 Review
Review

Affiliation:
1Gene Therapy Research Unit, Children’s Medical Research Institute, Faculty of Medicine and Health, The University of Sydney and Sydney Children’s Hospitals Network, Westmead NSW 2145, Australia
ORCID: https://orcid.org/0009-0000-7241-2280
Affiliation:
1Gene Therapy Research Unit, Children’s Medical Research Institute, Faculty of Medicine and Health, The University of Sydney and Sydney Children’s Hospitals Network, Westmead NSW 2145, Australia
ORCID: https://orcid.org/0000-0002-0876-6292
Affiliation:
1Gene Therapy Research Unit, Children’s Medical Research Institute, Faculty of Medicine and Health, The University of Sydney and Sydney Children’s Hospitals Network, Westmead NSW 2145, Australia
2Discipline of Child and Adolescent Health, Sydney Medical School, Faculty of Medicine and Health, The University of Sydney, Westmead NSW 2145, Australia
ORCID: https://orcid.org/0000-0002-6213-5627
Affiliation:
1Gene Therapy Research Unit, Children’s Medical Research Institute, Faculty of Medicine and Health, The University of Sydney and Sydney Children’s Hospitals Network, Westmead NSW 2145, Australia
2Discipline of Child and Adolescent Health, Sydney Medical School, Faculty of Medicine and Health, The University of Sydney, Westmead NSW 2145, Australia
3Institute of Endocrinology and Diabetes, The Children’s Hospital at Westmead, Westmead NSW 2145, Australia
Email: lara.graves@health.nsw.gov.au
ORCID: https://orcid.org/0000-0002-8476-220X
Explor Endocr Metab Dis. 2024;1:101–121 DOI: https://doi.org/10.37349/eemd.2024.00011
Received: January 22, 2024 Accepted: March 11, 2024 Published: July 09, 2024
Academic Editor: Charlotte Steenblock, University Clinic Carl Gustav Carus, Germany
The article belongs to the special issue The HPA Axis in Health and Disease
Congenital adrenal hyperplasia due to 21-hydroxylase deficiency leads to high morbidity and mortality, despite the availability of life-saving corticosteroid replacement therapy. Gene therapy represents a promising potential treatment for monogenic disorders such as congenital adrenal hyperplasia, overcoming the limitations of corticosteroid replacement approaches. Adeno-associated viral vectors are currently the leading vector for direct in vivo gene delivery. However, physiological properties of the adrenal gland limit the application of adeno-associated viral vector-based gene addition strategies. To achieve durable correction in the adrenal gland, gene editing must be employed to stably introduce a genetic modification into the CYP21A2 locus. The safety of this and other gene editing approaches could be greatly improved by using lipid nanoparticles for the delivery of editing machinery mRNA. While little data exists regarding adrenocortical lipid nanoparticle targeting, physiological features of this organ (such as high relative blood flow, fenestrated endothelium, and cholesterol uptake) indicate the promise of these delivery vectors for the treatment of monogenic diseases of the adrenal cortex. This review discusses the complexities of developing gene therapy for congenital adrenal hyperplasia and explores the viability of novel gene therapy strategies in this application.
Congenital adrenal hyperplasia (CAH) encompasses a group of seven monogenic endocrine disorders affecting the adrenocortical production of steroid hormones. CAH can lead to life-threatening adrenal crises, poor health outcomes, and low quality of life [1]. Steroid replacement therapy is the current standard of care for CAH, but it is unsatisfactory, in part due to inadequate biomarkers for disease monitoring [2]. Dosing regimens are unable to replicate the variability in endogenous corticosteroid levels, leading to both suboptimal treatment and a high incidence of side effects. Emerging pharmacological approaches are similarly imperfect [3]. CAH represents an excellent candidate for gene therapy. The most frequently used tools for in vivo gene delivery are recombinant adeno-associated virus (rAAV) vectors, which have been shown to transduce the adrenal cortex [4, 5]. While the application of rAAV-based gene addition strategies has demonstrated that phenotypic improvement can be achieved, rAAV genomes are predominantly episomal and so these approaches have been unable to provide a long-term benefit [4]. This is due to both the biology of adrenocortical cellular turnover and the choice of a gene addition strategy [3]. To provide a long-term benefit to individuals with CAH, a new durable strategy must be employed.
In contrast to gene addition approaches, gene editing permanently corrects pathogenic variants in the genome of a patient’s cells, allowing the cell to produce corrected progeny [6]. In the highly regenerative adrenal cortex, it will also be critical to target a gene editing therapy to the adrenocortical stem and/or progenitor cells, as the commonly targeted differentiated cells are relatively short-lived [7]. In this way, the adrenal cortex could be continuously repopulated with corrected cells, permanently improving the steroidogenic function of treated individuals.
However, gene editing faces challenges associated with the inadvertent creation of off-target double-stranded breaks (DSBs) in the host genome. Gene editing tools such as clustered regularly interspaced palindromic repeats/CRISPR-associated protein 9 (CRISPR/Cas9) carry the risk of creating off-target editing events, which can lead to cell cycle arrest and/or mutagenesis of host genes and the risk of eliminating any residual function in a hypomorphic allele if editing efficiency is inadequate [8]. While rAAVs have been used to carry both donor cassettes and editing reagents, their propensity to persist in the host cell for long periods increases the risk of off-target events [9, 10]. To improve safety, the gene therapy field is moving towards the use of lipid nanoparticles (LNPs) for the delivery of mRNA-encoded editing reagents [11–13].
There is a paucity of data regarding the efficacy of LNPs for delivering editing reagents to the adrenal glands. However, existing data hints at excellent prospects for adrenocortical LNP targeting [14–16]. This review outlines the prospects for a gene editing therapy for CAH using an LNP-AAV approach. The justification, practical aspects, and feasibility of such a strategy will be discussed in detail with a focus on the unique contexts of CAH and the adrenal glands.
