 Review
Review

Affiliation:
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China
†These authors share the first authorship.
ORCID: https://orcid.org/0009-0007-9350-8207
Affiliation:
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China
†These authors share the first authorship.
ORCID: https://orcid.org/0009-0006-7073-0465
Affiliation:
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China
2Chinese Glioma Genome Atlas Network (CGGA) and Asian Glioma Genome Atlas Network (AGGA), Beijing, China
†These authors share the first authorship.
ORCID: https://orcid.org/0000-0002-5229-3736
Affiliation:
1Department of Neurosurgery, Beijing Tiantan Hospital, Capital Medical University, Beijing 100070, China
3China National Clinical Research Center for Neurological Diseases, Beijing 100070, China
Email: liuweimingnsok@sina.com
ORCID: https://orcid.org/0000-0002-6245-6287
Explor Drug Sci. 2024;2:851–866 DOI: https://doi.org/10.37349/eds.2024.00077
Received: August 16, 2024 Accepted: October 31, 2024 Published: December 2, 2024
Academic Editor: Dazhi Liu, University of California Davis, Mirnova Therapeutics, Inc., USA
The article belongs to the special issue Leveraging the FDA-Approved Kinase Inhibitors to Treat Neurological Disorders
Traumatic brain injury (TBI) is a complex disease that leads to significant mortality and disability worldwide each year. TBI disrupts the normal activity of kinases and molecular signaling pathways, but the effective therapeutic methods for patients remain limited. Nowadays, kinase inhibitors approved by the Food and Drug Administration (FDA) mainly for cancer treatment have shown potential effects in TBI. Preclinical studies suggest their potential in promoting recovery. There are fewer randomized clinical studies that evaluate efficacy. We search the kinase inhibitors approved by the FDA and traumatic brain injury as keywords on websites and analyze associated research. This review explores the therapeutic efficacy of kinase inhibitors, identifies limitations that must be addressed in future research to advance the application of FDA-approved kinase inhibitors, and emphasizes their promising potential.
Traumatic brain injury (TBI) is a generally serious public health and social problem. Traffic incidents, falls, violence, and sports injuries are the major causes. In China alone, TBI results in 50,000 deaths annually [1], while in the US, similar trends are observed [2]. The cost of TBI adds up to 400 billion dollars, accounting for 0.5% of the gross world product [3]. The classification of TBI can divide this disease into different conditions to help therapeutic interventions exert the best effect. The courses of TBI are divided into three periods: acute phase (within 3 days), subacute phase (3 days to 3 weeks), and chronic phase (over 3 weeks). The severity of TBI is classified based on Glasgow Coma Scale (GCS) into mild (GCS 13–15), moderate (9–12), and severe (3–8) [3]. The primary injury and secondary injury of TBI are complex due to their diverse pathological processes. Primary injury directly relates to the external force of the brain damaging normal brain structures. Macroscopic and microscopic changes as hemorrhage, extrinsic compression from hematoma, contusion, traumatic axonal injury (TAI), neuronal injury, microvascular thrombosis, and demyelination deteriorate the injury. The harmful effect induced by primary injury will not terminate in a short time [2] and also trigger the secondary injury [4]. The secondary injury involves multiple biological responses leading to further brain injury. The injured neuronal cells will release excitatory neurotransmitters and toxic cytokines that aggravate the mitochondrial damage and neuron necroptosis [2]. The dysregulated mitochondria induce excessive reactive oxygen species (ROS) and elevate intracellular calcium. The calcium activates enzymes involved in kinase cascades like receptor-interacting protein kinases (RIPKs) 1 and 3 activated by tumor necrosis factor (TNF) linked to the progress of necroptosis [5]. Not only the increasing inflammatory cytokines but also the activated astrocytes and microglia play an important role in the disruption of the blood-brain barrier (BBB) and further brain edema [6]. The assorted mechanisms of secondary injury link to the neuron’s death and symptoms as cognitive deficits in patients.
The current treatment measures include hyperventilation, prophylactic hypothermia, hyperosmolar therapy, barbital coma therapy, and surgery, which can inhibit the progression of the disease. The complicated pathology and BBB primarily limit the therapeutic effect of the present treatments [7]. Nowadays, many other existing methods intended to mediate the above mechanisms have been applied in the TBI treatment. The changes of kinases have a strong link to pathological responses. Kinase inhibitors that regulate the activity of changed kinase after TBI have shown great neuroprotection in preclinical research.
Kinases regulate many cellular signaling cascades involved in cell growth, differentiation, proliferation, metabolism, transcription, angiogenesis, and apoptosis. 518 protein kinases and nearly 20 lipid kinases have been observed in humans. There are over 250 kinases expressed in the human brain [8]. The eukaryotic protein kinase (ePK) domain, which contains the ATP and substrate binding sites, is the shared highly conversed domain in many protein kinases. The main structures of ePK are the N-terminal lobe (N-lobe), C-lobe, and a linker that separates the lobes and contains important residues binding to ATP. The serine, threonine, or tyrosine residue in protein kinases can be phosphorylated. In the active state, the activation loop Asp-Phe-Gly (DFG) motif located on the C-lobe is lengthened from the ATP-binding site. The phosphate-binding loop of the N-lobe binds a phosphate [9, 10]. Many activated kinases are involved in the pathology of TBI. Kinase inhibitors have been applied in the study of neurodegenerative diseases. Today, kinase inhibitors treating TBI in preclinical research show a superior therapeutic effect. In the following, we will discuss preclinical research of kinase inhibitors approved by the Food and Drug Administration (FDA) applied in TBI and explore the possibility of further application in clinical practice.
