 Original Article
Original Article

Affiliation:
1Departamento de Biomiméticos para el descubrimiento de fármacos, Instituto de Química Médica (IQM-CSIC), 28006 Madrid, Spain
2Alodia Farmacéutica SL, 28108 Alcobendas, Spain
ORCID: https://orcid.org/0000-0002-8760-5621
Affiliation:
1Departamento de Biomiméticos para el descubrimiento de fármacos, Instituto de Química Médica (IQM-CSIC), 28006 Madrid, Spain
ORCID: https://orcid.org/0000-0002-4922-5345
Affiliation:
3Instituto de Investigación, Desarrollo e Innovación en Biotecnología Sanitaria de Elche (IDiBE), Universidad Miguel Hernández, 03202 Elche, Spain
ORCID: https://orcid.org/0000-0002-8035-0179
Affiliation:
4VIB Center for Brain and Disease Research, KU Leuven, 3000 Leuven, Belgium
ORCID: https://orcid.org/0000-0001-5526-5821
Affiliation:
3Instituto de Investigación, Desarrollo e Innovación en Biotecnología Sanitaria de Elche (IDiBE), Universidad Miguel Hernández, 03202 Elche, Spain
ORCID: https://orcid.org/0000-0002-2973-6607
Affiliation:
3Instituto de Investigación, Desarrollo e Innovación en Biotecnología Sanitaria de Elche (IDiBE), Universidad Miguel Hernández, 03202 Elche, Spain
Email: asia.fernandez@umh.es
ORCID: https://orcid.org/0000-0003-2741-1427
Affiliation:
1Departamento de Biomiméticos para el descubrimiento de fármacos, Instituto de Química Médica (IQM-CSIC), 28006 Madrid, Spain
Email: iqmg313@iqm.csic.es
ORCID: https://orcid.org/0000-0001-8833-4328
Explor Drug Sci. 2025;3:100882 DOI: https://doi.org/10.37349/eds.2025.100882
Received: September 10, 2024 Accepted: October 30, 2024 Published: January 13, 2025
Academic Editor: Fernando Albericio, University of KwaZulu-Natal, South Africa, Universidad de Barcelona, Spain
Aim: To expand the understanding of the structure-activity relationship within a family of amino acid-derived β-lactam TRPM8 (transient receptor potential melastatin channel, subtype 8) antagonists, this work investigated both the configuration-dependence of potency and selectivity, and explored strategies for increasing total polar surface area (TPSA).
Methods: Diastereoisomeric compounds derived from H-Phe-OtBu, and analogues incorporating differently substituted benzoyl groups, were synthesized by stereoselective solution pathways. Ca2+ microfluorometry assays were used for TRPM8 antagonist activity assessment, and then confirmed through electrophysiology (patch-clamp assay). The pharmacological activity in vivo was studied on a mice model of oxaliplatin-induced peripheral neuropathy.
Results: For OtBu derivatives, a 3S,4S-configuration was preferred, while compounds with 2'R chiral centers show higher selectivity for TRPM8 versus transient receptor potential vanilloid, subtype 1 (TRPV1) than their 2'S-counterparts. N-terminal benzoyl derivatives, which increased TPSA values, resulted in equipotent compounds as previous prototypes, but also showed activity in other pain-related targets [TRPV1 and cannabinoid receptor, subtype 2 (CB2R)]. A selected N-benzoyl derivative displays antinociceptive activity in vivo.
Conclusions: The potency and selectivity of these β-lactam TRPM8 antagonists developed from amino acid derivatives depend not only on the configuration but also on the substituents at the 4-carboxy and at the N-benzoyl groups. Dual and multitarget compounds were discovered within this family of TRPM8 antagonists.
The transient receptor potential (TRP) melastatin, subtype 8 (TRPM8) channel, or TRP cation channel subfamily M member 8, belongs to the TRP superfamily of ion channels [1, 2]. TRPM8 is often termed the “cold receptor” or “menthol receptor” due to its activation by cool temperatures and menthol [3–5]. When TRPM8 channels are activated, cations, principally calcium ions, enter the cell. This influx leads to cellular depolarization, generating electrical signals and contributing to the sensation of cold or the cooling effect associated with menthol. TRPM8 channels are predominantly expressed in sensory neurons, where they play a crucial role in perceiving cold temperatures and initiating responses to cold stimuli [6, 7]. Activation of TRPM8 channels contributes to the sensation of cold and can influence several physiological processes, such as pain perception and thermoregulation, among others [8–10]. This channel has been implicated in neuropathic pain conditions, such as oxaliplatin (OXP)-induced and diabetic peripheral neuropathies [11–14]. Different studies have shown that both activation and inhibition of TRPM8 can modulate these neuropathic pain responses [15–18]. Beyond pain, TRPM8 channels have been implicated in various pathological conditions, including different types of cancer [19–23], bladder hypersensitivity [24, 25], dry eye disease [26–29], and pulmonary diseases [asthma and chronic obstructive pulmonary disease (COPD)] [30–33]. Therefore, the modulation of TRPM8 activity may have broader implications for managing all these pathological conditions. Since the discovery of the TRPM8 channel, innumerable modulators with a wide variety of chemotypes have been described [3, 8, 10, 21, 34, 35]. TRPM8 antagonists, such as PF-05105679 and AMG333, progressed into clinical trials for the treatment of cold pain hypersensitivity and migraine symptoms, respectively [36–38]. However, these trials were discontinued after phase I, due to important side effects [39, 40], emphasizing the challenges in developing new TRPM8 modulators either with enough selectivity or with good tolerability. In this respect, the topical or local administration of TRPM8 modulators could represent therapeutic alternatives to avoid central side effects [41, 42].
In our continued exploration of amino acid-derived β-lactams, a few years ago, we discovered a new family of potent and selective TRPM8 antagonists [17]. These compounds can be considered peptidomimetics of previously elaborated dipeptides with modest, non-selective TRPM8 antagonist activity [43]. A prototype within the β-lactam family, compound 1a (Figure 1), displays antinociceptive activity in different preclinical models of neuropathic pain [44]. Our previous discoveries highlighted also the configuration-dependent potency of benzyl ester derivatives [45]. In the ongoing investigation of this family of TRPM8 antagonists, we focused first on the isomers of compound 1a (I) to provide deeper insights into whether or not the influence of the configuration follows a general trend within these derivatives. Secondly, there is a specific interest in developing analogues with increased total polar surface area (TPSA) to enhance the safety profile of these compounds for topical use. Optimizing TPSA can influence properties such as solubility and membrane permeability, which could be crucial for the development of compounds intended for topical administration. In this respect, monobenzyl derivatives, such as compound 2a (Figure 1), showed somewhat increased TPSA numbers compared to the dibenzyl prototype 1a [46]. Here we proposed the exploration of 2'-N-benzoylated analogues of 2a (II), which introduces an additional oxygen atom that supposedly will contribute to enlarging the TPSA values.
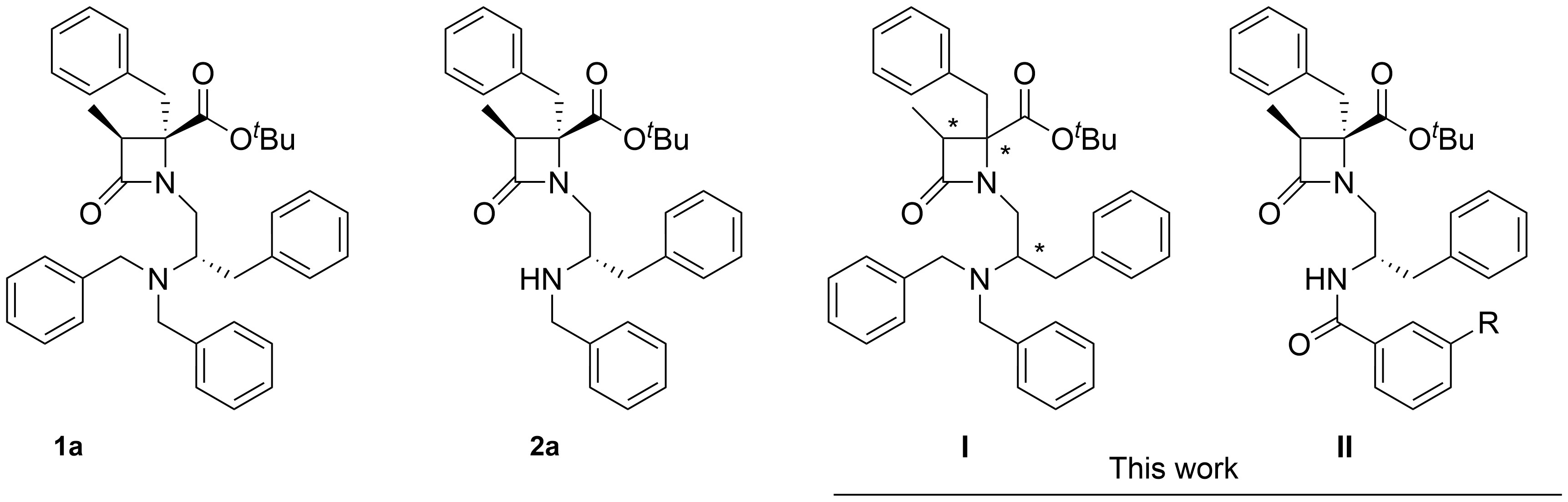
Structure of model compounds 1a and 2a, and analogues synthesized and investigated in this work. * indicates different configurations of the chiral centers
General procedures: reactions were monitored either by thin-layer chromatography (TLC, Silica gel 60 F254) and/or by analytic high performance liquid chromatography (HPLC, e 2695, Waters Alliance) (UV detection, 220 nm). Flash columns, filled with silica gel Merck 60 were used for chromatographic separations. A reversed-phase column, Sunfire C18 (4.6 × 50 mm, 3.5 µm), a flux of 1 mL/min, and mixtures of CH3CN, phase A, and H2O, phase B, both containing 0.01% formic acid, were used for analytical HPLCs. Mass spectra, in electrospray, positive mode, were obtained on a spectrometer (2998 PDA, QDa detector AcquityQDa, Waters). Electrospray ionization-high-resolution mass spectrum (ESI-HRMS) was recorded on an instrument (6520 Q-TOF, Agilent). Optical rotation for final compounds was measured in a polarimetry (PTC 262, P2000 Polarimeter, Jasco). Nuclear magnetic resonance (NMR) spectra were recorded in a spectrometer de 400 MHz (Avance III HD, Brucker) or spectrometer de 300 MHz (Ultrashield, Brucker), operating at 400 and 75 MHz for 1H and 13C experiments, respectively (with chemical shifts expressed in ppm and coupling constants in Hz). The general procedures described for the preparation of intermediates 6–8 and final compounds 1b–d are those indicated in reference [44]. Compounds 6a and 7a [44], as well as 10a [46] were prepared as previously described. Other new synthetic intermediates are described in the Supplementary materials. Final compounds were obtained in at least 95% purity.