In > 90% of cases, CAH is due to deficiency of the 21-hydroxylase (21OH) enzyme [17], and classical CAH occurs in 1:14,000–1:22,000 live births [18, 19]. 21OH deficiency is caused by mutations in the CYP21A2 gene [20]. 21OH is a cytochrome P450 enzyme that catalyses multiple reactions in the steroidogenic pathway. In the zona glomerulosa, 21OH converts progesterone to 11-deoxycorticosterone as part of aldosterone synthesis, and in the zona fasciculata, 17-hydroxyprogesterone is converted by 21OH to 11-deoxycortisol for cortisol synthesis [21]. Deficiency in 21OH therefore affects the production of both aldosterone and cortisol, leading to a build-up of the upstream metabolites of these reactions [22]. Reduced cortisol-mediated negative feedback on the hypothalamus and anterior pituitary gland leads to overproduction of adrenocorticotropic hormone (ACTH), stimulating hyperplasia and hypertrophy of the adrenal cortex, a feature that gives CAH its name [23]. ACTH stimulation also leads to excess 17-hydroxyprogesterone and progesterone, which are then shunted through to the 17-hydroxylase pathway, resulting in overproduction of adrenal androgens and consequential virilisation [1].
CAH phenotypes correlate with the level of residual 21OH function. Presentations are divided into two main groups; classical CAH, which encompasses both salt-wasting (SW) and simple-virilising (SV) phenotypes, and non-classical CAH (NCCAH) [24]. Although divided into these theoretically discrete groups, the phenotype occurs on a continuous spectrum, and most have compound heterozygosity with the phenotype tending to correlate with the less severe allele [25, 26]. Classical SWCAH occurs in patients with up to 1–2% residual enzyme activity and is the most severe phenotype [18]. In SWCAH, the production of aldosterone and cortisol is insufficient to sustain life, and there is an overproduction of adrenal androgens. Disease phenotypes become progressively less severe with increased residual enzyme activity. Higher levels of residual enzyme activity (up to 5–10%) result in the SVCAH phenotype, where aldosterone production is (generally) sufficient but severe cortisol deficiency and androgen overproduction remain [18]. However, relative aldosterone deficiency may still occur in people with SVCAH [27]. NCCAH occurs with up to 50% residual 21OH activity [28–30]. In NCCAH, sufficient cortisol can be synthesised to adapt to most physiological stressors, though there may occasionally be mild cortisol deficiency [29]. Adrenal crises are often precipitated by physiologically stressful events such as infectious illness [31, 32], and were found to be the leading cause of all-cause mortality in people with CAH [33].
Overproduction of adrenal androgens is a hallmark of all forms of 21OH deficient CAH, with effects most evident in female infants with classical CAH, who experience varying levels of genital virilisation evident at birth [34]. In childhood, adrenal androgen overproduction can lead to premature adrenarche and precocious puberty, as well as rapid growth and tall stature in childhood with early growth plate closure and short final height [35]. Women also experience fertility issues, hirsutism, and acne, while men are at risk of developing testicular adrenal rest tumours [36].
Aside from the immediate effects posed by a lack of sufficient corticosteroids, individuals with CAH experience a wide range of symptoms from both CAH itself and the treatment required. Even with optimal treatment, there are higher rates of adverse long-term outcomes than the general population. Overall, there is an increased risk of death from all-cause mortality, with estimates of mean age of death ranging from 6.5 to 18 years earlier than the general population [33, 37]. Significant mental health comorbidities can also present, with a higher lifetime incidence of depression in individuals with CAH [37]. Additionally, adrenal crises precipitated by physiological stress cannot always be prevented by increased steroid dosing [38].
Life-saving corticosteroid replacement therapy was first introduced in the 1950s [39–41], and this remains the current standard treatment for classical CAH [2]. Deficiencies in aldosterone and cortisol are corrected by administering fludrocortisone and hydrocortisone, respectively [2]. Hydrocortisone is administered 3–4 times daily in children, complemented by mineralocorticoid replacement when required. In adults, treatment is similar, though hydrocortisone may be swapped for longer-acting glucocorticoids for regimen simplification [2]. Treatment goals include the avoidance of adrenal crises, as well as reducing the overproduction of adrenal androgens by decreasing ACTH stimulation of steroidogenesis, particularly in women [42]. Steroid doses are increased during times of physiological stress (such as illness and surgery), and mildly supraphysiological doses are used to suppress androgen overproduction [2].
However, current treatment is unsatisfactory for several reasons. Steroid replacement therapy results in a variety of negative side effects such as short stature, loss of bone mineral density, and fertility issues [1]. Natural glucocorticoid hormone levels fluctuate significantly with physiological demand and circadian rhythm [43]. Given the early timing of the peak and the short half-life of hydrocortisone, it is very difficult to appropriately mimic physiological variation in cortisol levels [1]. Thus, both under- and over-treatment commonly occur within a single day. Overtreatment increases the incidence of negative side effects, while undertreatment risks adrenal crisis and cannot adequately control androgen overproduction [1].
Novel treatments for CAH include delayed-release oral corticosteroids, co-opting insulin pumps for continuous administration of hydrocortisone, and drugs to suppress adrenal androgen overproduction [44–46]. Delayed-release hydrocortisone (Efmody®, marketed for individuals with CAH over 12 years of age) has not been shown to be superior to standard therapy [47]. The use of an insulin pump for delivery of a continuous infusion of subcutaneous hydrocortisone may be able to replicate the circadian rhythm more accurately, but this solution is likely too expensive and complex for widespread use [48], and there is a risk of adrenal crisis in cases of pump failure. Finally, drugs to suppress adrenal androgen production (such as abiraterone acetate, which inhibits 17-hydroxylase) [46] are being trialled in pre-pubertal children (NCT02574910). While these approaches may be useful to reduce the effects of androgenisation, corticosteroid therapy is still required, and they cannot be used in pubertal adolescents or adults.
