 Review
Review

Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
1Department of Medicine, Liaquat University of Medical and Health Sciences, Jamshoro 76090, Pakistan
†These authors contributed equally to this work.
Affiliation:
2Department of Medicine, Dow University of Health Sciences, Karachi 74200, Pakistan
†These authors contributed equally to this work.
Affiliation:
3Department of Medicine, Khairpur Medical College, Khairpur Mir’s 66020, Pakistan
†These authors contributed equally to this work.
Affiliation:
4Research Institute for Collaborative Development, Kathmandu 44600, Nepal
†These authors contributed equally to this work.
Email: iambibekgiri@gmail.com
ORCID: https://orcid.org/0009-0003-0100-9111
Explor Drug Sci. 2025;3:100890 DOI: https://doi.org/10.37349/eds.2025.100890
Received: August 24, 2024 Accepted: November 30, 2024 Published: February 25, 2025
Academic Editor: Marcello Iriti, Milan State University, Italy
Ischemic stroke (IS) is a leading cause of death globally. IS occurs due to a blockage of cerebral arteries, leading to neuronal injury, tissue death, and brain infarcts. This induces lack of oxygenation to the brain which induces neuroinflammation, characterised by interactions involving molecules which can exacerbate brain damage but also aid recovery through processes like microglial phagocytosis. Post-stroke depression (PSD) affects 30–33% of stroke survivors, complicating recovery with various symptoms. The pathophysiology of PSD involves disruptions in the glutamatergic and monoaminergic systems, the gut-brain axis, and neuroinflammation. Agomelatine, an atypical antidepressant, can potentially treat both IS and PSD. It acts as a melatonin receptor agonist and a serotonin receptor antagonist, enhancing dopamine and norepinephrine availability in the prefrontal cortex. Agomelatine’s neuroprotective, anti-inflammatory, antioxidative, and antiapoptotic properties have been demonstrated in research, where it reduces reactive oxygen species (ROS) levels and activates the Nrf2 pathway, promoting antioxidative enzyme expression. Additionally, it prevents microglial activation by inhibiting the toll-like receptor 4 (TLR4)/nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) pathway, thus reducing inflammation. This review examines the pathophysiology of IS and PSD, highlighting agomelatine’s multifaceted therapeutic potential. Agomelatine’s distinct pharmacological profile and minimal side effects make it a compelling candidate for IS and PSD treatment, necessitating further exploration to optimise stroke management and improve patient outcomes.
Globally, stroke ranks as the second most common cause of death and the primary cause of disability. The incidence of stroke is rising as the number of people over 65 grows globally at a rate faster than that of any other age group [1]. The two types of stroke are referred to as ischemic stroke (IS) or hemorrhagic stroke (HS). While IS being more prevalent accounts for 87% of all cases [2]. It is caused by a transient or permanent obstruction of the cerebral arteries. IS if not managed can lead to various complications such as cerebral tissue death and brain infarcts [3]. A severe lack of blood flow to the brain following an IS results in an inadequate supply of oxygen, which ultimately causes neuronal death. The cause of ischemic injury in cerebral capillaries is inflammatory responses at the blood-endothelial interface. The pathophysiology of tissue injury in cerebral infarction is greatly dependent on inflammatory interactions including adhesion molecules, cytokines, chemokines, and white blood cells [4]. An acute stroke is followed by secondary neuroinflammation caused by mediators which induces more damage and ultimately results in cell death [5]. Neuroinflammation despite causing potential damage as severe as cell death, is also found to have positive impact by enhancing recovery. The process of post-ischemic inflammatory response is self-limiting and is regulated by numerous mediators that actively suppress inflammatory responses including the elimination of dead cells, the creation of anti-inflammatory environment, and production of pro-survival factors that promote tissue healing and reconstruction [6]. The duration from the onset of stroke, the degree of ischemia, systematic blood pressure, vascular symptoms, and the placement of infarcts are among the factors that affect infarct size and the severity of the neurological damage following IS episodes [3]. Stroke survivors have various physical limitations and an increased risk of psychiatric issues in addition to health challenges. The major post-stroke challenges such as paralysis, aphasia, dysphagia, epilepsy, and cognitive difficulties result in significant loss of healthy life [7].