The title compound was obtained in 61% (0.1 g) from 8b, as a syrup. Eluent: 8% of EtOAc in hexane. HPLC: retention time (tR) = 5.02 min (gradient from 50% to 95% of phase A, in 10 min). [α]D = 28.73 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.33–7.17 (m, 16H, Ar), 7.14 (m, 2H, Ar), 7.02 (m, 2H, Ar), 3.62 (d, J = 13.7 Hz, 2H, NCH2), 3.58 (m, 1H, H1'), 3.54 (d, J = 13.8 Hz, 2H, NCH2), 3.34 (m, 1H, H2'), 3.13 (dd, J = 14.5, 6.9 Hz, 1H, H1'), 3.08 (q, J = 7.6 Hz, 1H, H3), 2.87 (dd, J = 13.8, 8.1 Hz, 1H, H3'), 2.82 (d, J = 14.5 Hz, 1H, 4-CH2), 2.74 (dd, J = 13.8, 6.4 Hz, 1H, H3'), 2.63 (d, J = 14.5 Hz, 1H, 4-CH2), 1.33 (s, 9H, CH3, tBu), 1.13 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 170.1 (COO), 169.8 (C2), 140.4, 140.1, 136.1, 129.9, 129.8, 129.1, 128.5, 128.3, 128.2, 127.1, 126.9, 126.0 (Ar), 82.8 (C, tBu), 68.6 (C4), 59.1 (C2'), 54.0 (NCH2), 53.2 (C3), 41.8 (C1'), 40.9 (4-CH2), 35.3 (C3'), 28.1 (CH3, tBu), 10.8 (3-CH3). MS(ES)+: 589.47 [M+H]+. The exact mass calculated for C39H44N2O3: 588.3352, found 588.3362.
The title compound was obtained in 69% yield (0.416 g) from 8c. Eluent: 11% to 20% of EtOAc in hexane. HPLC: tR = 5.22 min (gradient from 50% to 95% of phase A, in 10 min). [α]D = +26.67 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ identical to its enantiomer 1b (see Supplementary materials, Figure S1). 13C-NMR (101 MHz, CDCl3): δ identical to its enantiomer 1b. MS(ES)+: 589.47 [M+H]+. The exact mass calculated for C39H44N2O3: 588.3352, found 589.3423.
The title compound was obtained in 60% yield (0.279 g) from 8d, as a syrup. Eluent: 8% of EtOAc in hexane. HPLC: tR = 2.17 min (gradient from 50% to 95% of phase A, in 5 min). [α]D = +3.117 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ identical to its enantiomer 1a [44] (see Supplementary materials, Figure S2). 13C-NMR (101 MHz, CDCl3): δ identical to its enantiomer 1a [44]. MS(ES)+: 589.47 [M+H]+. The exact mass calculated for C39H44N2O3: 588.3352, found 588.3347.
Compound 1d (0.2 g, 0.34 mmol) was treated with 4 M HCl in dioxane and stirred at room temperature (rt) until the disapearance of the starting material (TLC and analytical HPLC). The reaction mixture was evaporated to dryness and purified in column chromatography using a mixture of ethyl acetate and dicloromethane (1:5) to afford the corresponding carboxylic acid (0.145 g, 80%). 0.06 g of this carboxylic acid (0.113 mmol) in dry dicloromethane (5 mL) was treated with isobutylamine (13 µL, 0.135 mmol), PyBrop (0.07 g, 0.135 mmol), and TEA (19 µL, 0.135 mmol). Stirring was continued overnigth at rt. After evaporation of the solvent, the crude mixture was successively washed with 1 M HCl, 10% NaHCO3, and brine. The organic phase was dried over Na2SO4 and evaporated, followed by purification in column chromatography using a mixture of ethyl acetate and dicloromethane (1:12). The title compound was obtained in 75% yield (0.055 g). Finally, the compound was lyophylized in H2O:acetonitrile (1:2) in the presence of 0.1 M HCl (0.34 mL), and relyophylized in H2O:acetonitrile (1:2).
HPLC: tR = 7.34 min (gradient from 15% to 95% of phase A in 10 min). [α]D = +25.10 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 12.52 (br s, 1H, NH+), 10.31 (br s, 1H, NH), 7.55 (m, 3H, Ar), 7.45 (m, 7H, Ar), 7.17 (m, 3H, Ar), 6.92 (t, J = 7.3 Hz, Ar), 6.72 (m, 4H, Ar), 6.51 (d, J = 7.4 Hz, 2H, Ar), 4.64 (d, J = 12.3 Hz, 1H, NCH2), 4.56 (d, J = 16.1 Hz, 4-CH2), 4.34 (d, J = 12.4 Hz, 1H, H1'), 4.22 (br m, NCH2), 4.05 (br s, 1H, H2'), 3.77 (dd, J = 15.9, 11.3 Hz, 1H, H1'), 3.47 (q, J = 7.5 Hz, 1H, H3), 3.40 (br d, J = 12.8 Hz, NCH2), 3.21 (m, 1H, CH2, iBu), 3.13 (m, 1H, CH2, iBu), 3.0 (br s, 1H, H3'), 2.93 (d, J = 16.0, 1H, 4-CH2), 2.66 (dd, J = 0 15.9, 5.3, 1H, 4-CH2), 2,54 (t, J = 12.3, 1H, H3'), 2.03 (m, 1H, CH, iBu), 1.18 (d, J = 7.6 Hz, 3H, 3-CH3), 0.92 (d, J = 6.7 Hz, 3H, CH3, iBu), 0.91 (d, J = 6.6 Hz, 3H, CH3, iBu) (Supplementary materials, Figure S3). 13C-NMR (75 MHz, CDCl3): δ 173.2 (C2), 172.8 (4-CONH), 135.5, 133.6, 131.8, 130.9, 130.1, 130.0, 129.5, 129.1,128.5, 128.2, 127.4, 127.0 (Ar), 71.4 (C4), 56.8 (C2'), 56.7 (C3), 55.3 (NCH2), 52.3 (C1'), 42.8 (CH2, iBu), 40.2 (4-CH2), 32.2 (C3'), 28.4 (CH, iBu), 20.95, 20.9 (CH3, iBu), 20.3 (CH3, iBu), 10.4 (3-CH3) (Supplementary materials, Figure S3). MS(ES)+: 588.38 [M+H]+. The exact mass calculated for C39H45N3O2: 587.3512, found 587.3492.
To a solution of the substituted 2'-NH2 β-lactam derivative 10a (0.180 mmol) in dry DCM (2 mL), TEA (0.180 mmol, 25 µL) was added. After cooling at 0°C, propylene oxide (0.270 mmol, 19 µL) and the corresponding benzoyl chloride (0.270 mmol) was added. When the reaction was completed, the solvent was evaporated to dryness and the resulting crude was purified on a silica gel column, using the eluent system indicated in each case. Alternatively, the corresponding carboxylic acid was coupled to 10a using PyBrop (0.22 mmol) and TEA (0.4 mmol) and stirred overnight at rt. After evaporation, the reaction mixture was dissolved in EtOAc and washed with 0.1 M HCl, 10% NaCO3H, and brine. The organic phase was dried over Na2SO4 and evaporated. The residue was purified on a silica gel column, using the indicated eluent system.
The title compound was obtained in 87% yield (0.05 g) from 10a and benzoyl chloride, as a syrup. Eluent: 0% to 9% of MeOH in DCM. HPLC: tR = 8.72 min (gradient from 40% to 95% of phase A in 10 min). [α]D = 39.61 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.94 (d, J = 7.9 Hz, 1H, 2'-NH), 7.80 (dt, J = 7.0, 1.5 Hz, 2H, Ar), 7.41 (m, 3H, Ar), 7.31–7.16 (m, 8H, Ar), 7.09 (dt, J = 7.2, 3.7 Hz, 2H, Ar). 4.65 (m, 1H, H2'), 3.45 (d, J = 14.5 Hz, 1H, 4-CH2), 3.33 (dd, J = 14.4, 7.5 Hz, 1H, H1'), 3.15 (dd, J = 14.6, 4.8 Hz, 1H, H1'), 3.13 (q, J = 7.3 Hz, 1H, H3), 3.09 (dd, J = 13.6, 7.0 Hz, 1H, H3'), 3.06 (d, J = 14.5 Hz, 1H, 4-CH2), 2.87 (dd, J = 13.9, 7.1 Hz, 1H, H3'), 1.42 (s, 9H, CH3, tBu), 1.21 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.3 (COO), 170.2 (C2), 166.9 (CONH), 138.1, 135.1, 134.6, 131.3, 129.9, 129.4, 128.8, 128.6, 128.5, 127.6, 127.3, 126.6 (Ar), 83.9 (C, tBu), 68.7 (C4), 53.5 (C3), 50.4 (C2'), 46.6 (C1'), 40.9 (4-CH2), 38.6 (C3'), 28.2 (CH3, tBu), 11.0 (3-CH3). MS(ES)+: 513.29 [M+H]+. Exact mass calculated for C32H36N2O4: 512.2675, found 512.2687.