Gene therapy involves the use of nucleic acid or nucleic acid analogue to modify disease by regulating, restoring, replacing, or removing the expression of a gene. Due to the unsatisfactory treatment options and the monogenic basis of CAH, gene therapy treatments are being explored to restore or correct the defective CYP21A2 gene. An ideal gene therapy approach for CAH would allow synthesis of corticosteroids under complete physiological control, allowing serum concentrations to adjust to biological requirements [3]. CAH is also a good candidate for gene therapy due to the large phenotypic improvements conferred by very minimal increases in 21OH activity. For example, SWCAH and SVCAH phenotypes differ by only 1–2% in total 21OH activity [18]. As such, restoration of even a small amount of total 21OH activity by gene therapy could confer a large clinical benefit. Furthermore, the adoption of a ‘one-and-done’ treatment for CAH is made more feasible by the widespread implementation of newborn screening programs [2, 49], providing a unique window of opportunity for the administration of curative treatments prior to the onset of adrenal crisis.
Recombinant adeno-associated virus is the leading vector for efficient in vivo gene delivery [50]. Vectorology exploits the ability of a virus to insert genetic material into a host cell nucleus. AAV is a parvovirus first discovered in 1965 [51], which is 20–25 nm in diameter and consists of a protein capsid encapsulating a 4.7 kb single-stranded DNA genome [52]. The AAV genome consists of two genes, rep and cap, which are flanked by inverted terminal repeat sequences that are required for genome packaging during viral assembly [52]. In rAAV vectors, rep and cap are removed and replaced with a therapeutic cassette which contains the transgene, and may contain other regulatory elements such as promoters, enhancers, and polyadenylation sequences. The resultant virus is replication-incompetent while retaining the capacity to efficiently deliver its cargo to the host cell nucleus.
rAAV vectors are a flexible platform for gene delivery. A crucial advantage of AAV is the ability for the rAAV2 genome to be packaged into different capsid variants in a process known as pseudo-serotyping. As the capsid directs tissue tropism, a variety of species-specific tissue-targeted vectors can be created with relative ease [53]. A wide variety of naturally occurring AAV capsid variants have been discovered [52] and, by directed evolution or mutagenesis of capsid sequences, novel variants can be generated to enhance interaction with a target tissue [54]. The liver is particularly amenable to rAAV-mediated gene delivery. This is in part due to the physiological characteristics of the liver including fenestrated endothelium and high relative blood flow which provides opportunity for interaction with relevant endocytic receptors [55]. However, rAAV capsids have been engineered to target other organs, such as the lungs, brain, and heart [56].
In gene addition strategies, a replacement copy of a target gene is delivered with its own promoter and regulatory elements [52]. When rAAV is used, the vector genome containing the transgene remains predominantly episomal and in a stable cell population is continually expressed for at least 10 years [9]. However, episomal rAAV genomes are lost with cell replication, so rAAV gene addition is generally only stable in post-mitotic tissues such as the brain, heart, muscle, and adult liver [9, 57, 58].
Thus far, all published preclinical studies of CAH gene therapy have used gene addition strategies (Table 1). Most studies use the H-2aw18 mouse model, which accurately recapitulates CAH, with the exception of adrenal androgen overproduction as mice lack 17-hydroxylase expression in the adrenal cortex [59, 60]. Vector administration via intra-adrenal injection has been attempted [61, 62], but may not be a viable strategy due to technique difficulty [63], and low prospects for clinical translation. Delivering ectopic 21OH expression in muscle via local injection has not led to a significant measurable benefit [64]. Intravenous administration of rAAV vectors containing either the human or murine 21OH gene has shown promise, with biochemical phenotype correction observed post-treatment [4, 5]. ACTH, progesterone, and corticosterone levels can be improved, sometimes to near-wildtype levels [4, 5], by rAAV delivery of the human CYP21A2 gene [65]. When measured, improvements in adrenocortical morphology and adrenal mass were sometimes [62], but not always [5], observed.
Preclinical studies of gene therapy for CAH to date
| Vector and transgene | Model | Dose and route | Key outcome(s) | Longest benefit duration | Reference |
|---|---|---|---|---|---|
| AdenovirusCYP21A2 | H-2aw18 mice(Cyp21a1-/-) | 1 × 108 pfu/adrenalOpen bilateral intra-adrenal injections | CYP21A2 mRNA expressed in ~ 30% of cortex and medulla, peaking at 2–7 days and still detectable at 14 days.Normalised corticosterone production to 40 days.Improved progesterone/deoxycorticosterone ratio at 7 days but not at 40 days.Transduced adrenals still produced corticosterone ex vivo at 40 days.Improvement in adrenocortical ultrastructure at 7 days. | 40 days | Tajima et al. 1999[62] |
| RetrovirusCyp21a1 | H-2aw18 mice(Cyp21a1-/-) fibroblasts | 5 × 105 cells/mouseAutologous subcutaneous transplantation of ex vivo transduced fibroblasts | Four weeks post-implantation, 4 of 6 experimental mice had reduction in progesterone/deoxycorticosterone ratio. | 4 weeks | Naiki et al. 2016[64] |
| AAV2Cyp21a1 | H-2aw18 mice(Cyp21a1-/-) | 1 × 1011 vgc/mouseIntra-muscular injection | All mice (n = 4) had reduction in progesterone/deoxycorticosterone ratio (statistically insignificant).One mouse followed to 8 months retained “relatively low” progesterone/deoxycorticosterone ratio until 7 months. | 7 months | |