Depression is one of the most common post-stroke sequelae with prevalence of 30–33%. Anxiety, despondency, reluctance to communicate, and insomnia are common symptoms usually experienced by patients with post-stroke depression (PSD) and have negative impact on daily activities and post-stroke rehabilitation. Furthermore, it has been proposed that PSD is linked to higher risk of death in stroke survivors [7]. Symptoms of depression can appear at any point during recovery but majority of patients exhibit signs of depression three months after stroke. The precise pathophysiology of PSD is unknown. However, it has been demonstrated to be complicated and involve mechanisms such as disruption of glutamatergic and monoaminergic system, the gut-brain axis (GBA), and neuroinflammation [8]. Research indicates that currently, the most preferred treatment option for PSD is antidepressant regimen, which greatly reduces the reported depression symptoms [9]. Antidepressants are of following types: selective serotonin (5-HT) reuptake inhibitors (SSRIs), 5-HT and norepinephrine (NE) reuptake inhibitors (SNRIs), atypical antidepressants, 5-HT modulators, tricyclic antidepressants (TCAs), monoamine oxidase inhibitors (MAOIs), and N-methyl-D-aspartate (NMDA) antagonists [10].
Agomelatine is one of the most recent atypical antidepressants. It is classified as melatonin agonist and selective 5-HT antagonist. It functions as selective agonist of melatonin receptors MT1 and MT2 as well as antagonises 5-HT2C/5-HT2B receptor. The antagonistic action on 5-HT receptor boosts dopamine (DA) and NE availability in prefrontal cortex which has nootropic and antidepressant effects. In prefrontal cortex, agomelatine would optimise learning, consolidate long-term memory, and boost neuronal longevity. The capacity of agomelatine to alter glutamatergic neurotransmission in areas linked to mood and cognition is another significant characteristic. Agomelatine functions as an antidepressant, psycho-stimulant, and promoter of neural plasticity through its synergistic action as a melatoninergic agonist and antagonist of 5-HT2C. It also regulates cognitive symptoms and resynchronises circadian cycles in patients with autism, ADHD, anxiety, and depression [11].
Agomelatine treatment reduced the protein levels of components of the nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3 (NLRP3) inflammasome, and trigger toll-like receptor 4 (TLR4) or nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway. Furthermore, following cerebral ischemia injury, agomelatine inhibited pyroptotic cell death and microglial activation. These findings imply that agomelatine reduces inflammation and lessens brain damage by blocking the TLR4/NLRP3 signalling pathway, which in turn prevents microglial activation [12]. Agomelatine significantly increased the expression of heme oxygenase-1 (HO-1), antioxidative enzymes, and superoxide dismutase (SOD) activity, which is mediated by the Nrf2 pathway. Because agomelatine has antioxidant qualities and suppresses apoptosis, it protects the brain from cerebral ischemia or reperfusion injury. As a result, agomelatine may one day be developed as a possible treatment for IS [13].
In this review, we will describe the pathophysiology of IS and PSD. Role of agomelatine as neuroprotective, anti-inflammatory, antioxidative, and antiapoptotic agent, its safety profile, contraindications, and current limitations. In IS the most preferred treatment is reperfusion therapy which includes intravascular thrombolysis and thrombectomy. However, these treatments have narrow therapeutic window. Hence, there is need for more profound neuroprotective agents for management of IS. SSRIs which are mostly used in PSD can cause headaches, sleep disturbances, tremors, and bleeding. However agomelatine has fewer adverse effects showing reduced risk of insomnia, somnolence, and blurred vision.
