 Review
Review

Affiliation:
1Bone Research Program, ANZAC Research Institute, The University of Sydney, Concord, NSW 2139, Australia
Email: eugenie.macfarlane@sydney.edu.au
ORCID: https://orcid.org/0000-0003-3608-3285
Affiliation:
1Bone Research Program, ANZAC Research Institute, The University of Sydney, Concord, NSW 2139, Australia
ORCID: https://orcid.org/0000-0001-5899-9660
Affiliation:
1Bone Research Program, ANZAC Research Institute, The University of Sydney, Concord, NSW 2139, Australia
2Department of Endocrinology and Metabolism, Concord Repatriation General Hospital, Sydney, NSW 2139, Australia
ORCID: https://orcid.org/0000-0002-2701-378X
Explor Endocr Metab Dis. 2024;1:191–212 DOI: https://doi.org/10.37349/eemd.2024.00016
Received: April 17, 2024 Accepted: July 04, 2024 Published: August 16, 2024
Academic Editor: Marijn Speeckaert, Universitair Ziekenhuis Ghent, Belgium
The article belongs to the special issue The Fountain of Youth: Decoding the Hormonal Regulation of Aging
Stress hormones, namely glucocorticoids, have diverse actions throughout the body in regulating development, tissue metabolism, inflammation, circadian rhythms, and skeletal homeostasis. While endogenous glucocorticoid levels are important to support bodily homeostasis, chronically elevated levels can cause damage to tissues and drive diseases including bone loss (i.e., osteoporosis), myopathy (i.e., sarcopenia) and metabolic disturbances (i.e., glucose intolerance, diabetes, and abnormal fat accrual). There is substantial evidence that basal glucocorticoid levels increase during ageing while at the same time the amplitude of the diurnal variation in glucocorticoid secretion decreases. However, the significance of these changes for skeletal health is not well understood and has only recently been studied in more detail. Evidence from genetically modified mouse models indicates that changes in glucocorticoid signaling associated with ageing induce bone loss, sarcopenia and drive osteoarthritic joint disease. These studies provide important insights into the role of glucocorticoids in age-related skeletal diseases which will aid in the development of novel treatments especially needed for osteoarthritis which disproportionally affects the elderly.
Glucocorticoids are essential for life and play a critical role in nearly all physiological processes including somatic development, immunity, tissue metabolism, skeletal integrity, mineral homeostasis, and the regulation of circadian rhythms throughout the body. Under normal conditions, basal circulating glucocorticoid levels exhibit robust daily and seasonal rhythms to support a variety of homeostatic functions. In response to stress, however, blood glucocorticoid concentrations can quickly rise, reaching levels several times above baseline. While transient increases in glucocorticoid levels are important for survival in circumstances of infection or the “flight or fight” response, chronically elevated levels are associated with a plethora of adverse health effects including bone loss, myopathy, abnormal adipose tissue deposition, impaired glucose tolerance, and the development of osteoarthritis [1–3]. With advancing age, circulating as well as local, tissue-specific glucocorticoid levels exhibit distinct changes, characterized by the loss of normal diurnal rhythmicity and a modest increase in basal concentrations [4, 5]. However, the significance of these changes for skeletal health is not well understood and has only recently been studied in more detail. The development of transgenic and glucocorticoid receptor knockout mouse models that disrupt glucocorticoid signaling have provided important insights into how glucocorticoids drive skeletal diseases during ageing. This review provides an updated summary on studies that have disrupted glucocorticoid signaling in mice to investigate the implications of ageing-related changes in glucocorticoid physiology on osteoporosis, sarcopenia, and osteoarthritis.
Glucocorticoids are steroid hormones synthesized and secreted by the adrenal glands under the direct control of the hypothalamus and pituitary within a negative feed-back loop known as the hypothalamic-pituitary-adrenal (HPA) axis (Figure 1). In addition, the body’s master clock, which is located in the suprachiasmatic nucleus (SCN) of the brain, also modulates the activity of the HPA axis, so that under normal conditions adrenal glucocorticoid secretion follows a diurnal rhythm of approximately 24 hours [6, 7]. In humans, circulating glucocorticoid levels peak in the early morning, with a second smaller peak in the late afternoon, followed by a nadir during the night. In contrast, nocturnal rodents exhibit only one daily peak in the evening upon awakening. The pivotal role of the central master clock in maintaining diurnal glucocorticoid rhythmicity is evidenced by the observation that disruption of normal SCN signaling in rats abolishes the diurnal variation in circulating glucocorticoid levels [6, 8, 9].