The changes of kinases induce more detrimental biological consequences than favorable results after injury. In acute TBI, activated protein kinase B (Akt/PKB) and PKC affect the normal neuronal function by reducing the expression of glutamate transporter-1 (GLT-1) that mainly in astrocytes which clears glutamate from the synapse to maintain the extracellular glutamate homeostasis [11]. Activated homeodomain interacting protein kinase 2 (HIPK2) participates in neuron apoptosis [12]. Src family kinases (SFK) in the early phase directly bind N-methyl-D-aspartic acid (NMDA) receptors and mediate brain edema. Nuclear factor kappa B (NF-κB) activation in the acute phase induces neuroinflammation through the elevation of interleukin (IL)-1β, IL-6, and TNF-α [13]. In the chronic course, upregulated death-associated protein kinase 1 (DAPK1) mediates a conformation-specific cis-phosphorylated form of tau induction which contributes to neurodegeneration [14]. Necroptosis mediated by activated RIPK1 and RIPK3 is mainly linked to chronic trauma brain damage [15]. Repetitive mild or moderate TBI is the underlying mechanism of chronic traumatic encephalopathy (CTE). The activation of c-Jun N-terminal kinase (JNK), Akt, extracellular signal-regulated protein kinase 1/2 (ERK1/2) [16], and glycogen synthase kinase-3β (GSK-3β) [17] are associated to the accumulation of p-tau that initiates the neurological deficits and cognitive impairment. Figure 1 shows the kinases linked to the increased p-tau in CTE.
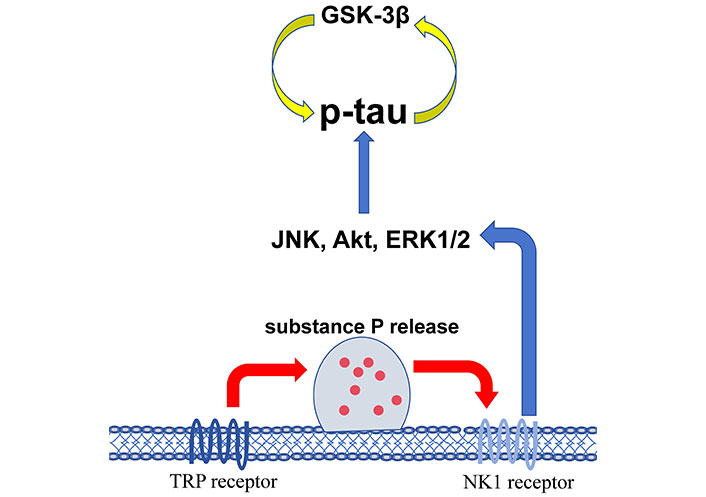
The kinases associated with the accumulation of p-tau in chronic traumatic encephalopathy (CTE). GSK-3β: glycogen synthase kinase-3β; JNK: c-Jun N-terminal kinase; Akt: protein kinase B; ERK1/2: extracellular signal-regulated protein kinase 1/2; TRP: transient receptor potential; NK1: neurokinin-1
In the mild TBI (mTBI), elevated GSK-3β may induce depressive behavior [18]. Activated dual leucine zipper kinase (DLK) is responsible for the death of layer V neurons [19]. Mesenchymal to epithelial transition factor (MET) activation promotes microglia activation and inflammatory cytokines release [20]. The distribution of spleen tyrosine kinase (SYK) is mainly consistent with microglia which may induce neuroinflammation [21]. Kinase alterations display different biological functions in moderate TBI. Increasing PTEN-induced putative kinase 1 (PINK-1) associates with mitochondrial dysfunction [22] after the injury. Activated PKA promotes tau hyperphosphorylation [23] and impairs autophagic flux with ERK2 [24] which results in harming recognition memory. Increased PKC disrupts the BBB integrity [25]. Activated Fyn, c-Src, Rho-associated coiled-coil-containing protein kinase (ROCK) influences hippocampal neuronal cell death linked to spatial memory loss [26]. Leucine-rich repeat kinase 2 (LRRK2) and ERK are involved in depression after moderate injury [27, 28]. Rho kinase degenerates nerve cells in moderate TBI [29]. The activated Janus kinase (JAK)/STAT pathway regulates γ-aminobutyric acid (GABA) type A receptor (GABAAR) related to memory and vestibular motor dysfunction [30] in severe TBI. Activated JNK in injured axons accelerates p-tau [31]. Increasing NF-κB activated by MAPK is related to inflammation in the injured brain [32]. The change of kinases is vital for the development of TBI so that the kinase inhibitors targeted on specific mechanisms have enormous prospects in TBI therapy.