The title compound was isolated in 63% yield (0.066 g, from 10a and 3-phenylbenzoyl chloride) as a syrup. Eluent: 16% to 33% of EtOAc in hexane. HPLC: tR = 9.10 min (gradient from 50% to 95% of phase A in 10 min). [α]D = +11.75 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 8.07 (t, J = 1.8 Hz, 1H, Ar), 8.02 (d, J = 7.8 Hz, 1H, 2'-NH), 7.76 (dt, J = 7.9, 1.3 Hz, 1H, Ar), 7.70 (ddd, J = 7.8, 1.9, 1.1 Hz, 1H, Ar), 7.64 (m, 2H, Ar), 7.46 (m, 3H, Ar), 7.36 (m, 1H, Ar), 7.30–7.17 (m, 8H, Ar), 7.10 (m, 2H, Ar), 4.67 (m, 1H, H2'), 3.46 (d, J = 14.4 Hz, 1H, 4-CH2), 3.36 (dd, J = 14.4, 6.8 Hz, 1H, H1'), 3.16 (dd, J = 14.3, 3.4 Hz, 1H, H1'), 3.15 (q, J = 7.2 Hz, H3), 3.11 (dd, J = 13.7, 7.0 Hz, 1H, H3'), 3.06 (d, J = 14.5 Hz, 1H, 4-CH2), 2.89 (dd, J = 13.9, 7.2 Hz, 1H, H3'), 1.40 (s, 9H, CH3, tBu), 1.21 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.4 (COO), 170.2 (C2), 167.0 (CONH), 141.5, 140.5, 138.1, 135.3, 135.1, 130.0, 129.9, 129.4, 129.0, 128.9, 128.8, 128.6, 127.7, 127.6, 127.3, 126.6, 126.3, 125.9 (Ar), 84.0 (C, tBu), 68.7 (C4), 53.5 (C3), 50.7 (C2'), 46.5 (C1'), 40.9 (4-CH2), 38.7 (C3'), 28.2 (CH3, tBu), 11.0 (3-CH3). MS(ES)+: 589.58 [M+H]+. Exact mass calculated for C38H40N2O4: 588.2988, found 588.2970.
The title compound was isolated in 84% yield (0.08 g) from 10a and 3-fluorobenzoyl chloride. Eluent: 16% to 33% of EtOAc in hexane. HPLC: tR = 7.28 min (gradient from 50% to 95% of phase A in 10 min). [α]D = 38.96 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 8.02 (d, J = 7.9 Hz, 1H, 2'-NH), 7.54 (m, 2H, Ar), 7.36 (td, J = 8.0, 5.6 Hz, 1H, Ar), 7.29–7.14 (m, 9H, Ar), 7.09 (m, 2H, Ar), 4.63 (m, 1H, H2'), 3.47 (d, J = 14.5 Hz, 1H, 4-CH2), 3.34 (dd, J = 14.4, 6.6 Hz, 1H, H1'), 3.16 (q, J = 7.9 Hz, 1H, H3), 3.14 (dd, J = 14.4, 3.1 Hz, 1H, H1'), 3.08 (dd, J = 13.9, 7.2 Hz, 1H, H3'), 3.06 (d, J = 14.5 Hz, 1H, 4-CH2), 2.87 (dd, J = 13.9, 7.1 Hz, 1H, H3'), 1.44 (s, 9H, CH3, tBu), 1.22 (d, J = 7.9 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.4 (COO), 170.3 (C2), 165.6 (CONH), 162.8 (d, J = 246.8 Hz, C3''), 138.0, 137.0 (d, J = 6.8 Hz, C1''), 135.1, 130.0 (d, J = 7.8 Hz, C5''), 129.9, 129.4, 128.8, 128.6, 127.6, 126.6, 122.7 (d, J = 3.0 Hz, C6''), 118.2 (d, J = 21.3 Hz, C4''), 114.6 (d, J = 22.8 Hz, C2'') (Ar), 84.0 (C, tBu), 68.8 (C4), 53.5 (C3), 50.6 (C2'), 46.5 (C1'), 41.0 (4-CH2), 38.5 (C3'), 28.2 (CH3, tBu), 11.0 (3-CH3). MS(ES)+: 531.48 [M+H]+. Exact mass calculated for C32H35FN2O4: 530.2581, found 530.2569.
The title compound was obtained in 82% yield (0.081 g) from 10a and 3-methoxybenzoyl chloride. Eluent: 16% to 33% of EtOAc in hexane. HPLC: tR = 6.87 min (gradient from 50% to 95% of phase A in 10 min). [α]D = 38.01 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.91 (d, J = 7.8 Hz, 1H, 2'-NH), 7.40 (dd, J = 2.7, 1.5 Hz, 1H, Ar), 7.36 (dt, J = 7.7, 1.3 Hz, 1H, Ar), 7.32–7.16 (m, 9H, Ar), 7.09 (m, 2H, Ar), 7.00 (ddd, J = 8.1, 2.7, 1.1 Hz, 1H, Ar), 4.63 (m, 1H, H2'), 3.83 (s, 3H, OCH3), 3.45 (d, J = 14.4 Hz, 1H, 4-CH2), 3.33 (dd, J = 14.4, 7.0 Hz, 1H, H1'), 3.14 (q, J = 7.5 Hz, 1H, H3), 3.14–3.08 (m, 3H, H1', H3'), 3.06 (d, J = 14.5 Hz, 1H, 4-CH2), 2.85 (dd, J = 13.9, 7.3 Hz, 1H, H3'), 1.42 (s, 9H, CH3, tBu), 1.21 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.4 (COO), 170.1 (C2), 166.8 (CONH), 159.8, 138.1, 136.1, 135.0, 129.9, 129.5, 129.4, 128.7, 128.6, 127.6, 126.6, 119.2, 118.0, 112.1 (Ar), 83.9 (C, tBu), 68.7 (C4), 55.5 (OMe), 53.4 (C3), 50.7 (C2’), 46.4 (C1'), 40.9 (4-CH2), 38.6 (C3'), 28.2 (CH3, tBu), 11.0 (3-CH3). MS(ES)+: 543.57 [M+H]+. The exact mass calculated for C33H38N2O5: 542.2781.
The title compound was isolated in 43% yield (0.045 g), from 10a and 3-acethoxybenzoic acid, as a syrup. Eluent: 16% to 50% of EtOAc in hexane. HPLC: tR = 5.88 min (gradient from 50% to 95% of phase A in 10 min). [α]D = 31.41 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.97 (d, J = 7.9 Hz, 1H, 2'-NH), 7.64 (m, 1H, Ar), 7.56 (m, 1H, Ar), 7.40 (m, 1H, Ar), 7.26 (m, 9H, Ar), 7.09 (m, 2H, Ar), 4.62 (m, 1H, H2'), 3.46 (d, J = 14.5 Hz, 1H, 4-CH2), 3.32 (dd, J = 14.4, 6.8 Hz, 1H, H1'), 3.15 (q, J = 7.5 Hz, 1H, H3), 3.19–3.08 (m, 2H, H1', H3'), 3.05 (d, J = 14.5 Hz, 1H, 4-CH2), 2.85 (dd, J = 13.9, 7.2 Hz, 1H, H3'), 2.30 (s, 3H, OCOCH3), 1.43 (s, 9H, CH3, tBu), 1.21 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.4 (COOtBu), 170.2 (C2), 169.4 (OCOCH3), 165.8 (CONH), 150.9, 138.0, 136.2, 135.0, 129.9, 129.5, 129.4, 128.8, 128.6, 127.6, 126.6, 124.7, 124.4, 121.0 (Ar), 84.0 (C, tBu), 68.7 (C4), 53.5 (C3), 50.7 (C2'), 46.5 (C1'), 41.0 (4-CH2), 38.6 (C3'), 28.2 (CH3, tBu), 21.2 (CH3, OCOCH3), 11.0 (3-CH3). MS(ES)+: 571.59 [M+H]+. The exact mass calculated for C34H38N2O6: 570.2730, found 570.2736.
The title compound was isolated in 79% yield (0.066 g) from 10a and 3-dimethylaminobenzoic acid. Eluent: 16% to 33% of EtOAc in hexane. HPLC: tR = 6.63 min (gradient from 50% to 95% of phase A in 10 min). [α]D = 35.78 (c 1, CHCl3). 1H-NMR (400 MHz, CDCl3): δ 7.81 (d, J = 7.5 Hz, 1H, 2'-NH), 7.30–7.17 (m, 12H, Ar), 7.09 (m, 2H, Ar), 4.63 (m, 1H, H2'), 3.44 (d, J = 14.4 Hz, 1H, 4-CH2), 3.32 (dd, J = 14.3, 7.1 Hz, 1H, H1'), 3.14 (q, J = 7.6 Hz, 1H, H3), 3.14–3.08 (m, 2H, H1', H3'), 3.06 (d, J = 14.5 Hz, 1H, 4-CH2), 2.98 (s, 6H, N(CH3)2), 2.84 (dd, J = 13.9, 7.3 Hz, 1H, H3’), 1.42 (s, 9H, CH3, tBu), 1.20 (d, J = 7.5 Hz, 3H, 3-CH3). 13C-NMR (75 MHz, CDCl3): δ 171.3 (COO), 170.1 (C2), 167.6 (CONH), 150.6, 138.2, 135.5, 135.1, 130.0, 129.5, 129.4, 129.2, 128.7, 128.6, 127.6, 126.5, 115.1, 111.7 (Ar), 83.9 (C, tBu), 68.6 (C4), 53.4 (C3), 50.6 (C2'), 46.4 (C1'), 40.9 [4-CH2, N(CH3)2], 38.7 (C3'), 28.2 (CH3, tBu), 11.0 (3-CH3). MS(ES)+: 556.59 [M+H]+. The exact mass calculated for C34H41N3O4: 555.3097, found 555.3091.
For biological assays, human embryonic kidney (HEK) cell lines stably transfected with TRPM8 (kindly provided by Dr Viana’s laboratory, Instituto de Neurociencias, Elche, Spain), and free of mycoplasma, were used. This cell line was cultured in a monolayer, in Earle’s minimum essential medium with Earle’s salts supplemented with 10% fetal calf serum, and 1% nonessential amino acids. 2 mM L-glutamine, 100 µg streptomycin/mL, 100 U penicillin/mL, and 0.4 µg/mL puromycin (referred to as Puro-EMEM) were added and the culture was kept at 37°C in a humidified atmosphere of 5% CO2 in a suitable incubator (STERI-CyCLE CO2 Incubator Hepa Class 100, Thermo Electrón Corporation, Waltham, MA, USA). Cells were stored under liquid nitrogen and used for 15 generations from unfreezing. For fluorometric experiments, HEK-293 cells stably expressing rTRPM8 or hTRPM8 were detached by means of trypsin/EDTA solution, resuspended in DMEM-10% FCS, and seeded at a concentration of 4 × 104 cells/mL.
