inhibition was calculated in relation to 100% enzymatic activity. The IC50 value for bicyclic peptides was determined using GraphPad Prism 9.3.0 software. Measurements of enzymatic activity were performed in triplicate and are presented as the mean ± standard deviation (s.d).
A solution of 3.7 μM 3CL protease mixed with 37 μM covalent peptide inhibitors in a reaction buffer (50 mM Tris, 1 mM EDTA, 1 mM DTT, pH 7.3) was incubated at 37°C for 1 h. Following the completion of the reaction, 5 μL of the crude product was injected into a Waters Xevo G2-XS QTof mass spectrometer equipped with a C4 column. Total ion chromatography was integrated and deconvoluted to reveal the mass of the product.
The 3CL protease is a member of the α/β hydrolase superfamily and features a catalytic triad composed of Cys145 and His41 (Figure 1A) [17, 22]. As part of the chymotrypsin-like protease family, 3CL protease specifically hydrolyzes substrates following glutamine residues. The S1 pocket, surrounded by His163, Phe140, and Glu166, is responsible for the specific recognition of glutamine and similar residues at the P1 position of substrate sequences [8, 17]. The S2–S4 pockets, positioned downstream of the S1 pocket, further refine the substrate recognition mechanism [23]. The S2 site, composed of Met49, Met165, His41, and Tyr54, displays hydrophobic characteristics and specifically requires a Leu residue in the native peptide substrate. The S3 site, positioned at the edge and formed by Met165, Leu167, and Gln192, generally shows no strong preference for specific amino acids on its flat surface. The S4 site, situated within a canal formed by Leu167, Phe185, Met165, Gln189, and Gln192, is capable of accommodating short hydrophobic amino acids.
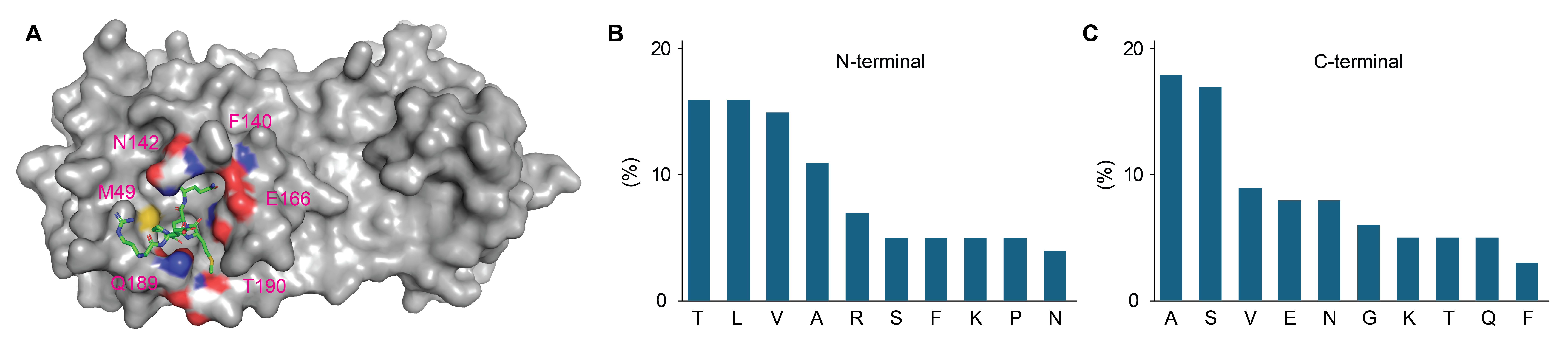
Illustration of the substrate binding site of 3CL protease and analysis of amino acid preferences of 3CL native substrate peptides. (A) 3CL C145A mutant in complex with nsp10-nsp11 cut site sequence (PDB ID 8DRX). The interactions between the residues and the substrate peptide were illustrated by coloring them according to atom type. Additionally, the corresponding residue names and their sequence numbers were indicated; (B) analysis of N-terminal residues of native substrate peptides recognized by 3CL protease; (C) analysis of C-terminal residues of native substrate peptides recognized by 3CL protease. 3CL: 3C-like
To facilitate the de novo design of 3CL protease inhibitors, we analyzed the native cleavage sites of the SARS-CoV-2 viral polypeptide. Examination of the S2–S4 peptides (Figure 1B) revealed significant interactions with Met49, Gln189, Thr190, and Met165. The hydrophobic center formed by Met49 and Met165 indicates a preference for hydrophobic amino acids such as valine and leucine on the N-terminal side of the substrate peptides. Additionally, hydrogen bond donor and acceptor residues, including Gln189, suggest a preference for residues such as threonine. On the opposite side of the catalytic center, the binding site is generally flatter, with residues like Asn142, Phe140, and Glu166 more exposed to the surface, allowing for interactions with the substrate (Figure 1C).