IS manifests acutely with neuronal dysfunction and cell death commencing within minutes of blood flow cessation. This interruption is attributed to two primary etiologies: embolic and thrombotic occlusion, where atherosclerotic plaque within cerebral arteries progressively narrows the lumen, ultimately leading to complete blockage [14]. The ischemic core rapidly undergoes irreversible cell death but the region surrounding the ischemic core called ischemic penumbra consisting of potentially degradable nutrient deprived cells but not yet succumbed to cellular demise, is the target of therapeutic interventions aimed at preventing further damage. The phenotypic manifestations, such as hemiplegia, paraplegia, dysarthria, and paresis, are the response of the body towards the cellular damage and they directly correlate with the location of the occluded artery and the corresponding brain region it supplies [15]. IS is characterised mainly by five mechanisms, excitotoxicity and related signalling pathways, oxidative stress, neuroinflammation, apoptosis, and autophagy.
The brain relies on uninterrupted blood flow for oxygen and nutrients. IS ceases this continuous blood supply by occluding cerebral artery, leading to insufficient blood flow and compromised energy production within the ischemic region [16]. This disrupts the ion channels (calcium ATPase, sodium/calcium exchange, and sodium/potassium ATPase) responsible for maintaining ion homeostasis [17]. Thus, calcium ions enter the neurons leading to the activation of enzymes which are dependent on calcium and they cause an excessive of glutamate [18]. This surge in extracellular glutamate overstimulates NMDA receptors (NMDARs) on postsynaptic neurons. NMDARs lead to the generation of reactive oxygen species (ROS) which in turn disrupts mitochondrial function and causes neuronal cell death [19]. In addition, excessive activation of NMDA receptors hinders neuronal plasticity, which has an impact on aging, memory, and learning and it potentially contributes to post-stroke cognitive decline [20]. In contrast to the excitotoxic pathway, some signalling pathways such as phosphatidylinositol 3-kinase (PI3K)-Akt signalling pathway and brain-derived neurotrophic factor (BDNF) act as a pro-survival mechanism in neurons [21, 22]. Extra-synaptic NMDARs frequently oppose the effects triggered by synaptic NMDARs due to their close connection to cell death-related signalling pathway [23].
Oxidative stress is an imbalance between oxidants and antioxidants. This imbalance harms brain cells by increasing susceptibility to free radical damage. IS worsens this imbalance by increasing oxidative metabolism, depleting antioxidants, and elevating pro-oxidants [24]. Disruption in calcium homeostasis [25], production of xanthine oxidase, NADPH oxidase enzymes, and disruption of mitochondria during ischemia leads to increased ROS production, decreased energy output, and the accumulation of stress signals. Furthermore, the activation of pro-apoptotic caspase, release of cytochrome c, and the opening of a membrane pore, lead to cell death [26].
Neuroinflammation is a complex series of inflammatory responses triggered by IS. This involves adaptive (lymphocytes) and innate immune cells (microglia). Necrosis and apoptosis are caused initially which later on initiate the release of chemokines, ROS, and cytokines ultimately leading to neuronal cell death. Microglia can become either pro-inflammatory cells worsening the ischemic injury or microRNAs acting as defenders mitigating stroke damage [27]. Additionally, IS often disrupts the blood-brain barrier (BBB) which further causes the infiltration of immune cells and exacerbates the inflammatory pathways [28].
IS triggers a cellular self-destruction called apoptosis. This process, triggered by both internal intrinsic events (nutrient/oxygen deprivation) and external events (inflammation), dismantles the cell in a controlled manner. Blood restriction during ischemia causes the cell to survive on inefficient energy resources through anaerobic pathway. Following an IS, cell death can occur via any five of the following mechanisms in addition to apoptosis: ferroptosis, phagocytosis, parthanatos, pyroptosis, or necroptosis [29].