| AAVRh10CYP21A2 | H-2aw18 mice(Cyp21a1-/-) | 2 × 1013 vgc/kgIntravenous injection | Correction in body weight up to 15 weeks.Correction of kidney renin expression at 18 weeks.Urinary progesterone near WT levels up to 15 weeks.Correction in stress response behavioural traits.No correction in adrenocortical morphology. | 18 weeks | Perdomini et al. 2017[5] |
| AAVRh10CYP21A2-HA | H-2aw18 mice(Cyp21a1+/+) | 6.5 × 1011 vgc/mouseIntravenous injection | Left adrenal showed waning CYP21A2 expression, from 65% positive cells (2 weeks) to 2.1% positive cells (32 weeks).Liver retained CYP21A2 expression at 32 weeks, while adrenal gland lost expression. | 16 weeks | Markmann et al. 2018[4] |
| AAVRh10CYP21A2-HA | H-2aw18 mice(Cyp21a1-/-) | 6.5 × 1011 vgc/mouseIntravenous injection | Progesterone significantly decreased at 2 weeks, no effect by 10 weeks.ACTH decreased from 2–8 weeks, no significant effect by 32 weeks. | 8 weeks | |
| AAVRh10CYP21A2 | Cynomolgus macaques | 5 × 1012, 1.5 × 1013, or 4.5 × 1013 vgc/kgIntravenous injection | Detectable vector genomes in adrenals and liver up to 24 weeks (high dose).Transgene expression increased from 4 to 12 weeks, decline evident at 24 weeks (high dose). | 24 weeks | Eclov et al. 2020[66] |
| AAV9CYP11B1 | Cyp11b1-/- mice | 1 × 1010 vgc/adrenalIntra-adrenal injection | All mice (n = 4) had significant decrease in serum deoxycorticosterone/corticosterone ratio at 4 weeks.Serum deoxycorticosterone/corticosterone ratio remained low in 2 mice followed to 5 months.No apparent transgene expression in adrenal glands of injected mice11B-hydroxylase activity did not improve (data not shown). | 22 weeks | Naiki et al. 2022[61] |
| AAV8CYP21A2 | H-2aw18 mice(Cyp21a1-/-) | 5 × 1011 vgc/mouseIntravenous injection | All mice (n = 10) normalised serum aldosterone and renal renin expression.All mice increased serum corticosterone and reduced adrenal hyperplasia. | 4 weeks | Graves et al. 2024[67] |
AAV: adeno-associated virus; ACTH: adrenocorticotropic hormone; CAH: congenital adrenal hyperplasia; pfu: plaque-forming units; vgc: vector genome copies; WT: wild-type
A nonhuman primate (NHP) study of an AAV capsid serotype 5 (AAV5)-CYP21A2 gene addition vector (BBP-631) developed by Adrenas Therapeutics led to a low but measurable expression of human 21OH [65]. Stable transgene expression for 6 months was reported [66], however, only 2 animals were followed up to this time point, both having received the highest vector dose studied (4.5 × 1013 vgc/kg), and declining mean transgene expression was already evident at 6 months. Furthermore, the reported data was not separated by sex, despite the inclusion of both sexes in the study. This would have masked any variations in transgene durability or therapeutic efficacy that may have resulted from sexual dimorphism, which is known to affect the adrenal glands in mice [7].
None of these approaches have shown durability of therapeutic benefit over time. In the study that best exemplifies this issue, serum progesterone and ACTH levels returned to near-normal for a short period post-treatment, but the effect was lost by 10 or 32 weeks, respectively [4]. Immunohistochemical staining showed progressive loss of 21OH expression, with 21OH-negative cells gradually repopulating the cortex in a centripetal manner. It is also possible that observed durability may have been a result of unintentional liver transduction, as liver-specific expression of 21OH following AAVRh10-CYP21A2 delivery has been shown to significantly improve biochemical phenotype in H-2aw18 mice [67].
Despite the evident difficulty of stably transducing the adrenal cortex with rAAV gene addition therapy [4, 62], BBP-631 has entered Phase I/II clinical trials (NCT04783181), representing the first-in-human administration of CAH gene therapy [65]. By January 2024, seven participants had been treated at four different dose rates with good safety outcomes [65, 68, 69]. One patient showed an increase in cortisol levels from 104.8 nmol/L at baseline to 234.5 nmol/L at 12 weeks post-injection [69]. Due to the known durability issues in previous adrenal-targeted gene addition strategies, the initiation of human clinical trials for BBP-631 has been criticised as premature [70]. The recurrent loss of therapeutic benefit observed across studies employing rAAV gene addition strategies, coupled with the timing and inward progression of this loss within the adrenal glands, foreshadows several critical issues with this approach to CAH gene therapy.
While rAAV gene delivery can have long-term clinical benefit in post-mitotic or slowly dividing tissues, the adrenal cortex is highly regenerative, undergoing constant cellular renewal [71], with proliferation of adrenocortical stem and progenitor cells at the periphery of the cortex [7, 72–74], inward movement of differentiated daughter cells [72, 75–77] which undergo lineage conversion to populate the deeper zones [78], and eventual apoptosis of the differentiated cells at the border of the medulla. While the presence of several populations of adrenocortical stem and progenitor cells has been confirmed, they are poorly characterised and cannot yet be readily isolated.
The regenerative nature of the adrenal gland has posed the largest challenge to achieving sustained benefit with an rAAV gene addition strategy for CAH. As discussed previously, while gene addition strategies have been applied to CAH in numerous preclinical studies [4, 5, 61, 62, 64, 66], none have achieved a duration of clinical benefit lasting beyond the cellular turnover time of the adrenal gland. The murine adrenal cortex is completely replaced after 3 months in the female mouse and estimated after 9 months in the male [71]. Adrenocortical turnover has been strongly implicated as the cause of waning therapeutic efficacy in preclinical gene therapy studies, exemplified by the inward movement and eventual disappearance of corrected cells at the corticomedullary border [4]. The sexual dimorphism of adrenocortical turnover in mice provides an additional explanation for the variable durability of effect between studies, as most existing work has not reported sex or has grouped analyses for both male and female mice.