Based on the preferred amino acids recognized by the 3CL protease, we generated random combinations of C-terminal amino acids in tri-residue peptide sequences and applied them to the first loop of the binding bicyclic peptides (Table 1). Concurrently, the preferred amino acids at the N-terminal end of the substrates were recombined for the second loop of the bicyclic peptide binders. To enable the alkylation reaction with a trivalent linker [24], three cysteine residues were inserted flanking the two generated random tripeptide sequences, thus cyclizing the peptide into a bicyclic form. A cysteine-reactive VS warhead was conjugated to the C-terminal end of the peptide to facilitate covalent interaction with the target protease. Additionally, a (3S)-γ-lactam alanine was inserted to mimic the interaction with the S1 pocket of the 3CL protease. Considering the predominantly hydrophobic nature of the substrate peptides and the narrow, deep active site of the 3CL protease, we also inserted an additional leucine residue as the P2 residue in some of the designed peptides.
Illustration of the structures of designed bicyclic peptides conjugated with a vinyl sulfone warhead. The primary amino acid sequences of the designed peptides were followed by the inhibition rate at a fixed concentration of 10 μM or 1 μM. Those with potent inhibition of > 80% were further characterized for their IC50
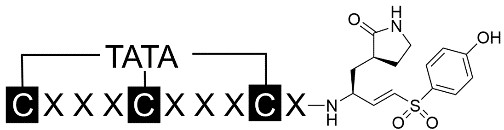 A series A series | 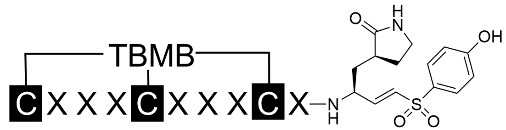 B series B series | ||
|---|---|---|---|
| Name | Amino acid sequences | Inhibition (%) | IC50 ± SD (nM) |
| BCP-1A | Ala-Cys-Val-Leu-Gln-Cys-Gly-Phe-Arg-Cys | 50.1% at 10 μM | - |
| BCP-1B | Ala-Cys-Val-Leu-Gln-Cys-Gly-Phe-Arg-Cys | 68.2% at 10 μM | - |
| BCP-2A | Ala-Cys-Ser-Gly-Phe-Arg-Cys-Arg-Val-Trp-Cys | 62.8% at 10 μM | - |
| BCP-2B | Ala-Cys-Ser-Gly-Phe-Arg-Cys-Arg-Val-Trp-Cys | 63.0% at 10 μM | - |
| BCP-3A | Ala-Cys-Ser-Gly-Phe-Arg-Pro-Cys-Arg-Val-Trp-Cys | 62.0% at 10 μM | - |
| BCP-3B | Ala-Cys-Ser-Gly-Phe-Arg-Pro-Cys-Arg-Val-Trp-Cys | 75.6% at 10 μM | - |
| BCP-4A | Ala-Cys-Ser-Gly-Phe-Arg-Cys-Arg-Pro-Val-Trp-Cys | 60.5% at 10 μM | - |
| BCP-4B | Ala-Cys-Ser-Gly-Phe-Arg-Cys-Arg-Pro-Val-Trp-Cys | 66.0% at 10 μM | - |
| BCP-5A | Ala-Cys-Arg-Gly-Ser-Gly-Cys-Pro-Asn-Ser-Thr-Cys | 53.4% at 10 μM | - |
| BCP-5B | Ala-Cys-Arg-Gly-Ser-Gly-Cys-Pro-Asn-Ser-Thr-Cys | 55.7% at 10 μM | - |
| BCP-6A | Ala-Cys-Gly-Ser-Gly-Arg-Cys-Ser-Gly-Val-Leu-Cys | 53.3% at 10 μM | - |
| BCP-6B | Ala-Cys-Gly-Ser-Gly-Arg-Cys-Ser-Gly-Val-Leu-Cys | 55.3% at 10 μM | - |
| BCP-7A | Ala-Cys-Ser-Gly-Thr-Arg-Cys-Ser-Gly-Phe-Leu-Cys | 50.1% at 10 μM | - |
| BCP-7B | Ala-Cys-Ser-Gly-Thr-Arg-Cys-Ser-Gly-Phe-Leu-Cys | 52.0% at 10 μM | - |
| BCP-8A | Ala-Cys-Ala-Gly-Arg-Cys-Pro-Ser-Ala- Cys-Leu | 82.2% at 1 μM | 357.43 ± 29.70 nM |
| BCP-8B | Ala-Cys-Ala-Gly-Arg-Cys-Pro-Ser-Ala- Cys-Leu | 96.8% at 1 μM | 153.63 ± 17.87 nM |
| BCP-8BNO | Ala-Cys-Ala-Gly-Arg-Cys-Pro-Ser-Ala- Cys-Leu | 8.7% at 1 μM | - |
-: no data. SD: standard deviation; TATA: 1,3,5-triacryloylhexahydro-1,3,5-triazine; TBMB: 1,3,5-tris(bromomethyl)benzene; BCP: bicyclic peptide; IC50: half-maximal inhibitory concentration
The mechanism of nucleophilic attack from the cysteine protease to the carbonyl bond of the substrate’s C-terminal amide was employed to generate covalent inhibitors of 3CL protease. The double bond of the VS was conjugated to the peptide’s C-terminal carbon atom. The formation of the γ-lactam side chain was achieved using Ritter reaction conditions, and the alkene was introduced via Horner-Wadsworth-Emmons reaction conditions (Figure 2) [17]. The P1 (3S)-γ-lactam side chain was used to create the P1-VS building block.