IS involves a cellular recycling program called autophagy in response to oxygen and nutrient deprivation. This process normally helps in degradation and recycling of damaged or unnecessary cellular components but in stroke, it plays a complex role [30, 31]. Oxygen deprivation activates hypoxia-inducible factor-1 (HIF-1), which promotes autophagy related genes [32]. Although autophagy has the potential benefit of removing damaged cellular components as a result aiding cell survival, excessive or prolonged activation can lead to the neuronal death and the excessive degradation of cellular components [33].
PSD is a common complication reported in IS survivors. It frequently hinders overall well-being, progress of recovery, and poor rehabilitation [34]. PSD pathogenesis involves a complex interplay between neuroanatomical changes, neuronal dysfunction, biochemical factors, and neurogenesis. The location of stroke lesions, biogenic amines, cytokine inflammation, and gene polymorphism play significant roles in the progression of PSD. IS lesions in critical brain regions such as the left frontal lobe and basal ganglia can disrupt the pathways that are responsible for controlling moods, hence leading to depression [35]. Furthermore, they can alter the neurotransmitter pathways including monoamines. Along with biological abnormalities, studies suggest that social and behavioural factors also play a pivotal role in development of PSD [36].
Additionally IS raises the levels of inflammatory cytokines that regulate the neuroendocrine stress system [37]. These elevated cytokines can suppress brain neurotrophic factors crucial for mood regulation and even contribute to cell death. As a result the immune system of PSD-affected individuals over produces the cytokines which enhances the effects of glutamate excitotoxicity resulting in the expansion of infarctions and the death of neuronal cells [38]. According to research, the stress induced by IS causes hypercortisolism. This cortisol surge decreases 5-HT transporters essential for efficient working of 5-HT, a key mood-regulating neurotransmitter [38]. The monoamine system, glutamatergic system, excitotoxicity, GBA, neuroinflammation, and abnormal neutrophil response all play important roles in the multimodal pathogenesis of PSD [39].
These interacting pathological processes are responsible for neuronal death in IS. Hence, these mechanisms can be targeted to help identify new neuroprotective strategies [40]. The two conventional approaches for managing PSD are pharmacotherapy and psychological therapy. The common antidepressant medications include MAOIs, TCAs, SSRIs, and SNRIs. A research regarding the use of antidepressant drugs to treat PSD [40], successfully demonstrated the effectiveness of nortriptyline in treating PSD. Researchers then provided evidence of citalopram’s effectiveness [41], which can significantly reduce depression symptoms in PSD patients within six weeks. According to accepted treatment regime, SSRI or SNRI should be used to treat PSD initially. If the first-line treatment doesn’t improve the symptoms, a tricyclic (such as nortriptyline) should be given. Certain TCAs, selectively block the transport of 5-HT and some are more active on NE. For many years, TCAs have been used as the most preferred antidepressant with potential benefits to treat PSD [42]. Over time, TCAs have resulted in a number of deleterious effects, including anticholinergic ones [43]. The effectiveness of these drugs in medical practice has been diminished by these adverse effects. Because senior people are more susceptible to these adverse effects and may feel them more severely, this situation is especially significant for them. Consequently, there has been little clinical use of TCAs, particularly in older persons where safety and tolerability are major problems.
An equally important strategy for managing PSD is psychological therapy. A common psychological therapy, cognitive behavioural therapy is considered a potent treatment in older PSD individuals [44]. In addition, repetitive transcranial magnetic stimulation (rTMS) is a non-invasive technique of altering brain physiology [45]. Rachid and Bertschy [46] determined that rTMS can improve depressive symptoms of PSD patients. While the available treatment options demonstrate efficacy in managing PSD, there is a recognised need for exploring alternative therapeutic options due to potential shortcomings such as variable treatment response and the emergence of side effects associated with these medications in some patients. Agomelatine with its recent efficacy in treating PSD and a distinct mechanism of action, presents itself as a promising candidate for further investigation in this regard [47].