Many kinase inhibitors are often classified into two types: ATP-competitive and non-ATP-competitive. The high intracellular levels of ATP and the highly conversed ATP-binding site restrict the binding affinity of ATP competitive kinase inhibitors which hinders the expected selective effect. To overcome this limitation, researchers are exploring novel targets beyond the ATP-binding site to develop non-ATP-competitive inhibitors that may offer greater selectivity [9]. Protein kinases as targets have been researched for 40 years. In the 1990s, fasudil inhibiting ROCK1 and ROCK2 was approved in Japan and sirolimus reached the market in the US. Imatinib approved by the FDA in 2001 was the breakthrough in the kinase inhibitors research [33].
Many kinase inhibitors have been applied in cancer therapy but there are no FDA-approved inhibitors applied for the clinical treatment of TBI [34]. The mechanism of TBI is complex which involves many kinase pathways and needs more research to find the medicine that can exert the maximum effect. Recently, many preclinical studies have shown better prognosis of TBI animal models after the treatment of kinase inhibitors. Compared to the other various kinase inhibitors, sirolimus [35–40], dasatinib [15, 41–43], imatinib [44–46], pexidartinib [47, 48], ruxolitinib [13, 49], abrocitinib [50], sunitinib [50], and trametinib [51–53] have shown great application potential as attenuating inflammation, promoting prognosis, increasing neurons in the preclinical research.
Rapamycin (sirolimus) is an inhibitor of the mammalian target of rapamycin (mTOR), which was approved by FDA in 1999. mTOR is a rapamycin sensitive serine/threonine protein kinase involved in cellular homeostatic functions such as translation, transcription, and metabolism which is composed of mTOR complex 1 (mTORC1) and mTORC2 [36, 37]. Rapamycin binds to FKBP12 as a result that allosterically inhibits mTORC1. mTORC2 is not sensitive to rapamycin but prolonged treatment of rapamycin (> 12 h) may block mTORC2 assembly [36]. The downstream molecule toll-like receptor 4 (TLR4), activated by mTOR, mediates glial phagocytic activity and inflammatory cytokines production resulting in neuroinflammatory response and brain damage post-TBI [37]. Phosphatidylinositol 3-kinase (PI3K)/Akt/mTOR signaling pathway leads to neuroinflammation by increasing the expression of IL-1β and TNF-α [38]. Rapamycin can suppress the activation of nucleotide-binding domain leucine-rich repeat and pyrin domain-containing protein 3 (NLRP3) inflammasome after TBI to block caspase-1 activation and reduce downstream inflammatory factors [39]. The neuroprotective effects of rapamycin extend beyond TBI, demonstrating potential therapeutic benefits in other neurodegenerative diseases, such as Alzheimer’s, Parkinson’s, and Huntington’s [40].
Dasatinib, approved by FDA in 2006, mainly inhibits Src, a kinase important to cell growth, cycle progression, development, and survival signaling pathways [41]. In TBI, oxidized delayed rectifier potassium (K+) channel KCNB1 can activate Src, leading to increased release of ROS [42]. Besides dasatinib, bosutinib and ponatinib are also Src inhibitors approved by FDA. RIPK1, RIPK3, and the downstream RIPK3-mediated phosphorylation of mixed lineage kinase domain-like protein (MLKL) involve in necroptosis. The neuroinflammation induced by RIPK connects with TBI. Emerging research indicates that dasatinib, foretinib, poziotinib, and pexmetinib can inhibit RIPK3 activity following TBI [15, 43].
Imatinib mainly inhibits platelet-derived growth factor receptor (PDGFR) related to Ras, MAPK, PI3K, Akt, PLCγ, and others [44, 45]. PDGF signaling pathway may compromise the integrity of BBB and lead to brain edema, neuroinflammation, neuronal death, and cognitive impairment [46]. Imatinib inhibits the activation of the cascade via binding to the ATP-binding pocket of the receptor [44]. Sunitinib, sorafenib, pazopanib, and nilotinib are also PDGFR inhibitors [45].
Pexidartinib (PLX3397) exhibits high selectivity for stem cell factor and colony stimulating factor 1 receptor (CSF1R) belonging to the PDGFR family. CSF1R required for the development, and survival of microglia, neurogenesis, and neuronal survival [47] binds to CSF1 and IL-34 which are highly expressed in the brain. Pexidartinib can attenuate neuroinflammation by eliminating the elevated CSF1R post-TBI through modulating microglial activity [48].
Ruxolitinib, the first selective inhibitor of JAK1 and JAK2 approved by FDA, demonstrates neuroprotective effects [49]. Abrocitinib, approved by FDA in 2022, promotes fluid percussion injury (FPI) mice recovery through JAK inhibition [13]. Sunitinib exhibits protection in TAI models by inhibiting DLK and leucine zipper kinase (LZK) in recent research [50]. Trametinib is the highly specific ATP non-competitive inhibitor of mitogen-activated protein kinase kinase (MEK), the essential component of the MAPK signaling cascade. Trametinib can mitigate the neuroinflammation and cognitive deficits associated with elevated MEK expression following TBI [51–53].
In addition to the mentioned drugs, other medicines also have the application prospect based on their preclinical research findings. Midostaurin has demonstrated the ability to attenuate neuroinflammation and improve functional outcomes after spinal cord injury (SCI) [54]. Retinal pathology after brain injury is mostly identical to retinal detachment (RD). AR-13503, the active metabolite netarsudil, which is a ROCK inhibitor, has shown protection in RD [55]. There is also clinical research to demonstrate the effect of kinase inhibitors.