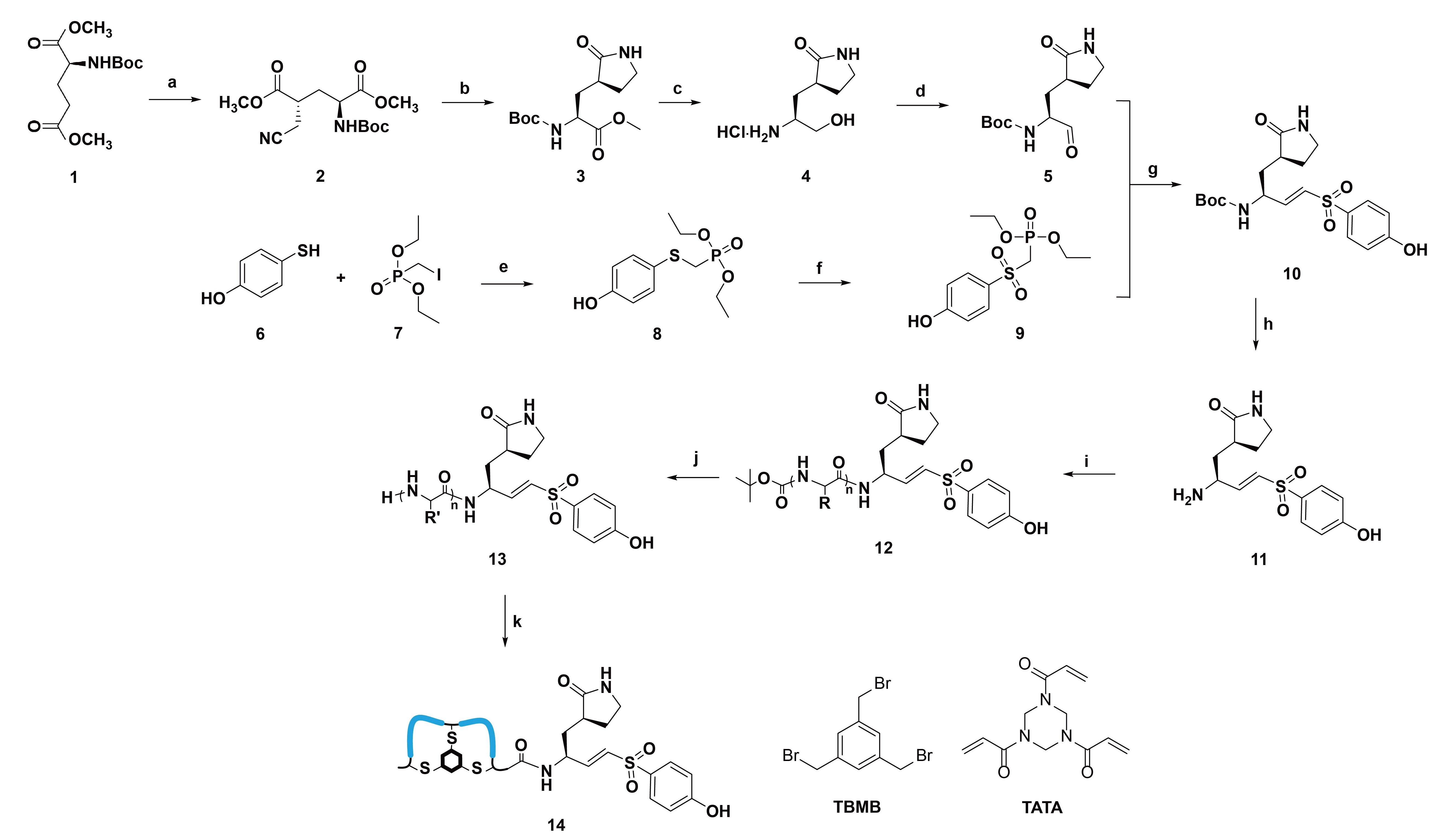
Synthetic procedure for bicyclic peptide conjugated with (3S)-γ-lactam alanine and vinyl sulfone at the C-terminus. Reaction conditions: (a) LiHMDS, THF, –78°C, bromoacetonitrile; (b) NaBH4, CoCl2·6H2O; (c) NaBH4; (d) DMP, DCM; (e) NaH, THF, rt; (f) m-CPBA, DCM, rt; (g) NaH, THF, rt; (h) TFA, DCM; (i) fully protected peptide, PyBOP, DIPEA, DCM; (j) TFA, EDT, water, thioanisole, phenol; (k) NH4HCO3 buffer, ACN, TBMB or TATA. LiHMDS: lithium bis(trimethylsilyl)amide; m-CPBA: m-chloroperoxybenzoic acid; rt: room temperature; TATA: 1,3,5-triacryloylhexahydro-1,3,5-triazine; TBMB: 1,3,5-tris(bromomethyl)benzene. The blue lines in the figure indicate the peptide loops
In the initial attempt to enable parallel peptide synthesis and streamline conventional SPPS of the designed peptides, a phenol group was introduced at one end of the building block to facilitate attachment to the solid phase during synthesis. Additionally, the N-terminal Boc protecting group was replaced with Fmoc to make it compatible with SPPS. The prepared building block was initially immobilized onto Wang resin to enable subsequent solid-phase peptide extensions. However, the low coupling efficiency between the hydroxyl group and the activated Wang resin resulted in insufficient resin loading for the following peptide synthesis. To overcome this, the P1-VS warhead block was deprotected at the amine group and coupled to an N-terminal Boc-protected and side chain fully protected peptide in solution, for achieving full conversion. The full-length peptide was then deprotected in a TFA cocktail cleavage solution and precipitated with diethyl ether to provide the crude peptide. This crude peptide was subsequently modified and cyclized using TBMB and TATA linkers (Figure 2) [24]. Following HPLC purification (Figure S9), the expected fraction with the desired mass corresponding to the bicyclic peptide-VS conjugate was obtained and dried for inhibition tests.