While these studies have clearly demonstrated the difficulty of achieving a durable effect, they have also shown that if a functional CYP21A2 gene is supplied in sufficient quantity to 21OH deficient animals, biochemical and phenotypic disease markers can be improved. The quantity of corrected cells needed for phenotypic benefit may be relatively low, as improvements have been observed in these studies with 39% adrenocortical cells corrected [5]. Therefore, if the hurdle of durability could be overcome, a highly effective CAH gene therapy could be developed.
To confer a permanent clinical benefit using gene therapy for CAH, it is likely that a gene editing approach targeting the adrenocortical stem and/or progenitor cells will be necessary (Figure 1) [3].
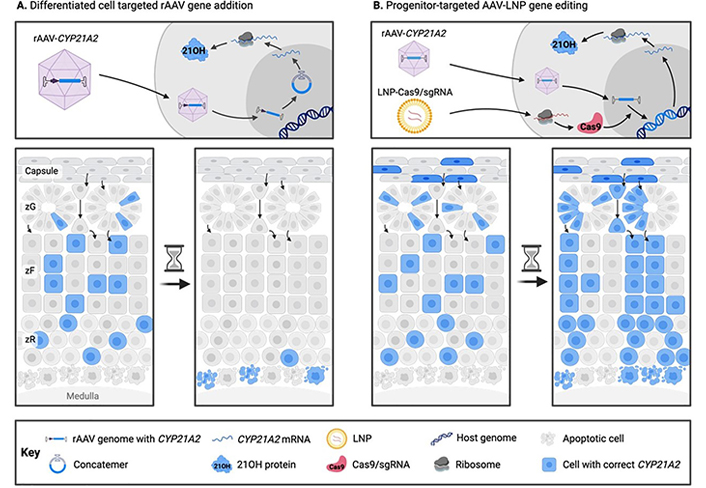
Comparison of traditional (A) and novel (B) gene therapy approaches for congenital adrenal hyperplasia. In traditional rAAV gene addition approaches targeting differentiated adrenocortical cells, clinical benefit as a result of transgene expression is gradually lost due to adrenocortical turnover and episome loss. In a proposed novel rAAV-LNP genome editing treatment for CAH, adrenocortical progenitor cells are stably corrected at the genomic level with a functional copy of CYP21A2, and thus the adrenal cortex is repopulated with corrected steroidogenic daughter cells. 21OH: 21-hydroxylase; CAH: congenital adrenal hyperplasia; LNP: lipid nanoparticle; rAAV: recombinant adeno-associated virus; sgRNA: single guide RNA; zF: zona fasciculata; zG: zona glomerulosa; zR: zona reticularis
Note. Adapted with permission from “Future directions for adrenal insufficiency: cellular transplantation and genetic therapies” by Graves LE, Torpy DJ, Coates PT, Alexander IE, Bornstein SR, Clarke B. J Clin Endocrinol Metab. 2023;108:1273–89 (https://academic.oup.com/jcem/article/108/6/1273/6972401). © Oxford University Press 2023.
The term “gene editing” can encompass several therapeutic strategies, which are selected based on the disease and mutation. In gene disruption, expression of a gene is reduced by creating a DSB which is repaired non-faithfully to induce a frameshift mutation [79]. In gene correction, a pathogenic variant in a gene is permanently corrected, either by alteration of a single base (base editing) or insertion of a replacement therapeutic gene cassette. Over 350 variants in the CYP21A2 locus span the promoter region and all 10 exons (and introns) [80], so the most viable strategy is to insert a partial or complete functional gene sequence into the host genome. To do so, a targeted DSB is induced in the genome in the presence of a “template” gene. The break is then repaired by endogenous mechanisms, and during the repair process the template gene is used to correct the mutation.
Early gene editing tools such as transcription activator-like effector nucleases (TALENs) and zinc-finger nucleases have been largely superseded by CRISPR/Cas based editing tools [6, 81]. Cas9 is part of the Cas family of proteins, which are programmable nucleases that use RNA guidance to target a site in the host genome. Cas9 has generally been used to create DSBs at the target site, though non-cutting or ‘nicking’ variants have also been developed [82]. The cutting activity of the Cas9 protein can be easily directed through the co-delivery of a single guide RNA (sgRNA), allowing targeting based on simple Watson-Crick base pairing rules. The only restriction on Cas9 targetable sites is that they must possess a protospacer adjacent motif, a short and enzyme-specific sequence that is required for Cas9-DNA binding [6].
CYP21A2, which encodes 21OH, lies in a highly complex region of the human genome on chromosome 6p21.3, as part of a tandem arrangement of repeated CYP21A and complement 4 genes. A non-functional pseudogene (CYP21A1-P), which has 98% exonic and 96% intronic homology with CYP21A2, is located approximately 30 kb from CYP21A2 [83–86]. The high degree of homology between gene and pseudogene results in the majority of CAH-causing mutations, as mutations from CYP21A1-P can be transferred to CYP21A2 or large regions may be deleted during crossing over [87]. Over 90% of pathogenic variants in CYP21A2 occur due to recombination events between CYP21A2 and CYP21A1-P, and it is this mechanism that explains the predominance of 21OH deficiency as a cause of CAH [87].
An important limitation in the use of CRISPR/Cas9 in CYP21A2 is that the chosen sgRNA binding site must be unique to CYP21A2 and not also present in CYP21A1-P. Selection of a unique and specific sgRNA targeting site will be critical to ensuring the safety of an editing strategy, as the creation of DSBs in both gene and pseudogene could result in the loss of the ~30 kb between them. This task is made more difficult by the high level of intronic and exonic homology between CYP21A2 and CYP21A1-P.