The kinase inhibitors in clinical trauma research are less. Baricitinib, a JAK inhibitor, is being studied in a phase II trial (NCT06065046) for the treatment of moderate and severe traumatic intracerebral hemorrhage and contusions. Imatinib has been explored the clinical effects on cervical SCI (NCT02363361). Rapamycin may be an optional interference for posttraumatic stress disorder (NCT01449955).
According to our investigation by searching on PubMed and Web of Science, there have been preclinical studies of kinase inhibitors approved by FDA on TBI. We discuss these medicines based on their effect as follows.
In the injured brain, microglia can exert neuroprotective effects for neurological recovery after TBI. However, activated microglia also produce pro-inflammatory molecule which hinders neural recovery [56, 57]. Increased glial cells and inflammatory molecules are the indicatives of neuroinflammation which often induces neurological impairments and neurodegeneration after TBI [58]. Rapamycin can reduce elevated IL-1β, TNF-α, and IL-18 after TBI [38, 39]. But one research result showed that rats pretreated with rapamycin (2 mg/kg, i.p.) 1 h before TBI could dramatically elevate the concentrations of TNF-α, IL-1β, and IL-6 [37]. Dasatinib reversed the elevation of RIPK3 phosphorylation-positive cells related to the necrosis induced by TNF [43]. The fewer number of glial fibrillary acidic protein (GFAP, a marker of astrocyte) positive cells were detected in the moderate TBI mice during treatment [42]. Ionized calcium binding adapter molecule 1 (Iba1, a marker of microglia) elevated in the pericortical, hippocampal cornu ammonis 1 region (CA1), dentate gyrus (DG), corpus callosum (CC), and posterolateral thalamus post-TBI which was altered after the treatment of dasatinib combined with quercetin [59]. Imatinib (200 mg/kg) treatment in the early phase markedly reduced the Iba1 immunoreactivity than the vehicle-treated group at postoperative day 28 [60]. Pexidartinib primarily decreased microglia at the dose of 290 mg/kg for 21 consecutive days before injury or starting 90 min after TBI for durations of 5 or 30 days. The inhibition of microglia is connected with better outcomes [53, 61]. Nicotinamide adenine dinucleotide phosphate oxidase 2 (NOX2) mediated by microglia is harmful in the chronic phase of TBI. The depletion of microglia treated by pexidartinib reduced the oxidative stress responses induced by NOX2. Pexidartinib treatment attenuated NLRP3 inflammasome as well [62]. Ruxolitinib, administered at a dose of 0.44 mg/kg, has been shown to inhibit the increase of caspase-1, caspase-3, caspase-8, and IL-1 in the acute phase following TBI, which is linked to the neuroprotection in functional tests like shorter latency in Morris water maze (MWM) and alleviated motor dysfunction in the wire-grip test [49]. The expression of inflammatory factors TNF-α, NOS2, IL-12, and GM-CSF was markedly reduced (P < 0.05) by the trametinib treatment [51]. In the acute phase of TBI, the level of NF-κB dropped after the abrocitinib treatment [13].
Additionally, microglia polarize to one of two distinct phenotypes after injury: M1 microglia promote the development of inflammation, while M2 microglia play a positive role in inhibiting inflammation [57]. The shift from M1 to M2 phenotype links to attenuate the inflammatory response and is crucial for recovery [13]. Abrocitinib promotes a shift in microglial polarization towards the M2 phenotype, which is typically associated with anti-inflammatory and tissue repair activities [13].
About 80% of TBI patients are diagnosed with post-traumatic epilepsy (PTE) within 2 years of their initial brain injury [63]. According to the pathogenic time of epilepsy after TBI, PTE is divided into early PTE (within a week) and late PTE (over a week). Necessary control of PTE can avoid disease deterioration.
In the brain, mTOR influences neuronal signaling and excitability which plays a role in the development of PTE. Compared to the vehicle, mice in the treatment of rapamycin (6 mg/kg/d, i.p.) injected 1 h after TBI continuing once daily for up to 4 weeks had a significantly lower rate of developing PTE (P < 0.05, Mantel-Cox log-rank test) and spontaneous seizure frequency (P < 0.05, Mann-Whitney U-test) [64]. An identical study was also conducted by Klein et al. [65] and the result was consistent. Berdichevsky et al. [66] utilized the rat hippocampal organotypic culture model of PTE to study long-term (four weeks) treatment of rapamycin on PTE observing a reduction sprouting from CA3 and DG areas. Rapamycin (3 mg/kg) injected 20–30 min after controlled cortical impact (CCI) injury in the study of Butler et al. [67] found that rapamycin treatment prevented the reduction of THIP-induced tonic GABAAR current amplitude in ipsilateral dentate granule cells (DGCs) at 1–2 weeks and 8–13 weeks post-injury. Prolonged rapamycin treatment for 8–13 weeks further reduced spontaneous inhibitory post-synaptic current (sIPSC) frequency in ipsilateral DGCs (P = 0.017) and altered the synaptic inhibition reduction of DGCs (P = 0.0064) ipsilateral to CCI injury [67]. Subchronic imatinib treatment can reduce the severity of seizures in CCI mice [60].