The synthesized peptide conjugates were tested for their ability to inhibit the in-house expressed and purified 3CL protease. Peptides at a final concentration of 10 µM were preincubated with 50 nM 3CL protease at 37℃ for 10 min, before adding 100 µM of the fluorogenic substrate (Dabcyl-KTSAVLQSGFRKME-Edans). The residual activity of 3CL protease after peptide inhibition was monitored by reading the fluorescent signal at Ex/Em 340 nm/490 nm for 10 min at 30-second intervals (Figure S10) [25].
The results indicated that the inhibition potency of peptides with the same primary sequence but cyclized with TBMB and TATA linkers did not differ significantly when their inhibition was moderate (BCP-1A to BCP-7B, Table 1). In contrast, BCP-8A and BCP-8B inhibited 3CL protease by 82.2% and 96.8%, respectively, at a concentration of 10 µM and were subjected to IC50 determination through serial dilutions. BCP-8A and BCP-8B showed differentiated inhibition of 3CL protease (Table 1), influenced by the cyclization linker, which is consistent with previous observations of conformational effects imposed by cyclization linkers [24]. To study the role of the warhead in 3CL protease inhibition, BCP-8BNO, lacking the P1-VS warhead, was cyclized with TBMB and tested for 3CL inhibition. BCP-8BNO did not inhibit 3CL protease at concentrations as high as 1 µM, revealing the critical role of the warhead in the inhibitory activity of the peptides. This indicates a covalent inhibition mechanism due to the electrophilic warhead.
To confirm the covalent inhibition of 3CL protease by the designed peptides, BCP-8A and BCP-8B were incubated with the protease, and the resulting complex was analyzed by MS following protein denaturation (Figure 3). This analysis revealed the covalent linkage between the peptides and 3CL, as evidenced by an increase in the mass of the 3CL protein corresponding to the molecular weight of the peptides. Encouraged by the observed potency and inhibition mechanism of the peptides, further optimization was performed to enhance their potency.