In CAH, the functionality of 21OH is attenuated to different extents depending on the mutation [30, 88]. As pathogenic 21OH variants can still have minimal residual function [88], a sgRNA selected for CAH gene editing should undergo extensive testing to ensure that it will not knock out the function of a potentially hypomorphic allele. Targeting the sgRNA deep within a non-coding sequence may decrease the chances of such an event.
Following a DSB, host DNA repair mechanisms repair the break. Several gene correction approaches have been developed that take advantage of these endogenous repair pathways, which can be broadly divided into non-homologous end joining (NHEJ) and homologous repair (HR) pathways (Figure 2). In NHEJ, the ends of a DSB are directly ligated in an efficient but error-prone process [89]. NHEJ is active throughout all stages of the cell cycle and is the most efficient DSB repair pathway [90]. NHEJ repair is mainly employed in gene disruption, where the generation of small insertions and deletions (‘indels’) is desirable to induce frameshift mutations [79]. NHEJ can also be exploited for gene correction, in the case where the most common predicted indel also happens to correct the mutation [91, 92]. HR repairs DSBs with much higher fidelity than NHEJ using a homologous template strand [89]. In the endogenous HR pathway, the sister chromatid that acts as a template is only present after DNA replication, so HR is restricted mostly to the late S/G2 phase [93]. In homology-directed repair (HDR) editing strategies, in addition to the CRISPR/Cas9 and sgRNA, a template gene flanked by sequences homologous to the target region is also delivered to act as a template for HR [94]. However, HDR strategies are inefficient in cells that are not actively dividing, restricting their efficient use in highly mitotic tissues [95].
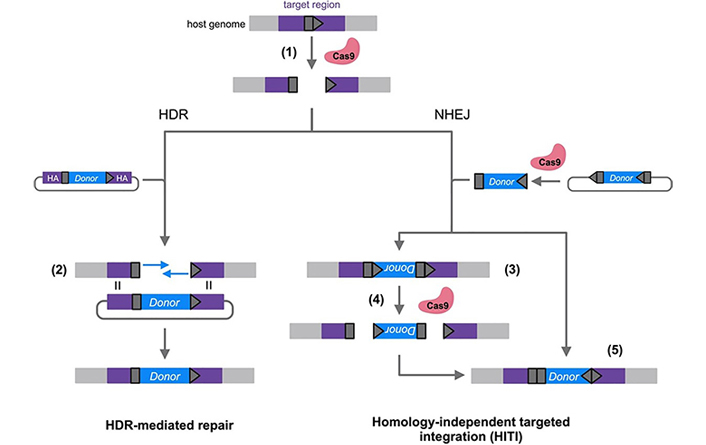
Comparison of homology-directed repair (HDR)-based and homology-independent targeted integration (HITI) gene editing strategies. A target site (dark grey pentagon) is selected in the host genome and Cas9 is directed to the site by a complementary single guide RNA (sgRNA). After a Cas9/sgRNA-induced double-stranded break (DSB) is made in the target site (1), host repair pathways can include both HDR and non-homologous end joining (NHEJ). HDR-mediated repair strategies exploit HDR by providing a donor vector with homology arms (HA) (purple) flanking the gene to be inserted (blue). This homology allows the donor sequence to be used as a template for the synthesis of repair DNA in the host genome (2). HITI strategies exploit the NHEJ pathway by including gRNA target sequences on either side of the donor cassette. When donor DNA is inserted in the incorrect orientation (3), the gRNA target sites are reconstituted and Cas9-mediated cutting can reoccur (4). When the donor is inserted correctly (5), the gRNA sites are destroyed, leaving the donor unable to be re-excised [96]
Note. Adapted with permission from “Genome editing in the human liver: Progress and translational considerations” by Ginn SL, Christina S, Alexander IE. Prog Mol Biol Transl Sci. 2021;182:257–88 (https://www.sciencedirect.com/science/article/abs/pii/S1877117321000417). © Elsevier 2021.
Homology-independent targeted integration (HITI) overcomes some of the limitations of HDR. The HITI approach was developed to exploit the more active NHEJ pathway while still allowing the insertion of a therapeutic gene [97]. The donor transgene is flanked by sgRNA binding sites that match the sites selected in the host genome. Both host genome and template DNA are cleaved by Cas9, and the donor fragment is inserted into the DSB of the host genome upon NHEJ repair. While the donor can be inserted in either direction, the sgRNA sites are designed so that they are reconstituted when the gene is inserted in the incorrect orientation and are destroyed when it is inserted correctly. Thus, incorrectly inserted genes are re-excised, while correctly inserted genes remain.
A HITI approach would be optimal for correcting CAH for a number of reasons. The CYP21A2 coding sequence is approximately 1.5 kb [83], compatible with the 4.7 kb packaging capacity of the rAAV backbone. This means it would be feasible to deliver and insert the entire CYP21A2 coding sequence, creating an opportunity for the development of a therapy that could treat most or all existing pathogenic variants. CAH has highly heterogenous causative variants, so this type of universal or near-universal treatment approach would be required for financial and practical feasibility. While HDR could also be used to insert a large transgene sequence, the increased efficiency of a HITI approach will be necessary to achieve clinical benefit, especially when considering the difficulty of adrenocortical progenitor cell targeting. The genetic heterogeneity of CAH also limits the practicality of HDR, as different therapies would need to be developed with homology arms that matched each individual’s set of genetic variants.
rAAVs are efficient for delivery of Cas9 editing reagents to a wide variety of tissues. However, episomal rAAV genomes can persistently express Cas9 within the host cell, leading to potential safety issues. In addition, rAAV genomes can integrate non-specifically into the host’s genome [98]. Therefore, delivery of Cas9/sgRNA sequences via rAAV is likely to result in long-term Cas9 expression and activity and off-target DSBs [10, 99]. This non-specific cutting is a safety risk, as it can lead to cell cycle arrest [100] and mutagenesis of off-target genes [8]. Furthermore, promoter/enhancer elements are necessary for Cas9/sgRNA expression from the episome, and the integration of these elements into the host genome creates further mutagenic risk [101]. Finally, a dual-AAV editing approach would necessitate a high total rAAV dose, as two distinct vectors must be used. In clinical trials using high-dose rAAV, immune responses and hepatotoxicity have been observed, which in some cases have led to participant death [102, 103].