The motor deficits can be alleviated by kinase inhibitors, as assessed by various behavioral tests. The grip test assesses the gross vestibulomotor function. At 24 h and 72 h following TBI, the scores of 31 mice injected rapamycin (2 mg/kg, i.p.) 30 min after injury were significantly improved in the grip test (P < 0.01, P < 0.05; respectively) [38]. Beam walking test assesses the forelimb and hindlimb locomotor activity after brain injury. 8 rats treated with rapamycin (3 mg/kg, i.p.) 4 h after daily and continuous mTBI for 5 days showed much fewer footslips compared to the injury group [68]. The results of rapamycin (0.5 mg/kg or 1.5 mg/kg, i.p.) treatment began at 1 h after cryogenic TBI (cTBI) were not identical. The performance of the 0.5 mg/kg rapamycin group was better than the 1.5 mg/kg group [69]. Huang et al. [51] treated mice with trametinib (1 mg/kg) for 7 consecutive days post-TBI which showed neuroprotection and fewer motor deficits. The longer latency of fall time in the rotarod test demonstrated trametinib improved motor function [51]. The latency time in the rotarod test was also longer as a result of dasatinib (25 mg/kg) injection 2 h after injury (P ≤ 0.039) [42]. Mice injected with imatinib (200 mg/kg) showed better performance in the probe trial as well (P < 0.01) [46].
The neurological severity score (NSS) evaluates the motor function, balance, and alertness ability of the experiment animal. The lower scores mean lower neurological deficits. The NSS scores of the TBI mice injected with rapamycin (2 mg/kg, i.p.) were significantly lower at 24 h and at 72 h following TBI (P < 0.01, P < 0.05; respectively) [70]. The scores of modified NSS (mNSS) were significantly lower after receiving rapamycin at the dose of 3 mg/kg in the rapamycin group post-TBI. The treatment of rapamycin combined with MCC950 showed a better outcome [39]. The NSS was lower in pexidartinib group mice showed CSF1R inhibition might result in better neurological outcomes [48].
Improving memory and cognitive functions is important for the patient’s recovery. Memory retention significantly improved in the MWM after dasatinib treatment (P = 0.025) [42]. Mice treated with dasatinib (5 mg/kg, i.g.) combined with quercetin (50 mg/kg, i.g.) for 3 consecutive days a week for 13 weeks showed a better performance in the novel object recognition (NOR) test [59].
Acute rapamycin treatment reduced the extent of tissue damage in the injured hippocampus [71]. Rapamycin treatment 30 min after TBI significantly decreased the apoptosis index (P < 0.05) and increased neurons (P < 0.05) in mouse brain cortices at 24 h after TBI [70]. The Fluoro-Jade B (FJB) staining result demonstrated that the amount of neuronal degeneration in the hippocampus decreased after rapamycin injection [64]. Rapamycin combined with MCC950 showed better neural protection [39]. Killifishes were treated with dasatinib combined with quercetin before injury and the number of mature neurons in the periventricular zone (P = 0.001) and parenchyma (P = 0.02) of the injured hemisphere was greater compared to the control group. After combined treatment, the number of neural progenitor cell (NPC) was also augmented in the injured dorsal ventricular zone [72]. FJC (the marker of neurodegeneration) progressively increased, which not only existed in the primary lesion, while the neuronal nuclear antigen (NeuN, the marker of the neuron) positive cell progressively decreased from 5 weeks to 4 months after TBI. Dasatinib with quercetin induced FJC intensity decrease and NeuN+ cell increase in cortex, CA1, DG, and LP of thalamus [59].
Ruxolitinib increased the volume of Nissl bodies which may reduce and even disappear when the neurons get injured, decreased degenerating cells, and dampened the decreased spared hemispheric volume. Ruxolitinib altered the change of decreased glutathione peroxidase 4 (GPX4) and increased TfR1 (P < 0.05) which relates to ferroptosis post-TBI. Inhibiting iron deposition by ruxolitinib exerts neuroprotection after TBI [73].
Trametinib increased the expression of oligodendrocytes, myelin basic protein (MBP), and myelin oligodendrocyte glycoprotein (MOG) associated with neuron repairing and remyelination [51]. Abrocitinib also reduced the extent of injury lesions and leakage of BBB. The HE staining and Nissl staining demonstrated increased neurons and TUNEL staining showed less neuronal apoptosis. The other better outcome was that abrocitinib did not exert harmful effect on the hemopoietic system [13]. PSD-95 and GluR1 (the marker protein of synaptic) were increased and the brain tissue loss caused by injury was reduced after pexidartinib treatment. CSF1R inhibition by pexidartinib attenuated neuronal apoptosis in the acute (for 3 days) and chronic (for 30 days) phases. RNAseq and gene set enrichment analysis (GSEA) also showed more genes related to dendritic spines and synapse function treated by pexidartinib [53, 61].
The reactive astrocytes in the early TBI are beneficial for preserving neural tissue and restricting inflammation. But in the chronic stage of TBI, the glial scar mainly consists of astrocytes which may inhibit neurogenesis. Rapamycin treatment (1 mg/kg, i.p.) 1 h post-CCI reduced astrogliosis in both sides of the hippocampus but did not alter the number of reactive astrocytes in the ipsilateral hippocampus 24 h after injury [71]. Rapamycin at the dose of 1.5 mg/kg markedly reduced the number of GFAP+ cells at 3 days following injury (P < 0.001). Rapamycin (0.5 mg/kg) significantly decreased the area and thickness of the glial scar (P < 0.01), increased GAP-43 (a marker of regenerating axons) expression (P < 0.05), and attenuated the expression of CSPG (P < 0.01) which is considered as inhibition of axon regeneration at 14 days post-injury [69]. Compared to rapamycin, dasatinib might not attenuate the glial scar [72].