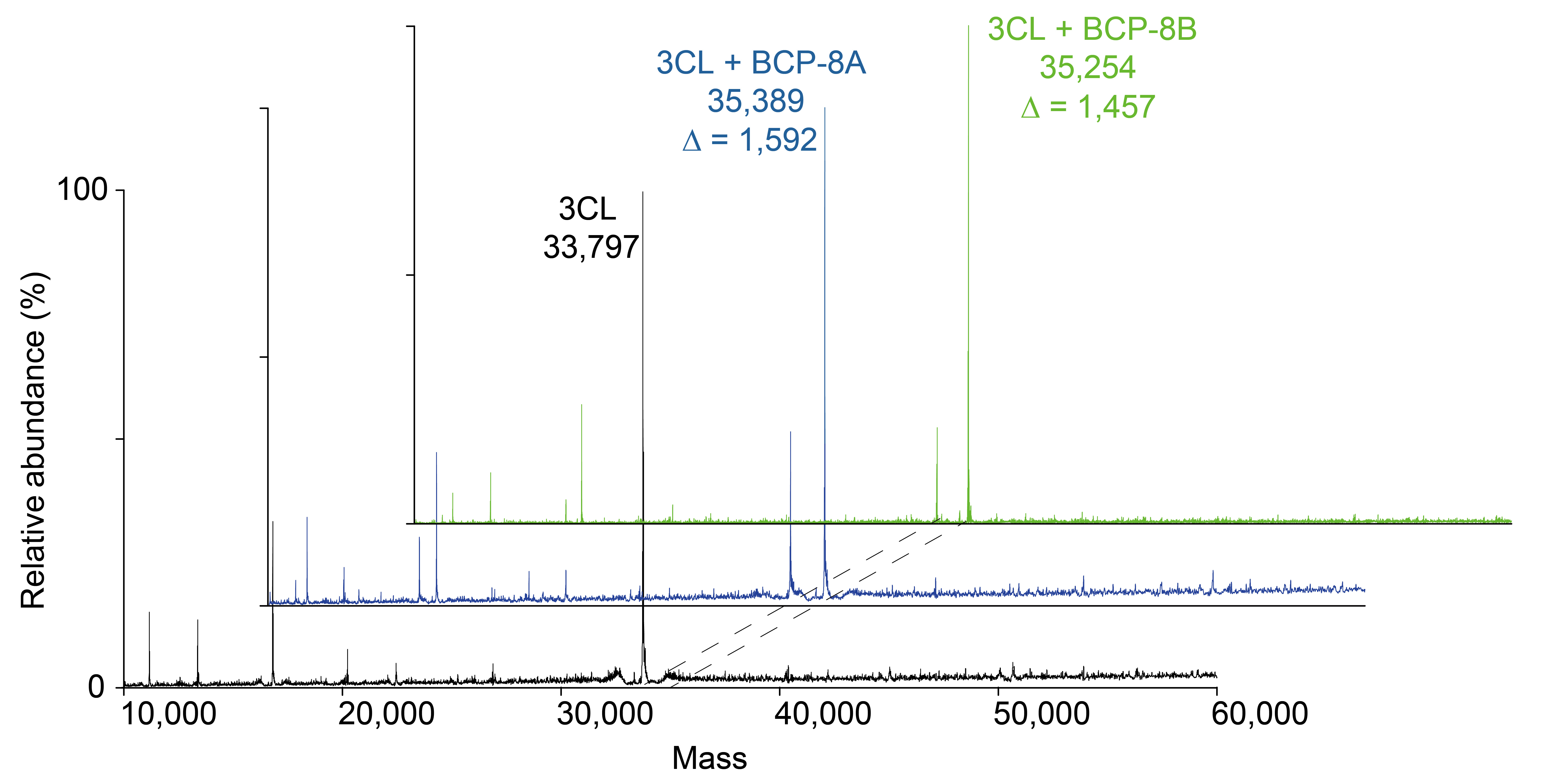
Mass spectrometry analysis of the 3CL protease and its covalent reaction with inhibitors BCP-8A and BCP-8B. The deconvoluted mass was indicated under the corresponding sample name, and the observed mass difference was also indicated for the protein-peptide complex samples. 3CL: 3C-like. ∆: mass shift
Based on BCP-8B, the peptide sequence was optimized to improve potency against the target 3CL protease. Internal non-alanine residues were individually replaced with alanine to study the role of each residue in inhibiting 3CL protease. Additionally, glycine-to-proline and proline-to-glycine mutations were introduced to preserve backbone angles in cases of cis-conformation peptide bonds (Table 2). The mutant peptides were tested for inhibition at a 1 µM concentration, and those with more than 90% inhibition were further quantified with serial dilutions to calculate the IC50.