The mechanism of action of agomelatine differs extensively from other antidepressants that are commercially available. Agomelatine exhibits dual behaviour as a potent agonist at melatonin MT1 and MT2 receptors and as a neutral antagonist at 5-HT2C receptors. The combined effects on melatonergic and 5-HT2C receptors are responsible for agomelatine’s psychotropic effects [48]. The pattern of binding of agomelatine differs from all other classes of antidepressants currently in use. The finding that agomelatine has no affinity (Ki > 10 μM) for the majority of screened receptors, including adenosine, adrenoceptors, DA, gamma-aminobutyric acid (GABA), muscarinic, nicotinic, histamine, excitatory amino acid, benzodiazepine, and sigma receptors, as well as sodium, potassium, and calcium channels, illustrates how its mechanism of action varies from that of other classes of antidepressants. Similarly, it has minor affinity for the 5-HT2C receptor family and does not recognise most 5-HT receptors but research indicates that agomelatine does interact with cloned human 5-HT2B receptors (pKi= 6.6) and bind to 5-HT2C receptors (pKi = 6.2) [49].
Cellular actions of agomelatine are not caused by melatonin agonists or 5-HT2C antagonists alone. It also causes increased glutamate release in the prefrontal and frontal cortices [50], increased expression of activity-regulated cytoskeleton associated protein in the frontal cortex [48], and increased division, and maturation, in the ventral hippocampus and prefrontal cortex. It also increases the expression of BDNF. Additionally, it has been shown that agomelatine resynchronizes the circadian cycles [51] particularly in suprachiasmatic nucleus (SCN) as a result they reduce depressive symptoms due to irregular sleep patterns. Agomelatine significantly raises DA and NE levels [52]. Unlike other antidepressants, it has little impact on extracellular 5-HT levels [53].
The pharmacokinetic profile of agomelatine indicates that agomelatine is swiftly and effectively absorbed (80%) when taken orally, with a time to maximum plasma concentration of one to two hours. With 95% plasma protein binding, the steady state volume of distribution is 35 L and dose-independent; however, with hepatic impairment, binding drops to 90%. Bioavailability is less than 5%, although it varies greatly amongst individuals and is higher in females. Systemic exposure rises up to 140 times with hepatic impairment, yet it is proportionate to therapeutic levels [54]. Although the bioavailability of agomelatine is less than 5%, it can be enhanced by using three routes; oral, transdermal, and intranasal. It was proposed that the oral bioavailability and, thus, the therapeutic impact may be enhanced by creating agomelatine loaded into nanostructured lipid carriers (NLCs) and targeting Peyer’s patch. The drug’s regulated release from NLCs may further contribute to its long-term effectiveness. Since NLCs have a second-generation lipidic nature, the M cells in Peyer’s patches can readily and directly absorb them. Because of their micro size, they can also bypass first pass metabolism. The results of the pharmacodynamic investigation showed that the agomelatine-NLCs were more effective than the marketed product and pure medication. The findings of the pharmacokinetic investigation showed that agomelatine-NLCs had higher maximum concentration (Cmax), area under the curve (AUC), and oral bioavailability when compared to a commercial product and a pure medication. Agomelatine-NLCs’ oral bioavailability rose by 6.5 times [55]. In addition to reducing first-pass drug metabolism, avoiding gastrointestinal irritations, maintaining drug activity, and minimizing side effects, transdermal drug delivery systems also improve patient compliance [56]. To improve transdermal drug permeation, various techniques are used, including the use of nanocarriers (e.g., microemulsions, nanoparticles, and liposomes), penetration enhancers, and physical techniques, such as iontophoresis, microneedles, and electroporation, either alone or in combination with other methods [57].
The nasal route was chosen to improve drug delivery to the brain by avoiding the hepatic first-pass metabolism and increasing absolute bioavailability. Solid lipid nanoparticles were used as a drug delivery technology to improve agomelatine permeability across the BBB, resulting in better brain delivery [58].