To measure the effectiveness of the compounds against TRPM8 activity we have used microfluorometry-based calcium flux assays with Fluo-4 NW Ca2+ dye and fluorescence as described previously [44]. Briefly, HEK cell lines stably transfected with TRPM8 were seeded in 96-well plates at a cell density of 40,000 cells. After 2 days, the medium was replaced with 100 µL of the dye-loading solution Fluo-4 NW supplemented with probenecid 2.5 mM and incubated 1 h at 37°C. The TRPM8 ion channel activity was measured using a POLASTAR plate reader (BMG Labtech, Ortenberg, Germany) setting the excitation wavelength at 485 nm and emission wavelength at 520 nm. The baseline fluorescence was recorded for 3 cycles before the addition of the vehicle, compound at different concentrations, and the antagonist [N-(3-aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride (AMTB), 10 µM]. DMSO, at the highest concentration used in the experiment, was added to the control wells. Fluorescence intensity was recorded during 7 more cycles and the agonist (Menthol, 100 µM) was added. Fluorescence intensity was recorded during 10 more cycles.
The Z-factor was calculated in each assay using the following equation:
Data are expressed as the concentration exerting a half-maximal inhibition (IC50) of agonist-induced (Ca2+)i elevation [or concentration exerting a half-maximal activation (EC50) of agonist-induced (Ca2+)i elevation] which was calculated using GraphPad Prism® software (GraphPad Prism 7, GraphPad Software, San Diego, CA, USA). The equation used was
Whole-cell patch-clamp recordings were conducted on HEK293-hTRPM8 cells seeded on 12 mmØ glass coverslips treated with poly-L-lysine solution (Sigma Aldrich, Spain) two days prior to the experiments [44]. The internal pipette solution consisted of 150 mM NaCl, 5 mM EGTA, 3 mM MgCl2, and 10 mM HEPES, adjusted to pH 7.2 with NaOH. The extracellular solution contained 150 mM NaCl, 6 mM CsCl, 1.5 mM CaCl2, 1 mM MgCl2, 10 mM D-glucose, and 10 mM HEPES, adjusted to pH 7.4 with NaOH.
The patch pipettes used in the experiment were created from thin-wall borosilicate capillary glass tubing and were pulled to a final resistance of 2–8 MΩ when filled with the internal solution. The pulling process was carried out using a horizontal flaming/brown Micropipette puller Model P-97 from Sutter Instrument. Recordings were obtained at a sampling rate of 10 kHz and subsequently low-pass filtered at 3 kHz. Any recordings that showed leak currents greater than 200 pA or series resistance exceeding 20 MΩ were excluded from the analysis.
During voltage-clamp recordings, the cells were held at a constant potential, and various modulators were applied as part of the experimental protocol. Menthol (100 µM) or 11a (10 µM; 2–4 min superfusion) were applied directly onto the cell under investigation by means of a multibarrel concentration-clamp device coupled to electronically driven miniature solenoid valves under the control of PatchMaster software (HEKA Electronics, Lambrecht, Germany).
Data were sampled at 10 kHz using an EPC10 amplifier with PatchMaster 2.53 software (HEKA Electronics, Lambrecht, Germany). The analysis was performed with PatchMaster 2.53 and GraphPad Prism 8.0 (GraphPad Software, San Diego, CA, USA). All measurements were conducted at 24°–26°C. Whole-cell patch-clamp experiments were analyzed as the percentage of activation of the TRPV1 channel. This was achieved by normalizing the ratio (p2/p1) of testing conditions to the ratio (p2/p1) of the control condition. GraphPad 8.0 was used for data analysis, a non-linear regression curve and EC50 were obtained by plotting log (agonist) vs. response fitting the curve to Hill equation:
All data were expressed as the mean ± standard error of the mean (SEM) (N = 3; n = 6–8).
The activity on TRPV1, TRPV3, TRPA1, and acid-sensing ion channel, subtype 3 (ASIC3) was subcontracted to Eurofins-CEREP or Eurofins-PANLABS, while TRPM3 antagonist activity was performed by the group of T. Voets (see [46] for more detailed explanation on these experiments).
C57BL/6JRccHSd female mice (15–23-week-old, 24–35 g; Harlan, The Netherlands) bred at the animal facility at Universidad Miguel Hernández de Elche (UMH, Elche, Spain) were used to assess the antinociceptive effects of 11a after the model of chemotherapy-induced neuropathic pain. Housing conditions were maintained at 21 ± 1°C and 55 ± 20% relative humidity in a controlled light/dark cycle (light on between 8:00 a.m. and 8:00 p.m.). Care was taken to minimize the number of animals used and the pain and stress they experienced. Animal experimentation procedures were conducted under the approval of the Institutional Animal and Ethical Committee at UMH (UMH.IDI.AFM.06.20), following the guidelines of the European Community (2010/63/EU), and the Committee for Research and Ethical Issues of the International Association for the Study of Pain [47].
Chemotherapy-induced sensitization was induced by repeated administration of OXP (ref#2623, Tocris, Bristol, UK). OXP was freshly prepared every day by dissolution in 5% dextrose in warmed distilled water at 37°C. It was administered intraperitoneally every other day for 5 days at 6 mg/kg and in a volume of 10 mL/kg, reaching a total dose of 18 mg/kg after the three injections.
The local effect of 11a or its vehicle was assessed 11 days after beginning the OXP treatment through subcutaneous intraplantar injection of 10 µg 11a or vehicle (2% Cremophor and 5% DMSO in saline) in the ventral side of the right hind paw, administered in a volume of 10 µL. This dose was chosen based on previous works [48], and considering the relative potency of the compound in the in vitro studies. Nociceptive sensitivity was evaluated 30, 60, and 120 minutes after the intraplantar injection with the acetone test.
Prior to conducting behavioral experiments, mice underwent a 2-day acclimatization period to the experimental conditions, during which they were handled and habituated to the male experimenter for a minimum of 2 minutes per day and mouse. Additionally, the animals were familiarized with each testing environment 2 hours per day, placed individually in Plexiglas® chambers (10 cm × 10 cm × 14 cm). Every day of evaluation, the mice spent an additional hour of habituation in the testing environment before the measurement of nociceptive sensitivity.
Mice were placed over a metal grid and allowed to habituate for approximately 1 h. Acetone (179124, Sigma-Aldrich) was applied in 20 µL drops onto the mid-plantar surface of the right hind paw by using a 200 µL pipette with a plastic tip manually curved. Responses were recorded using an iPhone SE camera (Apple, Cupertino, CA, USA) and quantification of paw-licking responses was conducted afterward by a blinded observer. The responses were measured for 1 min after acetone application, with a digital stopwatch (Xnote Stopwatch Version 1.63 2011 Dmitry Nikitin) and were averaged for the right hind paw after the intraplantar treatment. For each measurement, the paws were sampled three times, and the mean was calculated. The interval between each application of acetone was at least 3 min.
For the analysis of behavioral experiments, GraphPad Prism 9 was used. An ANOVA with two factors (within factor “OXP treatment”, between factor “11a vs. vehicle group”) and their interaction was first used to assess the effect of the OXP treatment and to assess possible baseline differences between treatment groups. A subsequent 2-way ANOVA was used to study the effects of 11a treatment (within factor “Time Point”, between factor “11a vs. vehicle group” and their interaction). Post-hoc Tukey’s multiple comparisons tests were run whenever the interaction was significant and differences were considered statistically significant when p value was below 0.05.
In a previous series of benzyl ester analogues of 1a, we showed that the TRPM8 antagonist activities were dependent on the configuration, with the 3R,4R,2'R isomer as the most potent isomer. To revise if the influence of the configuration follows a general rule in this series of β-lactams, we accomplished the synthesis and biological evaluation of the three diastereoisomers of 1a that are accessible following our previously described synthetic procedure (Figure 2).