The primary amino acid sequences of Ala scan peptides were listed with the inhibition rate at a fixed concentration of 1 μM. Those with > 90% inhibition were further characterized for IC50
| Name | Amino acid sequence | Inhibition (%) | IC50 ± SD (nM) |
|---|---|---|---|
| BCP-8B | Ala-Cys-Ala-Gly-Arg-Cys-Pro-Ser-Ala-Cys-Leu | 93.8% | 153.63 ± 17.87 nM |
| BCP-9B | Ala-Cys-Ala- | 66.2% | - |
| BCP-10B | Ala-Cys-Ala-Gly- | 46.6% | - |
| BCP-11B | Ala-Cys-Ala-Gly-Arg-Cys- | 81.8% | - |
| BCP-12B | Ala-Cys-Ala-Gly-Arg-Cys-Pro- | 69.7% | - |
| BCP-13B | Ala-Cys-Ala-Gly-Arg-Cys-Pro-Ser-Ala-Cys- | 3.4% | - |
| BCP-14B | Ala-Cys- | 10.4% | - |
| BCP-15B | Ala-Cys-Ala- | 99.5% | 62.00 ± 7.31 nM |
| BCP-16B | Ala-Cys-Ala-Gly-Arg-Cys- | 95.8% | 116.67 ± 7.79 nM |
-: no data. SD: standard deviation; IC50: half-maximal inhibitory concentration
Mutation of Gly4 to Pro (BCP-15B) and Pro7 to Gly (BCP-16B) improved the inhibition rate of the peptides (Table 2), indicating that a slight conformational change might be beneficial. The conformational change due to the mutation of Ala3 to Gly and the loss of side chains in other residues generally reduced the inhibitory activity of the peptides. Mutation of Leu11 to Ala (BCP-13B) resulted in a significant loss of potency. Considering its neighboring position with (3S)-γ-lactam, Leu11 was supposed to bind to the S2 pocket of the 3CL protease, which exhibits a strong preference for hydrophobic amino acids [26, 27].
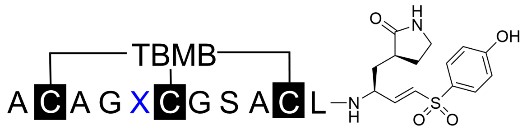
To further study the role of the side chains of Arg5, Ser8, and Ala9, additional mutant peptides were tested based on BCP-16B. The reason we chose BCP-16B is due to the presence of the flexible Gly4 and Gly7, which can potentially provide a more adjustable backbone and generate mutant peptides with the ideal conformation for inhibiting the 3CL protease. Replacement of Arg5 with various amino acids revealed that hydrophobic and negatively charged amino acids were less favorable. Amino acids with hydrophilic side chains were more favorable for binding the target, with substituting Arg5 with homo-arginine and serine showing the most potent inhibitory activity (Table 3). The results indicated that hydrogen bond donors and acceptors on side chains of different lengths can form networks of conserved hydrogen bonds that stabilize the binding interaction.
The primary amino acid sequences of the mutated peptides with variants at the fifth residue were listed with the inhibition rate at a fixed concentration of 1 μM. Those with > 90% inhibition were further characterized for IC50
 | |||
|---|---|---|---|
| Name | Amino acid sequence | Inhibition (%) | IC50 ± SD (nM) |
| BCP-16B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys-Leu | 95.8% | 116.67 ± 7.79 nM |
| BCP-17B | Ala-Cys-Ala-Gly- | 75.4% | - |
| BCP-18B | Ala-Cys-Ala-Gly- | 71.2% | - |
| BCP-19B | Ala-Cys-Ala-Gly- | 96.8% | 100.74 ± 16.29 nM |
| BCP-20B | Ala-Cys-Ala-Gly- | 80.1% | - |
| BCP-21B | Ala-Cys-Ala-Gly- | 67.4% | - |
| BCP-22B | Ala-Cys-Ala-Gly- | 99.3% | 79.15 ± 4.36 nM |
| BCP-23B | Ala-Cys-Ala-Gly- | 64.7% | - |
-: no data. SD: standard deviation; IC50: half-maximal inhibitory concentration
Replacements of Ser8 resulted in the discovery of several peptides with improved potency, indicating that serine is less ideal and that side chains of various sizes and hydrophobicities were acceptable for this site. Since both the sequence and surface shape of the target protein define the binding properties of peptide binders, the side chain at the eighth residue was supposed to bind to a more exposed and flexible pocket, and no hindrance effect could cause a dramatic loss of potency. By comparing various amino acid substituents, the introduction of hydrophobic amino acids (BCP-32B with Leu8 and BCP-33B with Ile8, Table 4) led to the discovery of the most potent peptide inhibitors, with IC50 values of 40.46 ± 6.35 nM and 48.17 ± 3.69 nM, respectively. This result is consistent with observations of P5 residue preferences of 3CL protease, showing tolerance for diverse amino acids, due to the flexible loop forming the S5 site and the conformational changes caused by inhibitor binding [26, 28].