Imatinib (200 mg/kg) was injected 45 min after TBI and Evans blue 24 h after TBI showed imatinib markedly decreased BBB leakage in the ipsilateral hemisphere. The apparent diffusion coefficient (ADC) value positively correlated with cerebral edema was significantly lower in the imatinib treatment (imatinib vs. vehicle, 0.799 mm2/s ± 0.046 mm2/s vs. 0.944 mm2/s ± 0.065 mm2/s) [46]. Different administration schedules of rapamycin as a dose of 3 mg/kg given 4 h after each mTBI [68], and a dose of 2 mg/kg administered 1 h before injury [37] showed contrary results in reducing cerebral edema.
Neurogenic heterotopic ossification (NHO) is mainly caused by traumatic SCI and TBI. Macrophages, lymphocytes, and other immune cells gather in the perivascular area of NHO lesions which may trigger the proliferation of progenitor cells in the early stage. Imatinib inhibits mast cell proliferation and the release of metalloproteases [74]. Rapamycin may also inhibit the development of NHO via the inhibition of hypoxia-inducible factor 1a (HIF1a) [74]. TAI is a common pathology of TBI. The degeneration of progressive axonal and somal retinal ganglion cells (RGCs) represents the TAI in the optic nerve and optic tract. The number of surviving RGCs increased from 1,500 cells/mm2 to nearly 2,300 cells/mm2 (P < 0.05, t-test) but did not reach the normal level through the treatment of sunitinib [50]. Table 1 summarizes the effects of kinase inhibitors in the preclinical research.
The preclinical research of kinase inhibitors in traumatic brain injury (TBI)
| Drug | Author/date | Injury model | Treatment | Pathological outcome | Clinical outcome |
|---|---|---|---|---|---|
| Sirolimus | Song et al. [38], 2015 | WDI mice | 2 mg/kg, i.p. 30 min after TBI | Neurons↑, IL-1β and TNF-α↓, microglial activation↓ | The NSS score↓, the score in the grip test↑ |
| Chen et al. [39], 2019 | WDI mice | 3 mg/kg, i.p. 3 h after TBI | NLRP3 inflammasome↓, mitochondrial dysfunction↓, cortical neuron↑, IL-1β↑, IL-18↓ | mNSS scores↓, brain edema↓ | |
| Jiang et al. [37], 2018 | CCI rats | 2 mg/kg, i.p. 1 h before TBI | TNF-α↑, IL-1β↑, IL-6↑ | mNSS score↑, brain edema↑ | |
| Nikolaeva et al. [71], 2016 | CCI mice | 1 mg/kg, i.p. 1h after TBI | Damaged neurons↓, astrogliosis↓, C-side GFAP positive cells↓ | Latency time of MWM↑ | |
| Ding et al. [70], 2015 | WDI mice | 2 mg/kg, i.p. 30 min after TBI | Neurons↑, apoptosis index↓ | NSS score↓ | |
| Wang et al. [68], 2017 | WDI rats for daily 5 days | 3 mg/kg, i.p. 4 h after each WDI | Apoptosis↓, autophagy↑, mitophagy↑ | Footslips in the beam walking↓ | |
| Fan et al. [69], 2018 | cTBI mice | Daily 0.5 mg/kg, i.p. at 1 h after cTBI | The infarct volume↓, glial scar↓, axon regeneration↑ | Footslips in the beam walking test↓ | |
| Daily 1.5 mg/kg, i.p. at 1 h after cTBI | GFAP+ cell↓ | Latency in rotarod↑ | |||
| Zhu et al. [35], 2014 | CHI mice | 4 μL 1.25 mmol/L or 12.5 mmol/L, ICV before CHI | - | 12.5 mmol/L worsened the performance in the hidden platform and probe trials↓ | |
| Berdichevsky et al. [66], 2013 | Accelerated in vitro model of post-traumatic epilepsy | At 3 days in vitro | Lactate↓, LDH↓, axon sprouting in CA3 and DG↓, surviving CA3 and CA1 neurons↑, ictal event↓ | - | |
| Guo et al. [64], 2013 | CCI mice | 6 mg/ kg, i.p. 1 h after TBI and continued once daily for 4 weeks | Neuronal death in the hippocampus↓, seizure frequency↓ | - | |
| Butler et al. [67], 2016 | CCI mice | 3 mg/kg, 20–30 min after injury and continued once daily | sIPSC frequency↓, synaptic inhibition current↑, THIP-induced tonic current↑ | - | |
| Dasatinib | Sun et al. [43], 2024 | WDI mice | 5 mg/kg, i.p. right after WDI | IL-6↓, RIPK3↓, necrosis↓ | - |
| Wang et al. [59], 2023 | CCI mice | D 5 mg/kg + Q 50 mg/kg 1 month after CCI for 13 weeks | IL-1β↓, IL-6↓, p16+ and p21+ astrocytes↓, p16+ and p21+ microglia↓, NeuN+ cell↑ | Defect in the Barnes maze test↓, depressive-like behavior in the Forced Swim↓ | |
| Yu et al. [42], 2016 | FPI mice | 25 mg/kg, i.p. 2 h after FPI | Positive cells in the typical lesion↓ | Latency in the rotarod↑ | |
| Van Houcke et al. [72], 2023 | Stab-wound injury killifish | D 5 mg/kg + Q 50 mg/kg, i.p. | Progenitors↑, NGPs↑, mature neurons↑ | - | |