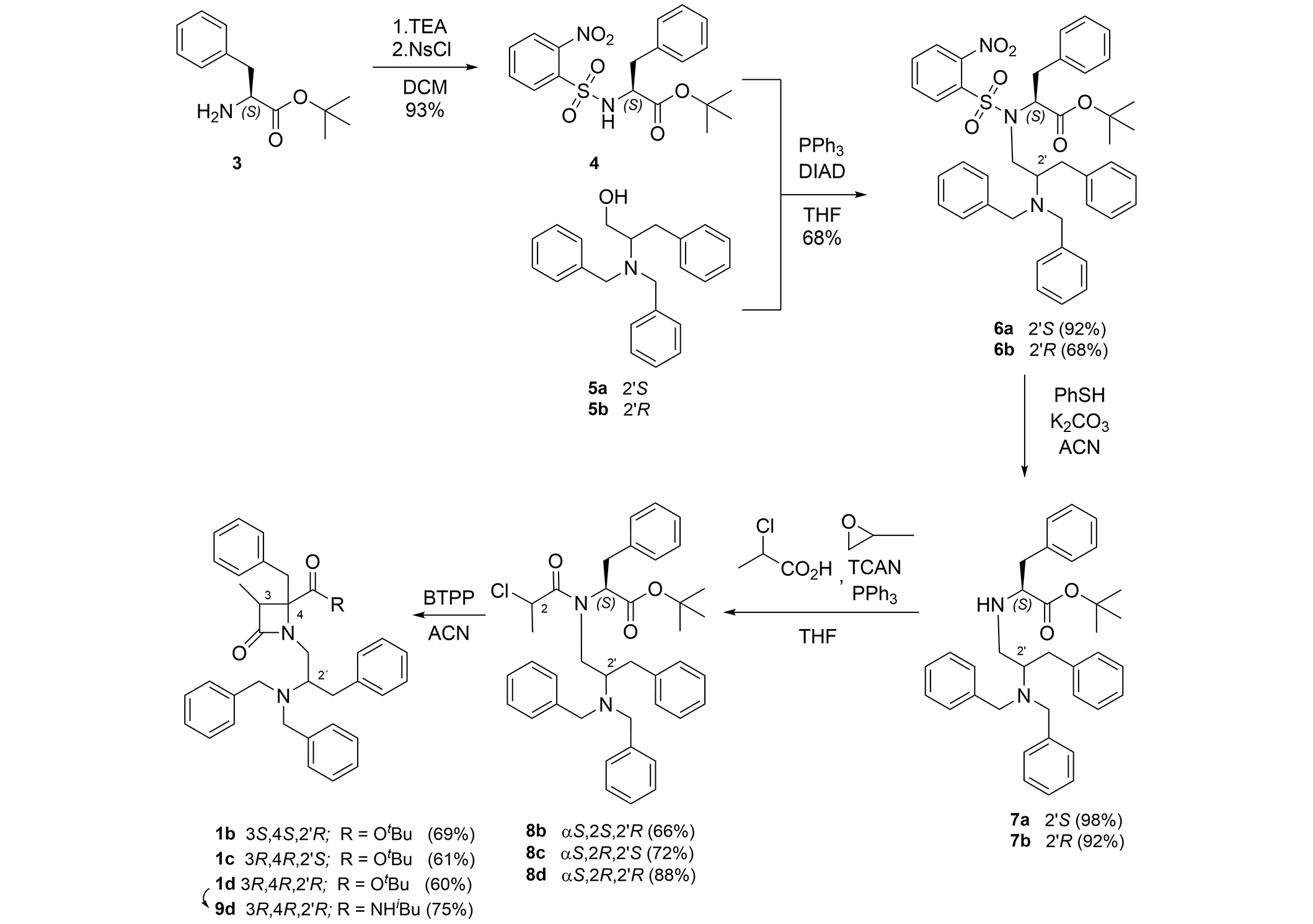
Synthetic procedure for the preparation of β-lactam 1b–d and 9d. BTPP: phosphazene base P1-tBu-tris(tetramethylene)
N-Ns-L-Phe-OtBu (4) [44] was the common initial precursor for the three stereoisomers 1b–1d. Thus, reaction of 4 with 2S- and 2R-(dibenzylamino-3-phenyl)propanol 5a and 5b gave compounds 6a and 6b. It should be noted that, when using the S-enantiomer of the alcohol (5a), the conjugate with the N-Ns-L-Phe-OtBu obtained in higher yield than in the case of the 2'R-diasteroisomer (5b). The removal of the nosyl group from 6a and 6b was carried out with PhSH in a basic medium, yielding the secondary amines 7a and 7b in almost quantitative yield. Acylation of 7a and 7b with 2(S)- and 2(R)-chloropropionic acid chloride afforded the three enantiopure chloropropanoyl derivatives 8b, 8c, and 8d. These intermediates were cyclized in the presence of phosphazene base P1-tBu-tris(tetramethylene) (BTPP) to the three desired diastereoisomeric β-lactams, 1b (3S,4S,2'R), 1c (3R,4R,2'S), and 1d (3R,4R,2'R). As previously described, the stereochemistry at the C3-C4 bond of the β-lactam ring is determined by the configuration of the precursor at the chloropropanoyl stereogenic center [49]. As expected, β-lactam 1d has a very close optical rotation value, but of the opposite sign, to that described for its enantiomer 1a [44]. The same occurs for β-lactams 1b and 1c, which are enantiomers between them. See supporting materials for a comparison of NMR spectra between enantiomeric compounds 1b and 1c, 1a and 1d (Figures S1 and S2, respectively). The treatment of compound 1d with TFA, followed by condensation of the resulting carboxylic acid derivative with iso-butylamine, using PyBrOP as the coupling agent, afforded amide derivative 9d, which was converted in the corresponding hydrochloride by treatment with 0.1 M HCl followed by lyophilization (see 1H- and 13C-NMR spectra in Figure S3).
The three new diastereoisomers of the model β-lactam 1a and compound 9d were first evaluated as TRPM8 antagonists in a calcium microfluorometry assay, working with HEK293 cells heterologously expressing the rTRPM8 channel (Table 1). During these experiments, menthol was used as the agonist (100 µM). Three separated experiments in triplicate were performed. Besides, AMTB and model compound 1a were used as prototype antagonists for comparison. The maximum calcium flow inhibition reached approximately 100% at 50 µM concentration for compounds 1b, 1c, and 9d, while it was 64% for analogue 1d, suggesting partial antagonism.
Antagonist activity of β-lactams 1a–d and analogue 9d at rTRPM8 channels in microfluorometry assays and patch-clamp experiments
| Compound | R | Configuration | Ca2+ microfluorometry | Patch clamp | ||
|---|---|---|---|---|---|---|
| IC50 (µM) | 95% confidence intervals | IC50 (µM) | 95% confidence intervals | |||
| 1a | OtBu | 3S,4S,2'S | 1.06 ± 1.21 | 0.72 to 1.55 | 0.97 ± 1.56 | 0.38 to 2.45 |
| 1b | OtBu | 3S,4S,2'R | 0.72 ± 0.12 | 0.64 to 0.80 | 1.21 ± 0.4 | 0.82 to 1.72 |
| 1c | OtBu | 3R,4R,2'S | 4.48 ± 0.80 | 3.62 to 5.56 | 2.94 ± 0.75 | 2.27 to 3.69 |
| 1d | OtBu | 3R,4R,2'R | 5.02 ± 1.50 | 3.76 to 6.77 | 3.34 ± 0.8 | 2.40 to 5.52 |
| 9d | NHiBu | 3R,4R,2'R | 0.41 ± 0.10 | 0.36 to 0.46 | 0.90 ± 1.25 | 0.57 to 1.42 |
| AMTB | NA | ND | 7.3 ± 1.5 | 8.82 to 7.85 | ND | ND |
ND: no determined; NA: not applicable; AMTB: N-(3-aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride; IC50: concentration exerting a half-maximal inhibition; rTRPM8: rat transient receptor potential melastatin channel, subtype 8; R: substituent at the 4-position; OtBu: tert-butyl ester; NHiBu: N-isobutyl amide
The results in Table 1 show that the four diastereoisomers of β-lactam 1 are able to block menthol-induced TRPM8 channel activation. The eutomer, the isomer that showed the best IC50 value, is the 3S,4S,2'R-configured compound 1b, while in the benzyl series, it was the 3R,4R,2'R-isomer. The distomer, or compound with lower TRPM8 antagonist activity, is the diastereoisomer of configuration 3R,4R,2'R 1d, enantiomer of 1a, just the contrary result to that in the previous series. The diastereoisomers 3R,4R,2'S 1c show a similar potency as 1d. A 2'R stereogenic center is preferred over the 2'S for β-lactams of 3S,4S configuration. Similar results have been obtained for the iso-butylamide derivatives. Thus, while the 3R,4R,2'R isomer 9d displayed submicromolar activity, the all S enantiomer reached a two-digits nanomolar antagonist potency [46]. Some compounds showed one order of magnitude larger antagonist activity than model AMTB.
On the other hand, in patch-clamp experiments, diastereoisomeric β-lactams with a tert-butyl ester at position 4 did not show the same trend in potency as that observed in calcium microfluorometry assays (Table 2, Figure 3, and Figure S4). While in the fluorimetry assay, the best blocker was the 3S,4S,2'R isomer (1b), in patch-clamp the highest blockade was obtained for the 3S,4S,2'S isomer 1a, although all values are quite similar. It should be noted that in the buffer used for the patch-clamp assays, all the O-tert-butyl and the 4-CONHiBu derivatives showed some solubility issues.
Antagonist activity of N-benzoylated derivatives 11a–16a in rTRPM8 channels (Ca2+ microfluorometry assay). Activation is induced by 100 µM menthol
| Compound | R | aTPSA, (A2) | cLogP | IC50 (µM) | 95% confidential intervals |
|---|---|---|---|---|---|
| 1a | NA | 49.85 | 9.88 | 1.06 ± 1.21 | 0.72 to 1.55 |
| 2a | NA | 58.64 | 7.63 | 3.06 ± 1.25 | 2.67 to 3.51 |
| 11a | H | 75.71 | 7.01 | 2.49 ± 1.45 | 1.10 to 5.66 |
| 12a | Ph | 75.71 | 8.90 | 1.14 ± 1.53 | 0.46 to 2.80 |
| 13a | F | 75.71 | 7.36 | 2.30 ± 1.29 | 1.34 to 3.95 |
| 14a | OMe | 84.94 | 7.29 | 3.21 ± 1.30 | 1.85 to 5.57 |
| 15a | OAc | 102.01 | 6.64 | 9.73 ± 1.28 | 5.74 to 16.48 |
| 16a | N(Me)2 | 78.95 | 7.37 | 2.54 ± 1.33 | 1.40 to 4.60 |
| AMTB | NA | ND | ND | 7.3 ± 1.5 | 8.82 to 7.85 |
a Calculated with ChemDraw 22.2.0. TPSA: total polar surface area; IC50: concentration exerting a half-maximal inhibition; rTRPM8: rat transient receptor potential melastatin, subtype 8; R: substituent at the benzoyl moeity; cLogP: logarithm of the partition coefficient between n-octanol and water; ND: no data; NA: not applicable
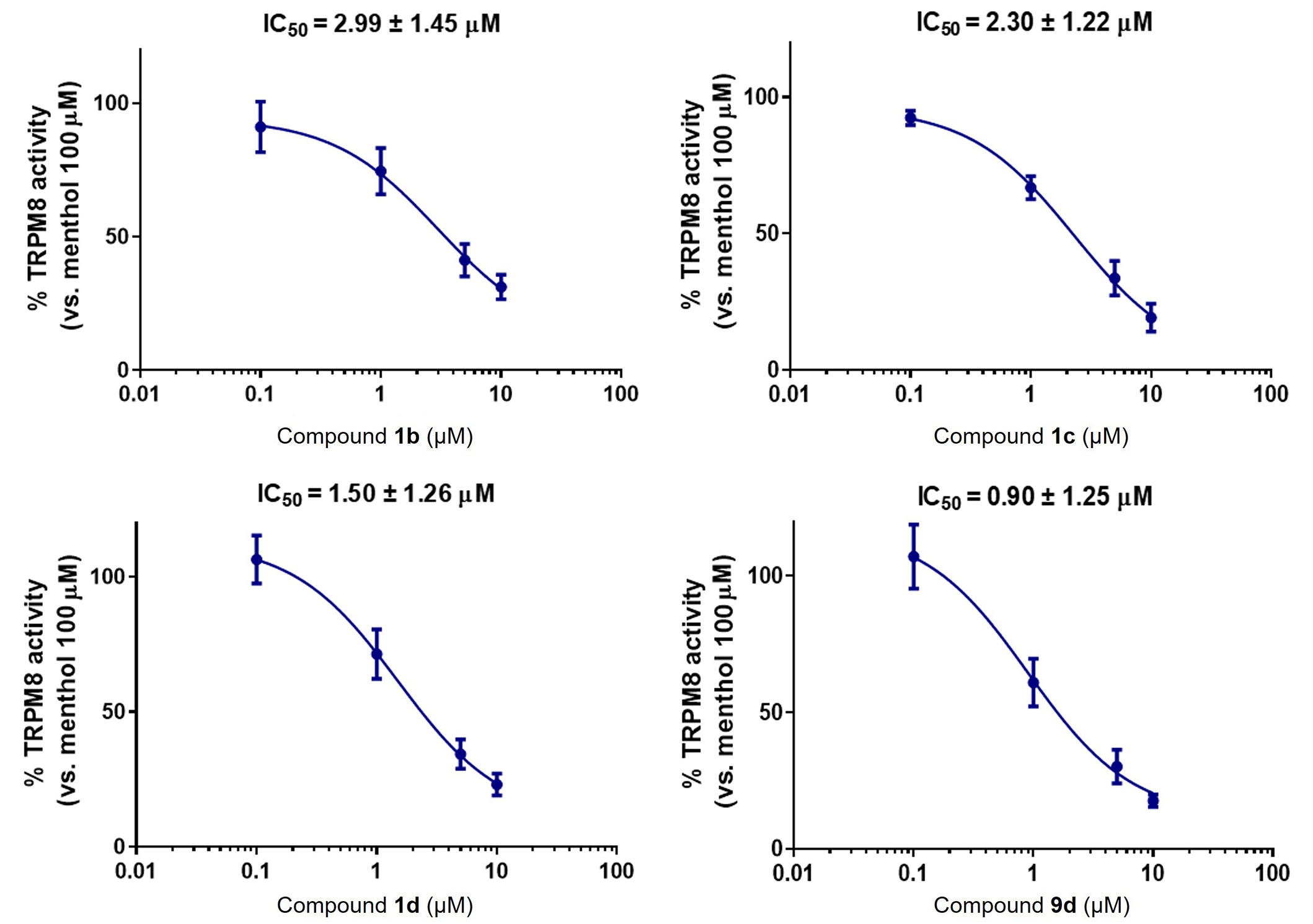
Compounds 1b–d and 9d block TRPM8-mediated responses evoked by menthol in rTRPM8-expressing HEK293 cells, in electrophysiology patch-clamp experiments. Concentration/response curves for rTRPM8 current blockade by the different compounds at a holding voltage of −60 mV. The solid line represents fits of the experimental data to the following equation:
The preparation of the N-monobenzyl derivatives, like 2a, generally proceeded in low yields and the increase of TPSA is very modest compared to 1a [44]. In an attempt to amplify the TPSA values of this family of β-lactam compounds, we considered the synthesis of 2'-N-benzoylated analogues. In this newly designed series, different substitutions at the phenyl ring of the benzoyl moiety, such as Ph, F, OMe, OAc, and N(Me)2 have been included to modulate TPSA figures, which increase in all cases, and possibly to fine-tuning the activity (11a–16a, Table 2). The meta position was selected for the substituents, as in previous work this location led to improved results with respect to the para position. The last compound, 16a, incorporates a tertiary amino group in the molecule, which can be protonated, to obtain compounds with enhanced solubility in aqueous medium, compared to the other substitutions that generally render highly hydrophobic derivatives.