Agomelatine is instantly metabolised, with hepatic cytochrome P450 1A2 (CYP1A2) enzyme orally accounting for 90% of the process. However, 10% of the metabolism also involves CYP2C9 and CYP2C19. Thus, caution be taken when agomelatine is given with drugs that trigger one or more of these enzymes to be inhibited or stimulated, such as fluvoxamine, ciprofloxacin, rifampicin, cigarettes, or oestrogens. When these drugs are used concurrently, agomelatine exposure reduces; yet, agomelatine exposure increases when oestrogens are administered. Its high clearance rate of 1,100 mL/min and its mean half-life of 1 to 2 hours are mostly due to urinary metabolites (80%). Agomelatine’s main metabolites, hydroxylated and demethylated, are quickly eliminated and are inert [54].
Apart from vertigo, agomelatine has been demonstrated to have a favourable side effect profile [59]. Additionally, no increase was observed in body weight and sexual dysfunction, which are prevalent with several kinds of antidepressants [60]. Although clinically significant increase in aminotransferases was observed in 2.4% and 4.5% of patients in the groups taking 50 mg of agomelatine, but not in the groups taking 25 mg. In the agomelatine 50 mg group, cholecystitis, gallbladder disease, or hepatic steatosis were more common in past medical histories. Thus, people with hepatic impairment should not use agomelatine. This indicates higher incidence of hepatobiliary disorders [61].
Agomelatin’s dual effect aids in the resynchronization of disturbed circadian rhythms and irregular sleep patterns. Antidepressant effects are ascribed to its effects on the blockage of 5-HT2C receptors as well as its sleep-promoting and chronobiotic properties, mediated via MT1 and MT2 receptors. Both NE and DA are released at the fronto-cortical dopaminergic and noradrenergic pathways upon the blockage of 5-HT2C receptors, which improves mood and cognition [62, 63]. Additionally, agomelatine also improves the clinical status of major depressive disorders [64]. A study showed that administration of agomelatine prolongs the duration of slow-wave sleep, decreases the duration of REM sleep, and decreases the motor activity at onset of the dark [65]. Agomelatine maintains the quality of sleep at night while improving mood during the day. This is because of its dual mode of action, whereby its melatonin-promoting action promotes sleep while its 5-HT2C antagonistic action counteracts the antihypnotic effects [66]. Therefore, the key element of agomelatine’s antidepressant action is its ability to enhance sleep efficiency and synchronise the sleep-wake cycle.
Agomelatine, like melatonin, serves as an antioxidant. Hence it can significantly reduce neuronal damage caused by ischemia by activating anti-apoptotic, and antioxidative pathways. Since IS generates ROS that disrupts antioxidant mechanisms, specifically Nrf2 signalling, which regulates antioxidant enzymes, activating Nrf2 promotes the expression of antioxidant enzymes which protect cells from the negative effects of ROS. Furthermore, administering agomelatine has been shown to improve neurological impairments and reduce brain infarction volume. A study found that agomelatine dramatically reduces ROS, malondialdehyde (MDA) levels, and lipid peroxidation in cerebral IS injury. Agomelatine administration significantly boosted Nrf2, HO-1, SOD, and glutathione peroxidase, and lowered glutathione levels in the ischemic brain, indicating that it modulates the Nrf2/antioxidant defence mechanism. Agomelatin also reduces pathological alterations and protects the ischemic penumbra [13].