The primary amino acid sequences of the mutated peptides with variants at the eighth residue were listed with the inhibition rate at a fixed concentration of 1 μM. Those with > 90% inhibition were further characterized for IC50
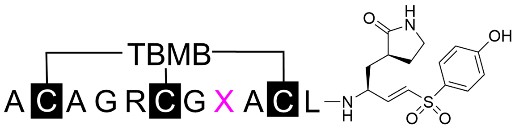 | |||
|---|---|---|---|
| Name | Amino acid sequence | Inhibition (%) | IC50 ± SD (nM) |
| BCP-16B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys-Leu | 95.8% | 116.67 ± 7.79 nM |
| BCP-24B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 98.8% | 80.56 ± 3.86 nM |
| BCP-25B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 57.78 ± 2.83 nM |
| BCP-26B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 88.8% | - |
| BCP-27B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 57.09 ± 4.79 nM |
| BCP-28B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 92.74 ± 14.33 nM |
| BCP-29B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 48.16 ± 6.28 nM |
| BCP-30B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 78.01 ± 4.70 nM |
| BCP-31B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 98.1% | 118.39 ± 17.34 nM |
| BCP-32B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 40.46 ± 6.35 nM |
| BCP-33B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 100.0% | 48.17 ± 3.69 nM |
| BCP-34B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 98.3% | 107.82 ± 11.47 nM |
| BCP-35B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 86.9% | - |
| BCP-36B | Ala-Cys-Ala-Gly-Arg-Cys-Gly- | 98.6% | 90.77 ± 7.41 nM |
-: no data. SD: standard deviation; IC50: half-maximal inhibitory concentration
Replacement of Ala9 with various amino acids exclusively reduced the inhibition potency of the peptides (Table 5), fitting with previous observations of P4 preferences for alanine with a small side chain [29], due to the crowded cavity formed by Leu and Pro at the bottom and Thr and Ala at the top. The hydrophobic nature of the S4 pocket also can’t tolerate the charged and polar amino acids and lead to the dramatic decrease of potency of the synthesized mutant peptide inhibitors.
The primary amino acid sequences of the mutated peptides with variants at the ninth residue were listed with the inhibition rate at a fixed concentration of 1 μM. Those with > 90% inhibition were further characterized for IC50
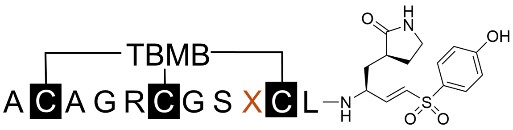 | |||
|---|---|---|---|
| Name | Amino acid sequence | Inhibition (%) | IC50 ± SD (nM) |
| BCP-16B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys-Leu | 95.8% | 116.67 ± 7.79 nM |
| BCP-37B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 7.5% | - |
| BCP-38B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 12.9% | - |
| BCP-39B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 11.4% | - |
| BCP-40B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 4.1% | - |
| BCP-41B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 26.0% | - |
| BCP-42B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 10.7% | - |
| BCP-43B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser- | 10.1% | - |
-: no data. SD: standard deviation; IC50: half-maximal inhibitory concentration
In the sequence of the designed peptide, no specific P3 residue was inserted. Due to the undefined nature of the S3 site of the 3CL protease, the side chain of the modified cysteine can act as a general amino acid for adapting to this pocket. Though this site has no selectivity, the extended structure of the acylated cysteine can serve as a flexible motif that avoids potential clashes.
The hydrophobic S2 site of 3CL protease is mainly formed by Met49 and Met165, and it highly prefers leucine for native substrate peptides. The deeper S2 pocket can also accommodate slightly longer and bulkier side chains. We chose to mutate Leu11 to several reported favorable P2 amino acids. The incorporation of phenylalanine (BCP-44B, Table 6) indeed improved the potency of the parental peptide. However, the mutation to 4-fluorophenylalanine led to an almost complete loss of inhibitory activity. This result indicates that the backbone of the bicyclic peptide may have adopted a more rigid conformation compared to native linear substrate peptides, and slight changes in the peptide structure may introduce dramatic clashes that can lead to significant changes in potency (Table 6).