To get these new compounds, the N-benzyl substituents of β-lactam 1a were removed by hydrogenolysis in the presence of HCl, and PdOH as the catalyst, giving rise to the fully N-deprotected amino analogue 10a, in its hydrochloride form. Further treatments of compound 10 with different substituted benzoyl chlorides, using propylene oxide as HCl scavenger, or with the corresponding benzoic acids and PyBrOP/TEA as the coupling system, allowed the preparation of compounds 11a–16a in moderate to very good yield (Figure 4).
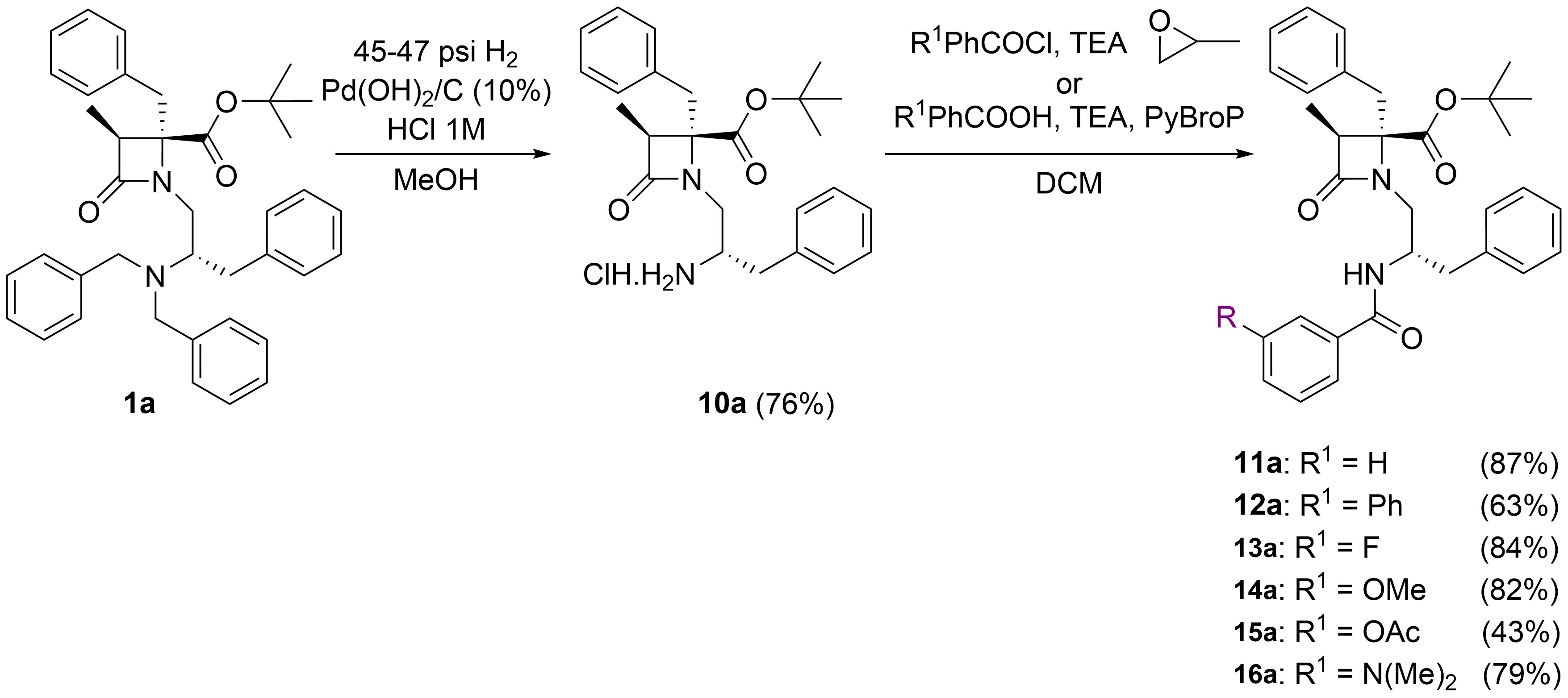
Synthetic procedure for the preparation of N-benzoylated derivatives 11a–16a from β-lactam 1a. The letter “a” after the numbers denotes 3S,4S,2'S configuration
The N-benzoylated β-lactams 11a–16a were evaluated as modulators of the TRPM8 channel in the calcium microfluorometry assays, as indicated above. Results in Table 2 are compared to those of AMTB and model compounds 1a and 2a. All N-monobenzoylated compounds displayed micromolar potency as antagonists of the TRPM8 channel (Table 2), similar to that of the reference dibenzylated compound 1a and the N-monobenzylated analogue 2a. Benzoylated derivatives have calculated TPSA values from 75 to 102 A2, being between 20 and 55 units higher than those of the reference compounds. Among this series, the lower IC50 values were achieved for compound 12a, with a m-Ph substitution, while slightly lower potency was observed for the m-F derivative 13a, the unsubstituted benzoyl derivative 11a, the m-N(Me)2 substituted analogue 16a and the m-OMe-containing compound. These four compounds increased the TPSA values among 17 and 25 A2 compared to the monobenzyl model compound 2b. Unluckily, the introduction of a m-acetoxy group, which greatly enhanced the TPSA figure up to 102 A2, showed an IC50 potency 6- and 3-fold lower than those of model compound 1a and the N-monobenzyl β-lactam 2a, respectively. In general, with the only exception of the 3-phenyl-substituted compound 12a, all benzoyl derivatives showed lower cLogP values than the monobenzyl analogue 2a, and especially than the dibenzyl model compound 1a.
To know more about the activity of this series of compounds as TRPM8 antagonists, calcium microfluorometry assays were performed using the HEK293 cell line that expresses the human isoform of the TRPM8 channel (hTRPM8) [44]. In this experiment, N-benzoylated β-lactams 11a and 12a showed slightly higher IC50 values in hTRPM8 compared to those of rTRPM8 (Table 3). This general tendency has been evidenced for previous β-lactam derivatives and other TRPM8 chiral antagonists [44, 50, 51].
Antagonist activity of β-lactams 11a and 12a in hTRPM8 (Ca2+ microfluorometry) and electrophysiology in rTRPM8 channels (patch-clamp assays). Activation is induced by 100 µM menthol
| Compound | Ca2+ microfluorometry assays | Patch-clamp assay | |||
|---|---|---|---|---|---|
| rTRPM8, IC50 (µM) | hTRPM8, IC50 (µM) | 95% confidence intervals | rTRPM8, IC50 (µM) | 95% confidence intervals | |
| 1a | 1.06 ± 1.21 | 1.74 ± 1.19 | 1.23 to 2.45 | 0.60 ± 1.66 | 0.20 to 1.76 |
| 11a | 2.49 ± 1.45 | 4.33 ± 1.13 | 3.39 to 5.55 | 0.51 ± 1.33 | 0.28 to 0.92 |
| 12a | 1.14 ± 1.53 | 2.08 ± 1.20 | 1.45 to 3.00 | NDa | ND |
a Solubility issue in the Patch-Clamp buffer. ND: no data. IC50: concentration exerting a half-maximal inhibition; rTRPM8: rat transient receptor potential melastatin, subtype 8; hTRPM8: human transient receptor potential melastatin, subtype 8
Electrophysiology studies, using the patch-clamp technique on single HEK293 cells expressing rTRPM8 channels, further confirmed the antagonist activity of compound 11a. As depicted in Figure 5, perfusion with 100 µM menthol induced a characteristic outward rectifying ion current pattern (blue line), with minimal current observed at negative potentials and a linear increase in current at positive voltages. However, when compound 11a was applied at a concentration of 10 µM, there was a noticeable reduction in menthol-induced activation (red line), particularly at depolarizing voltages. Dose-response relationships for compound 11a were established through triplicate experiments at various concentrations, employing a holding voltage of −60 mV. These experiments demonstrated that compound 11a effectively inhibited TRPM8-mediated responses induced by menthol, exhibiting similar potency to that of the model β-lactam 1a, with efficacy observed in the submicromolar range.