Due to the mounting issues identified in the use of dual-AAV Cas9 editing strategies, alternate delivery methods are sought. LNPs are composed of both lipids and nucleic acids, and measure 60–150 nm in diameter [104]. Originally developed to enhance chemotherapeutic drug delivery, LNPs have been more recently adopted for the delivery of nucleic acids such as silencing RNA (siRNA) and mRNA [105]. The clinical use of LNPs for mRNA delivery was accelerated by the recent development of LNP-mRNA SARS-CoV-2 vaccines [106].
To exert a therapeutic effect, mRNA must enter the cytoplasm of a target cell. As naked RNA is rapidly degraded in the bloodstream, LNPs are used to envelop the nucleic acids and protect them from degradation [107]. Additionally, LNP encapsulation allows mRNA molecules to cross the cell membrane, as they are otherwise too large and negatively charged to effectively do so [108, 109]. A successful LNP-mRNA therapeutic agent must reach its target organ while avoiding clearance, must interact with and be taken up by the correct cell type, and must then efficiently escape the endosome. Ideally, mRNA-containing LNPs for gene therapy should also be minimally immunogenic.
Modern LNP formulations consist of four lipid components: an ionisable lipid, a ‘helper’ phospholipid, cholesterol, and a poly(ethylene)glycol-functionalised (PEGylated) lipid. The identities and ratios of these lipid components can be tuned to optimise delivery characteristics (“passive targeting”), or the LNP can be functionalised with ligands specific to a given cell type or tissue (“active targeting”) [110].
In an LNP-rAAV gene editing approach, mRNA encoding the Cas9 protein is delivered via LNP (Figure 3). If required, a donor cassette is delivered via rAAV, as LNPs are currently inefficient for delivery of large DNA cassettes to the nucleus [111]. Guide RNA can either be delivered in the LNP or can be transcribed from the rAAV-delivered cassette.
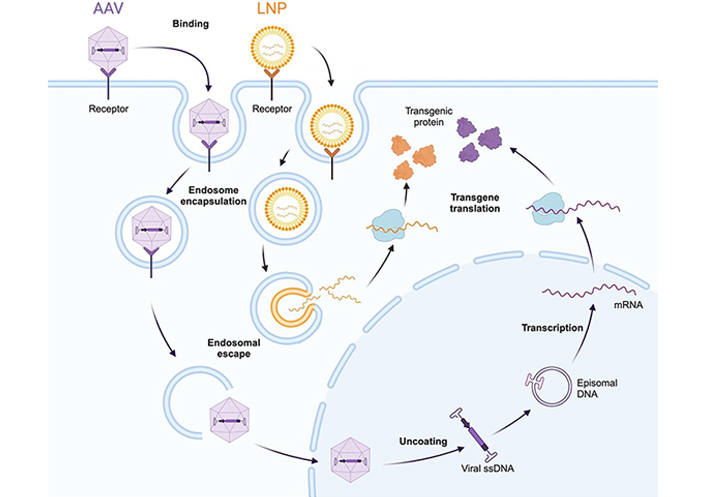
Cellular entry and nucleic acid delivery mechanisms of rAAV (purple) and LNP (orange) delivery vectors. Both vectors must be internalized via endosome encapsulation, and subsequently escape the endosome to enter the cytoplasm. rAAV vectors are still intact at this stage and go on to enter the nucleus, where the single-stranded viral genome is released and concatemerises to form a double-stranded episome. The episome is transcribed by host polymerase systems. Conversely, upon endosomal escape, LNPs deliver their mRNA cargo into the cytoplasm directly, where it is translated to protein. This mechanism bypasses the need for nuclear entry. AAV: adeno-associated virus; LNP: lipid nanoparticle
Note. Adapted from “AAV Vector Infection”, by BioRender.com (2023). Retrieved from https://www.biorender.com/template/aav-vector-infection
LNP-rAAV gene editing strategies have several advantages over dual-rAAV approaches.
LNPs do not have the same strict packaging capacity as AAVs [112]. For example, the coding sequence of SaCas9 will take up 3.1 kb of the total 4.7 kb rAAV packaging capacity, leaving minimal flexibility for the addition of promoter and regulatory elements. While the length of mRNA correlates with the number of encapsulated molecules [113], this varies with formulation parameters, so there is no strict packaging capacity. This increases the flexibility of LNP delivery.
The synthesis and purification of LNPs is much less labor-, time-, and cost-intensive than that of rAAVs, significantly reducing the cost per dose of a potential gene therapy [114, 115].
mRNA delivered by LNP is degraded quickly after entering the cytoplasm, so the protein is only expressed for hours to days [116], in comparison to the extended expression resulting from rAAV use. As the prolonged presence of DNA-encoded Cas9 protein in the host cell is more likely to lead to off-target cutting [10], delivery of Cas9 as an mRNA provides a safety benefit through shorter duration of expression.
It is possible that rAAV-delivered Cas9 DNA could integrate into the host genome [98], a risk which is removed with Cas9 mRNA delivery.
An rAAV transgene encoding Cas9 must contain a promoter to express it, and in the case of rAAV genome integration, this creates a further risk of insertional mutagenesis [101]. When both Cas9 and sgRNA are delivered via LNP, the strategy can become completely free from promoter/enhancer elements, greatly reducing mutagenic risk.
LNPs are much less immunogenic than rAAVs [117, 118]. Additionally, LNPs carry a much lower risk of pre-existing host immunity. The reduced immunogenicity also opens possibilities for repeat administration of the LNP, which is not currently possible for rAAVs [117].