| Imatinib | Su et al. [46], 2015 | CCI mice | 200 mg/kg, p.o. 45 min after TBI twice daily for 5 days | BBB leakage↓, lesion size↓, edema↓, tissue loss↓ | Deficit in MWM↓, the performance in probe trial↑ |
| Sakai et al. [60], 2021 | CCI mice | 200 mg/kg, p.o. 12 h and 2 h before CCI and twice daily for 4 days after CCI | Iba1 immunoreactivity↓, pilocarpine-induced seizure severity↓, pilocarpine-induced seizure interval↓ | - | |
| Pexidartinib | Wang et al. [61], 2020 | FPI mice | 290 mg/kg, p.o. for 21 days before FPI and continued after FPI | Microglia↓, infiltrated immune cells↓, apical dendritic spines↑, synaptic proteins↑, cell apoptosis↓, ER stress↓, neurite sprouting↑ | - |
| Wang et al. [47], 2022 | CCI mice | 290 mg/kg, p.o. 90 min after CCI for 5 days or 30 days | 5 days: CD68+ M/M↓, GFAP+ astrocytes↓, Iba1+ M/M↓, apoptosis marker Bax↓, Ki67 cells↓ | Weight↓, NSS↓ | |
| 30 days: brain tissue loss↓, NeuN+ neurons↑ in male mice, gene of immune processes and phagocytosis in male↑ | NSS↑ in male, motor deficits in rotarod test↑ in female | ||||
| Ruxolitinib | Chen et al. [73], 2021 | CCI mice | 0.44 mg/kg, i.p. 30 min after TBI | COX2↓, TfR1↓, GPX4↓, the shrinkage and hyperchromatic morphology of the Nissl bodies↑, degenerating cells↓, brain edema↓, iron-positive cells↓ | Motor dysfunction in the wire-grip test↓, latency in MWM↓, total distance in open field test↓ |
| Peng et al. [49], 2023 | CCI mice | 0.44 mg/kg, i.p. 15 min or 6 h after TBI | Lesion volume↓, caspase-1↓, caspase-3↓, caspase-8↓, IL-1β↓, neurodegeneration↓, maintain the homeostasis of cathepsin B (↑ in the early, ↓ in the following) | Vestibulomotor dysfunction↓, latency in MWM↓, total distance in open field test↓ | |
| Sunitinib | Welsbie et al. [50], 2019 | IA mice | 60 mg/kg, i.p. 24 h and 4h before IA, immediately once and daily for 3 weeks after IA | Retinal ganglion cells↑ | - |
| Trametinib | Huang et al. [51], 2020 | CCI mice | 1 mg/kg for 7 days after CCI | Oligo2-positive cells↑, myelin basic protein↑, myelin oligodendrocyte glycoprotein↑, IL-1β↓, CD86↓, TNF-α↓, NOS2↓, IL-12↓, GM-CSF↓, microglia↓ | Revisiting error time in eight-arm radial maze assay↓, latency in MWM↓, latency in rotarod assay↑ |
| Abrocitinib | Li et al. [13], 2022 | FPI mice | 10 mg/kg | The lesion size↓, hematoma↓, BBB integrity↑, cortical cerebral blood flow↑, neurons↑, apoptosis↓, neutrophils↓, microglia↓, M1 polarization↓, M2 polarization↑, IL-6↓, TNF-α↓, NF-κB↓, NLRP3↓, ASC↓, caspase-1↓, GSDMD↓, IL-1β↓, IL-18↓, pyroptosis↓ | mNSS score↓ |
↑: increase or promote; ↓: decrease or reduce; -: no relevant experiment. WDI: weight drop injury; IL: interleukin; TNF: tumor necrosis factor; NSS: neurological severity score; NLRP3: nucleotide-binding domain leucine-rich repeat and pyrin domain-containing protein 3; mNSS: modified NSS; CCI: controlled cortical impact; GFAP: glial fibrillary acidic protein; MWM: Morris water maze; cTBI: cryogenic TBI; ICV: intracerebroventricular; CHI: closed head injury; LDH: lactate dehydrogenase; CA: cornu ammonis; sIPSC: spontaneous inhibitory post-synaptic current; RIPK3: receptor-interacting protein kinase 3; D: dasatinib; Q: quercetin; FPI: fluid percussion injury; NGPs: non-glial progenitors; BBB: blood-brain barrier; Iba1: ionized calcium binding adapter molecule 1; ER: endoplasmic reticulum; M/M: microglia/macrophage; Bax: B-cell lymphoma 2-associated X protein; COX2: cyclooxygenase 2; GPX4: glutathione peroxidase 4; IA: impact acceleration; NF-κB: nuclear factor kappa B; i.p.: intraperitoneal; p.o.: oral; DG: dentate gyrus; NeuN: neuronal nuclear antigen
The dosage of the mentioned medicine is different due to the reference content. The previous study is the main reference source. The appearances of post-injury are mainly identical to neurodegenerative diseases and the mechanism may be consistent. Researchers conducted the above experiments with reference to neurodegenerative diseases. The molecular mechanism research can provide the basic evidence for further research. The dose of pexidartinib is based on the associated mechanism experiment [75].