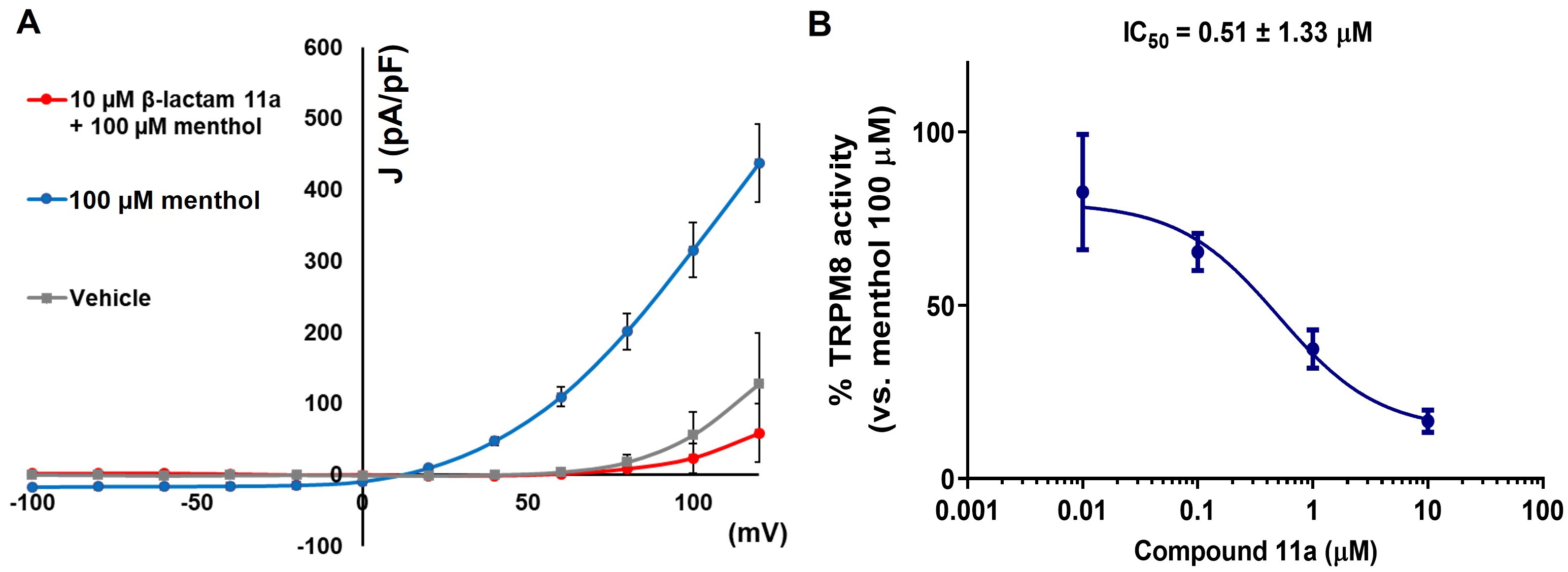
Compound 11a blocks TRPM8-mediated responses evoked by menthol in rTRPM8-expressing HEK293 cells. (A) Curves obtained after exposure to vehicle solution (grey trace), 100 µM menthol (blue trace), and 100 µM menthol + 10 µM 11a (red trace). Peak current data were expressed as pA/pF (to allow comparison among different size cells). Each point is the mean ± SEM of n = 15. (B) concentration/response curves for rTRPM8 current blockade by 11a at a holding voltage of −60 mV. The solid line represents fits of the experimental data to the following equation:
The effects of compounds 1a–1d, 9d, and 11a have been assessed on various ion channels and receptors involved in temperature sensing, nociception, and pain modulation. Specifically, the evaluation included TRP channels such as hTRPV1, hTRPV3, hTRPA1, and TRPM3, which are known to play crucial roles in temperature integration and nociception [52, 53]. Additionally, the investigation encompassed the hASIC3 channel, which is widely distributed in the peripheral nervous system and contributes to the excitability of primary sensory neurons [54].
As shown in Table 4, the configuration at the 2'-chiral center not only affects activity but also plays a significant role in selectivity. Specifically, the all-S diastereoisomeric compound 1a and the 3R,4R,2'S isomer 1c exhibited low to good ability to activate TRPV1 channels. In contrast, their enantiomeric analogues, the all-R counterpart 1d and the 3S,4S,2'R diastereoisomer 1b, did not exhibit significant agonist activity at this channel. Consistent with this observation, the all-R amide derivative 9d did not show perceptible TRPV1 activation. Conversely, the 3S,4S,2'S benzoyl derivative 11a demonstrated less selectivity.
Percentages of activation or inhibition in Ca2+ microfluorometry assays of TRPV1, TRPV3, TRPA1, TRPM3, and ASIC3 channels selected β-lactams (10 µM)
| Compound | Config | hTRPV1 agonism (%) | hTRPV1 antagonism (%) | hTRPV3 antagonism (%) | hTRPA1 agonism (%) | hTRPA1 antagonism (%) | hTRPM3 antagonism (%) | ASIC3 antagonism (%) |
|---|---|---|---|---|---|---|---|---|
| 1a | 3S,4S,2'S | 31.3 ± 3.6 | 19.4 ± 1.7 | –6.3 ± 5.9 | 0.4 ± 0.3 | 4.2 ± 1.1 | ND | 6.0 ± 0.9 |
| 1b | 3S,4S,2'R | 4.7 ± 3.6 | –9.4 ± 15.1 | –6.6 ± 2.9 | 0.4 ± 0.2 | –2.4 ± 7.5 | 8.9 ± 8.8 | –10.1 ± 8.9 |
| 1c | 3R,4R,2'S | 63.7 ± 3.1 | 52.6 ± 17.0a | 6.6 ± 2.5 | 0.7 ± 0.1 | –13.1 ± 8.8 | 7.5 ± 10.3 | ND |
| 1d | 3R,4R,2'R | –2.2 ± 3.2 | –1.5 ± 0.8 | 11.2 ± 3.0 | 0.2 ± 0.6 | –6.1 ± 4.9 | 8.5 ± 1.8 | ND |
| 9d | 3R,4R,2'R | 4.9 ± 0.6 | –8.8 ± 2.9 | 0.1 ± 0.9 | 1.3 ± 0.2 | 7.8 ± 0.6 | 10.6 ± 2.9 | ND |
| 11a | 3S,4S,2'S | 26.7 ± 3.1 | 46.4 ± 0.4a | 7.1 ± 0.4 | 1.1 ± 0.4 | 9.5 ± 3.4 | 19.9 ± 6.6 | 2.1 ± 6.2 |
hTRPV1: human transient receptor potential vanilloid, type 1; hTRPA1: human transient receptor potential ankirin, type 1; hTRPM3: human transient receptor potential melastatin, subtype 3; ASIC3: acid sensing ion cannel, subunit 3; IC50: concentration exerting a half-maximal inhibition; ND: no data. In all cases, data is from two experiments in duplicate. Agonism: assay for agonist activity. Antagonism: assay for antagonist activity (see Materials and methods). Agonists used for activation of the different channels: TRPV1 (capsaicin, 30 nM), TRPV3 [2-aminoethoxydiphenyl borate (2-APB), 30 µM], TRPA1 (allylisothiocyanate, 10 µM), TRPM3 (pregnenolone sulfate, 50 µM), ASIC3 (buffer, pH 5.5). Antagonists used as references: TRPV1 (capsazepin, IC50 1.3 × 10−7 M), TRPV3 (ruthenium red, IC50 2.5 × 10−7 M), TRPA1 (ruthenium red, 10 µM), TRPM3 (isosakuranetin, 10 µM; IC50 50 nM), ASIC3 (amiloride, 1 mM). ª The observed antagonism could be due, at least in part, to desensitization, since the compound is able to activate the receptor > 20%
Compound 11a was also evaluated for its ability to inhibit binding to calcitonin gene-related peptide receptor (CGRPR), cannabinoid receptor, subtype 2 (CB2R), and muscarinic receptor, subtype 3 (M3R), which are expressed in peripheral sensory neurons (Table 5). While model compound 1a was unable to displace radioligands from these receptors, the benzoyl derivative 11a showed a non-negligible affinity for the CB2R.
Percentages of inhibition of specific binding at CGRPR, CB2R, and M3R
| Compound | Displacement of radioligand binding (%) (compounds at 10 µM) | ||
|---|---|---|---|
| hCGRPR | hCB2R | hM3R | |
| 1a | −1.0 ± 4.3 | 7.3 ± 0.3 | −4.1 ± 3.1 |
| 11a | 14.3 ± 1.8 | 32.8 ± 2.8 | –3.9 ± 8.6 |
hCGRPR: human calcitonin gen-related peptide receptor; hCB2R: human cannabinoid receptor, subtype 2; hM3R: human muscarinic receptor, subtype 3. In all cases, data are from two experiments in duplicate. Radioligands used: hCGRPR: [125]hGCRPα, agonist hGCRPα (1 µM); hCB2R: [3H]WIN 55212-2, agonist (5 µM); hM3R: [3H]4-DMAP, agonist 4-DMAP (1 µM)
Given the inhibitory activity of compound 11a over TRPM8 and the antinociceptive effects of TRPM8 antagonists described in models of OXP-induced neuropathy [46, 48, 55], we conducted a behavioral experiment in mice to assess the antinociceptive effects of compound 11a after repeated OXP administration (Figure 6). OXP was intraperitoneally (i.p.) administered every other day for 5 days at 6 mg/kg, reaching a total dose of 18 mg/kg after the three injections. The acetone test, based on the duration of the paw-licking response after the application of acetone drops to the hind paws, revealed a significant enhancement in the nociceptive response to cold after OXP treatment (Figure 6, p < 0.01 before OXP vs. after OXP).