The primary amino acid sequences of the mutated peptides with variants at the eleventh residue were listed with the inhibition rate at a fixed concentration of 1 μM. Those with > 90% inhibition were further characterized for IC50
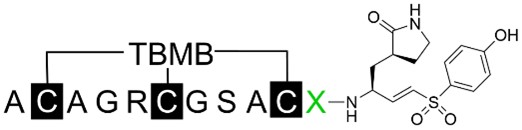 | |||
|---|---|---|---|
| Name | Amino acid sequence | Inhibition (%) | IC50 ± SD (nM) |
| BCP-16B | Ala-Cys-Ala-Gly-Arg-Cys- Gly -Ser-Ala-Cys-Leu | 95.8% | 116.67 ± 7.79 nM |
| BCP-44B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys- | 98.0% | 90.33 ± 3.95 nM |
| BCP-45B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys- | 93.6% | 155.77 ± 22.37 nM |
| BCP-46B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys- | 19.0% | - |
| BCP-47B | Ala-Cys-Ala-Gly-Arg-Cys-Gly-Ser-Ala-Cys- | 88.8% | - |
-: no data. SD: standard deviation; IC50: half-maximal inhibitory concentration
This study demonstrates a strategy for developing covalent inhibitors of 3CL protease by employing the rational design of bicyclic peptides and conjugation with an electrophilic VS warhead. Various technologies enabling the screening of monocyclic peptide warhead conjugates were reported, highlighting their efficiency in inhibitor discovery [30, 31]. For instance, cyclic peptides conjugated with reactive warheads have been incorporated into libraries constructed using techniques such as mRNA display [32], phage display [31], and DNA-encoded libraries [33]. Due to the challenges of generating combinatorial bicyclic peptides with a C-terminal electrophile for covalent targeting of proteases, we studied the approach of rational design of bicyclic peptide ligands.
The success of discovering bicyclic peptide binders relies not only on the primary sequence of peptides but also on their conformation, which is crucial for effective binding and inhibition. To increase the likelihood of identifying an active initial bicyclic peptide binder, the composition of 3CL substrate peptides was analyzed to provide information for the design of peptide binders. Random combinations of tripeptide sequences resulted in the discovery of several active bicyclic peptides. Among the initially obtained peptides, BCP-8A and BCP-8B, with primary sequences partially matching the residue preference of native substrates, exhibited moderate inhibitory activity. In contrast, totally random peptides that did not match the positional preferences of 3CL protease showed very weak inhibition.
This result underscores the challenge associated with designing bicyclic peptides without detailed information about the binding site properties of the target protein. In subsequent optimization, positional scanning with diverse amino acids was effective in achieving ideal inhibitors, resulting in the discovery that each position individually matched the preference of the 3CL protease. Looking ahead, we anticipate that the rational design strategy outlined in this study will complement existing peptide display toolkits and help expand the scope of cyclic and bicyclic peptide ligands for future chemical probe development and drug discovery.
1H NMR: 1H-nuclear magnetic resonance
3CL: 3C-like
ESI-MS: electrospray ionization mass spectrometry
HPLC: high performance liquid chromatography
IC50: half maximal inhibitory concentration
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
SPPS: solid-phase peptide synthesis
TATA: 1,3,5-triacryloylhexahydro-1,3,5-triazine
TBMB: 1,3,5-tris(bromomethyl)benzene
VS: vinyl sulfone
The supplementary materials for this article are available at: https://www.explorationpub.com/uploads/Article/file/100871_sup_1.pdf
We acknowledge the instrument facility of the Biotech Drug Research Center for providing support with mass spectrometry, protein expression, and plate reader. We also acknowledge the institutional center for Shared Technologies and Facilities of SIMM for providing services including NMR and MS data recording.
QW: Conceptualization, Validation, Formal analysis, Investigation, Data curation, Writing—original draft. YW and JL: Conceptualization, Data curation, Supervision. HL and SC: Conceptualization, Resources, Supervision, Writing—original draft, Writing—review & editing, Project administration, Funding acquisition, Resources. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the corresponding author, without undue restriction, to any qualified researcher upon request.
This study was supported by the Distinguished Young Scholars Program and the General Program of the National Natural Science Foundation of China (#22477128) awarded to SC. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Qiushi Chen ... Xuechen Li
Ping Zeng ... Lanhua Yi
Shoko Tanaka ... Kohei Sato
Feliciana Real-Fernandez ... Paolo Rovero
Chloé O. Sebilleau, Steven J. Sucheck
Qian Zhang ... Chunqiu Zhang
Suze Ma ... Qi Zhang
Yanyan Liao, Xuefeng Jiang
Jarais Fontaine, Jianfeng Cai
Oscar Noya ... Sandra Losada