Neurological abnormalities resulting from IS are largely caused by white matter injury, which calls for efficient repair techniques, including oligodendrocyte-driven re-myelination. Agomelatine maintains the integrity of the white matter following a cerebral IS and stimulates the development of oligodendrocyte precursor cells (OPCs) because of its mechanistic activation of the LRP1 and peroxisome proliferator-activated receptor γ (PPARγ) signalling pathways, which are essential for OPC development and post-stroke remyelination. Research indicates that 5-HT and melatonin antagonists provide protection against white matter damage following a stroke. Hence agomelatine with its dual melatonin receptor agonist and selective 5-HT receptor antagonist properties, is a potentially effective treatment. It alleviates long-term sensorimotor and cognitive deficits caused by stroke and decreased brain tissue loss [19].
It can suppress apoptosis demonstrating significant neuroprotective benefits. It enhances B cell lymphoma-extra large protein expression and prevents apoptotic neuronal cell death, thereby significantly reducing apoptosis by lowering Bax and cleaved caspase-3 levels [13]. Agomelatine inhibits TLR4/NLRP3 pathway-mediated microglial activation to produce an anti-inflammatory effect in IS [12]. IS increases the permeability of the BBB leading to the induction of inflammation through chemical mediators and the influx of inflammatory cells like macrophages [67]. Agomelatine deals with this by suppressing macrophage polarisation and invasion [68]. Furthermore, under hypoxic conditions, agomelatine has the capacity to prevent macrophage movement through endothelial cells by increasing the expression of claudin-5, an essential protein that keeps endothelial tight junctions intact and helps rebuild the integrity of the BBB. Additionally, agomelatine effectively decreases the elevated Evans blue staining, which was indicative of BBB breakdown caused by IS [69] (Figure 1).
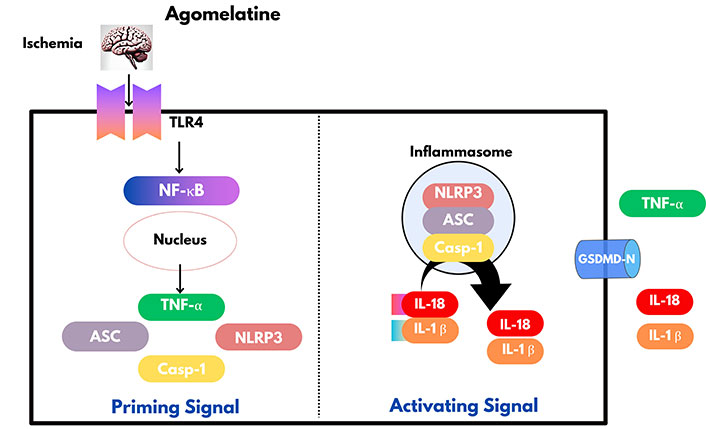
Agomelatine exerts an anti-inflammatory effect by inhibiting microglial activation through TLR4/NLRP3 pathway. ASC: apoptosis associated speck like protein containing a caspase activation and recruitment domain; Casp-1: caspase-1; GSDMD-N: N-terminus of gasdermin D; IL-18: interleukin-18; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3; TLR4: toll-like receptor 4; TNF-α: tumor necrosis factor-α. Created with canva.com
Several research and clinical trials have proved the efficacy of agomelatine in PSD. Researchers have demonstrated that agomelatine exhibits greater efficacy and fewer side effects in the treatment of PSD resulting in improved depressive symptoms, promoted recoverance of neural function, enhanced sleep quality, better cognitive function, increased motivation and interest, reduced anxiety and stress, and improved tolerability [70, 71].
There is a causal relation between the quality of sleep and the outcome of depression. Thus, improvement in sleep quality can directly improve the emotional status of patients suffering from depression therefore activation of MT1 and MT2 by agomelatine can improve patients’ sleep quality and wakefulness during daytime which in turn lessens the depressive symptoms [72, 73]. By agonizing melatonin receptors, agomelatine can also help in reducing oxidative stress and protecting neural cells [74].
Agomelatine increases the levels of DA and noradrenaline in PSD patients which induces therapeutic effects on depressive symptoms and improves mood, motivation, and cognitive function. Furthermore, agomelatine also reverses neuropsychiatric symptoms, such as anhedonia (inability to experience pleasure or enjoyment from activities that would normally be pleasurable) [72, 73, 75].