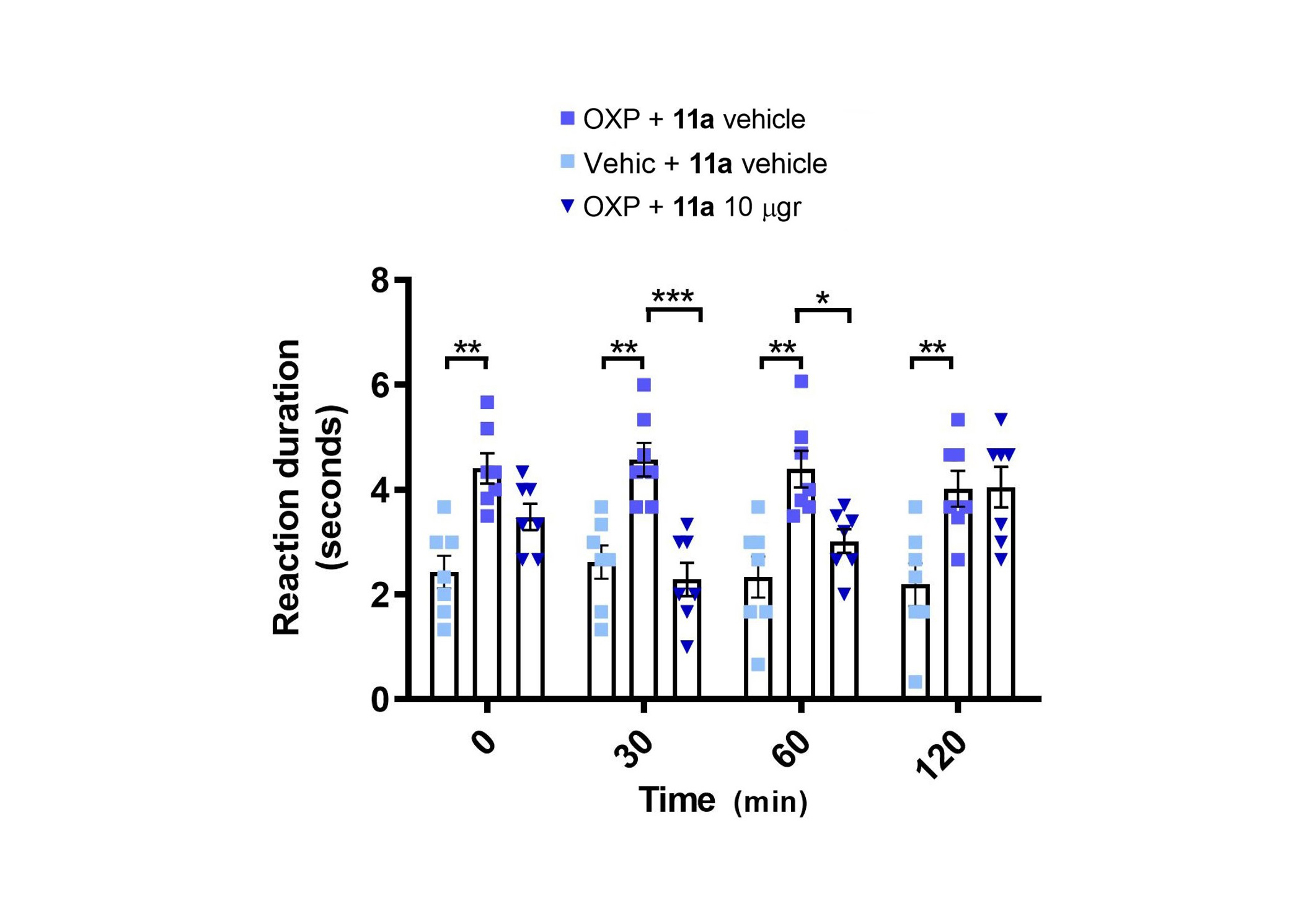
Local application of 11a alleviates oxaliplatin (OXP)-induced sensitivity to cold. Repeated administration of OXP increased the duration of responses to cold-induced by acetone application (** p < 0.01 before vs. after OXP, 2-way ANOVA). 11a alleviated oxaliplatin-induced cold sensitivity 30 and 60 min after its local subcutaneous administration in the paw (i.pl., *** p < 0.001 after OXP vs. 30 min after 11a, * p < 0.05 after OXP vs. 60 min after 11a). 2-way ANOVA followed by Tukey post-hoc test when appropriate. Bars represent average values and error bars represent standard error of the mean (SEM). Dots are the individual values of each mouse (N = 7)
After evidencing OXP-induced sensitization, we evaluated the antinociceptive effects of intraplantar administration of compound 11a (10 µg) or its vehicle in the acetone test, conducted 30, 60, and 120 minutes after treatment. Local subcutaneous application of compound 11a in the paw produced a significant antinociceptive effect in the acetone test, evident at 30 and 60 min after compound application (p < 0.001 at 30 minutes, p < 0.05 at 60 minutes, Figure 6). Collectively, these results suggest that 11a exhibits a significant antinociceptive effect following local application near the peripheral nerves.
After successful stereoselective preparation of the designed β-lactams, their evaluation in the Ca2+ microflorometry assay indicated some influence of the configuration on the channel modulation. The importance of stereochemistry has also been described for other structurally different families of chiral TRPM8 antagonists [48, 51]. However, for the tert-butyl ester derivatives described here, the importance of the absolute configuration does not follow the same tendency than that observed for benzyl ester analogues [45] in Ca2+ microfluorometry assays. In fact, a 2'R configuration is preferred over the 2'S in 3S,4S-cis-configured β-lactams, but not in the 3R,4R-cis-counterparts. All together, these results suggested that both the configuration and the substituents at the 4-carboxylate group dictate the final antagonist potency. In addition, the 2'R configuration afforded compounds with higher selectivity for TRPM8 channels than the corresponding 2'S diastereoisomers. These findings suggested that the 2'-configuration in these compounds is crucial for the discrimination between TRPM8 and TRPV1 channels, with the 2'R-configuration being the preferred orientation for TRPM8 selectivity. Actually, diastereoisomer 1c (3R,4R,2'S) could be considered a dual TRP channel modulator, displaying both agonist activity at TRPV1 and antagonist properties at TRPM8.
As for benzoyl derivatives, most compounds show fairly-higher blockade of menthol-induced TRPM8 channels than AMTB, and are very similar to those of model β-lactam di- and monobenzyl analogues 1a and 2a, respectively, with negligible influence of the substituents on the benzoyl moeity. Further electrophysiology experiments confirmed the antagonist activity in this new series of compounds.
The injection of OXP in mice increased the expression level of TRPM8 channels in primary afferents, which was correlated to cold allodynia and cold pain, which can be reduced by pharmacological blockade of TRPM8 channels [56, 57]. Using this in vivo model, we demonstrated that compound 11a displays antinociceptive activity similar to that described for related β-lactam analogues [44, 46]. A noteworthy feature of compound 11a is its ability to act on other drug targets, such as TRPV1 or CB2R, although less effectively than TRPM8. This broader spectrum of action suggests a potential utility in the treatment of pain associated with the activation of these channels, thus expanding the therapeutic scope beyond TRPM8-mediated conditions. Although drug selectivity has long been considered for therapeutic leads and pursued in medicinal chemistry programs, it is now recognized that mild cross-reactivity with structurally similar receptors may enhance the therapeutic value of drug candidates, as pathological conditions may involve a variety of channels. For example, this multichannel activity may be beneficial in painful conditions, such as OXP-induced neuropathy, where there is a contribution from TRPV1, TRPM8, and TRPA1 channels [58]. The ability to hit multiple targets with a single compound suggests that compound 11a may be a promising therapeutic candidate for further development in this condition, as well as other conditions such as migraine and neuropathic pain, where multiple thermosensory channels are involved [59]. In support of this hypothesis, as indicated, the local application of compound 11a showed significant antinociceptive activity and attenuated OXP-induced cold allodynia. Its local anti-allodynic activity paves the way for the development of this compound for the treatment of this disabling condition in cancer patients. In this sense, topical treatments may represent an optimal approach for the treatment of OXP-induced neuropathy, as they minimize systemic interference with oncological treatments. This opens new opportunities for developing multitarget agents for pain control, warranting further investigation.
AMTB: N-(3-Aminopropyl)-2-[(3-methylphenyl)methoxy]-N-(2-thienylmethyl)benzamide hydrochloride
ASIC3: acid-sensing ion channel, subtype 3
HEK: human embryonic kidney
HPLC: high performance liquid chromatography
NMR: nuclear magnetic resonance
OXP: oxaliplatin
rt: room temperature
TLC: thin-layer chromatography
TPSA: total polar surface area
tR: retention time
TRP: transient receptor potential channel
TRPA1: transient receptor potential ankyrin 1
TRPM8: transient receptor potential melastatin, subtype 8
TRPV1: transient receptor potential vanilloid, subtype 1
The supplementary materials for this article are available at: https://www.explorationpub.com/uploads/Article/file/100882_sup_1.pdf.
We thank Jessy Medina for technical assistance.
CME and TV: Investigation, Formal analysis, Writing—review & editing. MAB and AMP: Investigation, Writing—review & editing. AFM: Supervision, Funding acquisition, Writing—review & editing. AFC: Supervision, Funding acquisition, Writing—original draft, Writing—review & editing. RGM: Conceptualization, Supervision, Funding acquisition, Writing—original draft, Writing—review & editing. All authors read and approved the submitted version.
Rosario González-Muñiz, the Editorial Board Member of Exploration of Drug Science and Guest Editor of the special issue—Mimicking Nature: Biomimetics as Tools for Diagnosis and Therapeutics, had no involvement in the journal review process of this manuscript. The other authors declare that they have no conflicts of interest.
Animal experimentation procedures were conducted under the approval of the Institutional Animal and Ethical Committee at UMH (UMH.IDI.AFM.06.20), and approved by the Generalitat Valenciana (2021/VSC/PEA/0089).
Not applicable.
Not applicable.
Most data from this paper are included in this manuscript and the supplementary materials. Additional data could be obtained from the authors upon request.
This research was funded by FEDER [MCIN/AEI/10.13039/501100011033]; Una manera de hacer Europa [PID2021-126423OB-C21, to AFM and AFC], [PID2021-126423OB-C22, to RGM]; Generalitat Valenciana [PROMETEO/2021/031, to AFM]; CSIC [202180E073, to RGM]; and Comunidad de Madrid [IND2017/BMD7673, to RGM]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