Unlike cancer therapy, the specific target kinase has not precisely been observed in TBI patients. The complex kinase cascades exert different influences in the early or late phase of disease. The mentioned kinases are not involved in just one pathway, as a result that diverse downstream kinases may induce converse biological results. Finding the most suitable target kinase in the development of TBI is a challenging problem. Multiple kinase inhibitors may need further research to confirm the most suitable one [34].
The highly selective BBB combined with protective efflux systems limits many xenobiotics penetrating into the brain parenchyma [76]. Imatinib showed effect in the brain cancer preclinical study but failed in clinical research due to poor brain intake to a certain extent [77]. Nowadays, only a small number of kinase inhibitors have been reported that are BBB permeable in neurodegenerative disease. Many companies have reformed kinase inhibitors with BBB penetration for which there may be higher medicine concentration in the lesion [78] to lessen the inhibition of BBB.
The safety and side effects of kinase inhibitors have been verified in cancer, autoimmune diseases, hematological disorders, and so on. The previous studies indicate that side effects such as gastrointestinal events, fluid retention, muscle cramps, fatigue, and hepatotoxicity may appear when patients are on long-time treatment of imatinib. Dasatinib may also cause pleural effusion, pulmonary hypertension, headache, and dyspnea [79]. Sunitinib has been proved to increase RGCs after TAI [50] but also relates to cognitive deficit, memory decrease, cortical and hippocampal neuronal degeneration, and hippocampal autophagic machinery in the normal experimental models [80]. The associated contents have not been fully confirmed in the nervous system diseases. TBI patients are in vulnerable condition. The clinical symptoms may be identical to the adverse events of these drugs. The clear classification between clinical appearance and adverse drug reaction needs more exploration. Further experiments are needed to avoid side effects in the treatment of kinase inhibitors.
The complex progression of TBI inhibits the excessive production of drug efficacy. Multiple target kinase inhibitors require more exploration to achieve better effects. Various drug delivery methods, such as biomaterials, have been used in many diseases. Improving the levels of kinase inhibitors in the injured brain tissue may promote the regeneration of neurons and alleviate neuroinflammation. The new delivery methods have been studied in many diseases, especially cancers. The novel ways which deliver medicine to brain tumors are the templates for TBI. Certain nanomaterials can be employed as a carrier for medicine [81]. The combination of kinase inhibitors and nanomaterial may have great prospects in clinical practice. Improving outcomes and avoiding untoward reactions meanwhile may be a considerable question. Further efficacy needs more studies to confirm. Compared to the current conservative treatment, the cost of kinase inhibitors is higher. The more clinical practice may help to determine the most affordable price.
Compared to their application in the acute phase, the use of kinase inhibitors in chronic and subacute TBI is less. More research is needed to determine the optimal timing and duration of administration. The kinase inhibitors mentioned in the research are mainly those approved by the FDA over 10 years ago. The newly confirmed drug that targets the same kinases can be considered for preclinical research.
The pathology of TBI is complex, involving many biological processes. Kinases link pathological changes together as they are involved in many cascades that are activated after the primary or secondary injury. The mentioned drugs such as sirolimus, dasatinib, imatinib, pexidartinib, ruxolitinib, abrocitinib, sunitinib, and trametinib have shown promise in preclinical models, and their translational application in clinical practice remains untested. Additionally, the variability in kinase pathways across TBI severities is complex. The most suitable kinase inhibitor needs more studies to find and confirm. The treatment with kinase inhibitors is supported by animal experiments, and more research is needed to test the medicine’s efficacy in humans.
Akt/PKB: protein kinase B
BBB: blood-brain barrier
CA1: hippocampal cornu ammonis 1 region
CCI: controlled cortical impact
CSF1R: colony stimulating factor 1 receptor
DG: dentate gyrus
DGCs: dentate granule cells
ERK: extracellular signal-regulated protein kinase
FDA: Food and Drug Administration
IL: interleukin
JAK: Janus kinase
mTBI: mild traumatic brain injury
mTOR: mammalian target of rapamycin
mTORC1: mammalian target of rapamycin complex 1
NF-κB: nuclear factor kappa B
NHO: neurogenic heterotopic ossification
N-lobe: N-terminal lobe
NSS: neurological severity score
PDGFR: platelet-derived growth factor receptor
PTE: post-traumatic epilepsy
RGCs: retinal ganglion cells
RIPKs: receptor-interacting protein kinases
ROCK: Rho-associated coiled-coil-containing protein kinase
SCI: spinal cord injury
TAI: traumatic axonal injury
TBI: traumatic brain injury
TNF: tumor necrosis factor
DG: Writing—original draft. YS: Data curation, Writing—review & editing. ZW: Writing—review & editing, Supervision. WL: Conceptualization, Writing—review & editing, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Angela Asir R V ... Izhak Michaelevski
Hassan Aliashrafzadeh ... Da Zhi Liu