There is growing proof that the widespread use of agomelatine is linked to liver damage [76]. Reportedly, agomelatine causes pathological liver injury and raises liver weight and coefficient [77]. Consequently, the European Medicines Agency (EMA) declared that “hepatotoxic reactions” linked to the usage of agomelatine since significant increases in liver enzyme levels were observed. Most commonly encountered side effects were mild nausea and dizziness. Age over 50, female sex, polypharmacy, and liver illness have all been implicated as potential risk factors for the development of agomelatine induced hepatotoxicity [78]. Hence, monitoring liver enzyme levels in all patients has been recommended by the EMA [78] before beginning treatment, and upon each followup. Hence, the regular usage of agomelatine raises apparent concerns regarding all of these liver-related disorders.
The agomelatine despite being approved in certain significant areas has not been approved by FDA [79]. A major limitation of agomelatine is the lack of long-term studies on tolerability and safety [80]. Another drawback is its inability to raise 5-HT levels, unlike other antidepressants. Despite several clinical trials, it was inefficient in reducing gastrointestinal side effects and sexual dysfunction [81]. Studies indicate that agomelatine is ineffective in older patients (75 years and above), additionally, its use has not been established in paediatric population and teenagers [79].
In summary, IS is a significant global health issue due to its widespread occurrence, complications, and impact on patient’s quality of life. The condition involves complex mechanisms like excitotoxicity, oxidative stress, neuro-inflammation, apoptosis, and autophagy, leading to severe neuronal damage. PSD, a common complication of IS, further hampers recovery by altering brain structure, neurotransmitter levels, and inflammatory processes, resulting in a poor prognosis.
Agomelatine, an atypical antidepressant, shows promise in treating both IS and PSD. It works as a melatonin receptor agonist and selective 5-HT antagonist, offering neuroprotective benefits such as reducing oxidative stress, preventing cell death, and controlling neuroinflammation. Additionally, agomelatine improves sleep, mood, and cognitive function, making it a potential treatment for PSD. However, more research is needed to fully understand its safety, particularly regarding liver health, and its effectiveness in older adults and children. Exploring the combination of agomelatine with other treatments could enhance patient outcomes, but further studies are essential to confirm its role in treating PSD.
5-HT: serotonin
BBB: blood-brain barrier
BDNF: brain-derived neurotrophic factor
CYP1A2: cytochrome P450 1A2
DA: dopamine
EMA: European Medicines Agency
GBA: gut-brain axis
HO-1: heme oxygenase-1
IS: ischemic stroke
MAOIs: monoamine oxidase inhibitors
NE: norepinephrine
NLCs: nanostructured lipid carriers
NLRP3: nucleotide-binding domain, leucine-rich-containing family, pyrin domain-containing-3
NMDA: N-methyl-D-aspartate
NMDARs: N-methyl-D-aspartate receptors
OPCs: oligodendrocyte precursor cells
PSD: post-stroke depression
ROS: reactive oxygen species
rTMS: repetitive transcranial magnetic stimulation
SNRIs: serotonin and norepinephrine reuptake inhibitors
SOD: superoxide dismutase
SSRIs: selective serotonin reuptake inhibitors
TCAs: tricyclic antidepressants
TLR4: toll-like receptor 4
During the preparation of this work, the author(s) used the canva.com to generate the image/figure. After using the tool/service, the author(s) reviewed and edited the content as needed and take(s) full responsibility for the publication's content.
AA, ARS, SHA, UAS, SKB, UN, KW, FL, and MU: Data curation, Investigation, Conceptualization, Methodology, Writing—original draft. BG: Investigation, Project administration, Visualization, Supervision, Writing—review & editing. All authors have equal contributions to this study, and they have approved the final version of this manuscript.
The authors declare that there are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