There is no published work which describes the deliberate targeting of LNPs to the adrenal cortex, either by rational design or screening approaches. The adrenal gland is not frequently included in the analysis of LNP biodistribution and functional activity, leaving a paucity of data regarding adrenocortical targeting prospects. However, several studies have reported incidental adrenal LNP transfection.
Several lipid-based nanocarrier formulations (“lipidots”) composed of soybean oil, PEGylated lipid, phospholipids, and esterified cholesterol were tested for delivery of fluorescent dye or a radioactive tracer to steroidogenic organs in female mice [16]. The adrenal glands and ovaries had levels of radioactive LNP association equivalent to or above that of the liver, depending on the formulation. Upon histological examination of the adrenal glands following fluorescent dye delivery, it was found that the border of the medulla had a bright signal, and patches of dim fluorescence were visible throughout the zona fasciculata and zona glomerulosa. The ovaries, which share an embryological origin with the adrenal cortex, had the highest signal in several tested formulations, which increased in a dose-dependent manner [16]. The authors hypothesised that low-density lipoprotein (LDL) mimicry contributed to the steroidogenic organ targeting of the nanoparticles, as cholesterol uptake is required for steroidogenesis and the adrenal expresses high levels of the LDL receptor (LDLR) [119, 120].
OnpattroTM (patisiran) is an approved LNP-siRNA therapeutic for the treatment of hereditary transthyretin-mediated amyloidosis and is formulated to deliver the therapeutic cargo to the liver [105]. In preclinical rat studies, radiolabelled liver-targeted LNPs were found to associate with the adrenal gland, with evidence that they received between 0.03% and 0.14% of the total LNP dose [15]. In NHPs, adrenocortical vacuolation was reported in a repeat-dosing toxicity evaluation 6 weeks post-treatment [15]. Adrenocortical vacuoles often represent an accumulation of excess steroidogenic precursors, including cholesterol [121]. While it is possible that the vacuolation seen in this study represented an accumulation of LNP-derived lipids, vacuolation can also occur due to external stressors or HPA axis perturbation [122]. Regardless, this study provides evidence that the OnpattroTM LNP formulation (or products of its degradation) interacts with the adrenal cortex.
Further evidence of adrenal LNP tropism has arisen from the testing of a liver-targeted LNP base-editing approach for the treatment of familial hypercholesterolaemia [14]. Base edits of PCSK9 occurred in the adrenal gland at a rate of 1–5%, and the left and right adrenal glands were the 2nd and 3rd most edited off-target organs, respectively. While the ionisable lipid is proprietary, the formulation used in this study appears to closely mimic the OnpattroTM formulation in terms of lipid ratio and the identity of the other three components [116].
Given that most evidence of LNP activity in the adrenal gland has come from studies of liver-targeted LNPs, it is unsurprising that several characteristics hypothesised to improve LNP uptake are shared by both the liver and the adrenal. Both organs contain a fenestrated endothelium, a feature of the blood vessels which facilitates the passage of nanoparticles from the bloodstream into the organ [123, 124]. Both organs receive a high proportion of blood flow relative to their size, with the adrenal gland (in rats) receiving 7× total blood flow relative to its size and the liver (in humans) receiving 10× [125–127], increasing the likelihood of LNP interactions. Finally, ApoE uptake is high in both organs, and they share a high expression of LDLR [119, 128]. Given that nanocarriers formulated without cholesterol could still associate strongly with the adrenal gland [16], it is possible that ApoE-independent mechanisms may also contribute to adrenal LNP accumulation. While data on adrenal tropism of LNPs is minimal, extensive data supports liver tropism of these along with other nanoparticles. The similarities between these organs, along with the supporting data from studies outlined above, show that the adrenals are promising but under-studied target organs for LNP-mRNA therapeutics.
The work examined in this review points toward the promise of CAH and the adrenal glands as targets for an LNP-AAV gene editing therapeutic. Prior work has shown that rAAV-based CYP21A2 gene addition can restore normal physiology in 21OH deficient animal models. However, durability remains the largest hurdle to clinical viability. A successful LNP-AAV gene editing approach targeted toward the adrenocortical stem and progenitor cells would overcome this hurdle, while improving greatly on the safety outcomes of dual-AAV editing approaches. The increasing adoption of LNP-AAV gene editing strategies, coupled with improvements in the genetic and phenotypic characterisation of CAH, will continue to provide valuable information in the application of these approaches to CAH.
21OH: 21-hydroxylase
ACTH: adrenocorticotropic hormone
CAH: congenital adrenal hyperplasia
Cas9: CRISPR-associated protein 9
CRISPR: clustered regularly interspaced palindromic repeat
DSB: double-stranded break
HDR: homology-directed repair
HITI: homology-independent targeted integration
HR: homologous repair
LDL: low-density lipoprotein
LNPs: lipid nanoparticles
NCCAH: non-classical congenital adrenal hyperplasia
NHEJ: non-homologous end joining
rAAV: recombinant adeno-associated virus
sgRNA: single guide RNA
SVCAH: simple virilising congenital adrenal hyperplasia
SWCAH: salt-wasting congenital adrenal hyperplasia
Figures were created in Biorender.com.
EBVD: Conceptualization, Writing–original draft, Writing–review & editing. SLG and IEA: Validation, Writing–review & editing, Supervision. LEG: Conceptualization, Writing–review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
EBVD was supported by the Research Training Program Scholarship, Australian Government. EBVD was additionally supported by the Children’s Medical Research Institute PhD Scholarship. LEG was supported by the Yass Memorial Scholarship, Children’s Medical Research Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Stefan R. Bornstein ... Waldemar Kanczkowski
Amanpreet Kaur Kalsi ... Jai Bhagwan Sharma
Waldemar Kanczkowski ... George P. Chrousos
John Milton, Alexander Churilov
Laura C. A. van der Zwet ... Tom Deboer