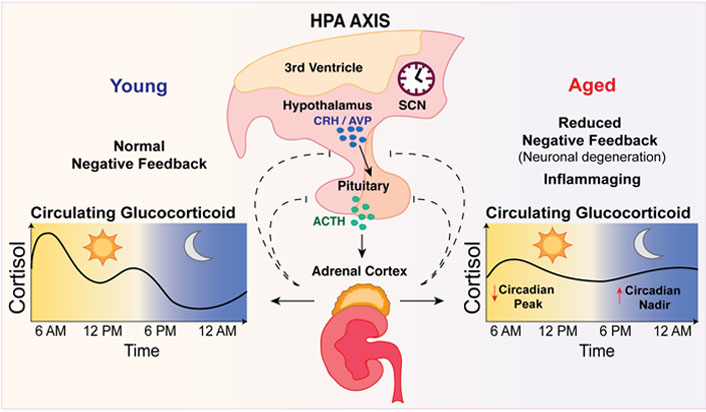
Circulating glucocorticoid levels and rhythmicity becomes disturbed with ageing. Glucocorticoids (i.e., cortisol) are synthesized by the adrenal glands under the control of the hypothalamic-pituitary-adrenal (HPA) axis and the body’s master clock in the suprachiasmatic nucleus (SCN). Physiological stimuli including light, stress, inflammation, illness, and exercise, stimulate the hypothalamic paraventricular nucleus to release corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) into the anterior pituitary gland, triggering the release of adrenocorticotropic hormone (ACTH). ACTH drives release of glucocorticoids from the adrenals into the circulation [10, 11]. In young adults, circulating glucocorticoid levels peak in the morning upon awakening, with a smaller peak in the afternoon and lowest levels observed around midnight [6, 7]. In the elderly, glucocorticoid rhythmicity becomes markedly disturbed whereby the morning peak in circulating glucocorticoids occurs earlier and is reduced, with higher cortisol secretion at night [4, 5]. This occurs from blunted and delayed negative feedback inhibition of ACTH secretion [12–14]
In addition to circadian time cues (i.e., light exposure), various other stimuli including stress, inflammation, illness, or exercise can stimulate the hypothalamic paraventricular nucleus to release corticotropin-releasing hormone (CRH) and arginine vasopressin (AVP) into the hypophysial portal vessels that connect to the anterior pituitary gland [10, 11]. In the anterior pituitary, CRH and AVP bind to their respective receptors and trigger the release of adrenocorticotropic hormone (ACTH), which stimulates the synthesis of glucocorticoids in the adrenal zona fasciculata, and their release into the circulation [10–14]. However, glucocorticoid release can also occur through direct SCN-autonomic innervation of the adrenals via the splanchnic nerve [15].
As well as the classical 24-hour circadian rhythm, glucocorticoids are also secreted in a pulsatile fashion, displaying an ultradian rhythm of short-lived bursts of glucocorticoid release from the adrenals [16–18]. The pulsatile frequency of glucocorticoids is 90 min in humans and 60 min in rodents, likely due to differences in the biological half-lives of cortisol and corticosterone [19]. It has been proposed that the ultradian pulsatility in glucocorticoid secretion occurs due to delays between the activation of the HPA axis and the secretion of glucocorticoids, as well as delays in the negative feedback of glucocorticoid inhibition on the hypothalamus and pituitary [20].
At the tissue level, glucocorticoids freely diffuse across the cell membrane and into the cytoplasm where their concentrations are further finetuned by the actions of 11β-hydroxysteroid dehydrogenase (11β-HSD) isoenzymes 1 and 2 (Figure 2). 11β‐hydroxysteroid dehydrogenase type 1 (11β-HSD1), also known as a cortisone reductase, interconverts inactive cortisone into its active form, cortisol. Conversely, 11β-HSD2 catalyzes the conversion of cortisol into inactive cortisone [21]. While 11β-HSD1 is widely expressed in most tissues including the liver, adipose and bone, expression of 11β-HSD2 is confined to mineralocorticoid target tissues such as the kidneys, colon, and salivary glands [22]. In such tissues, 11β-HSD2 primarily acts to prevent cortisol preferentially binding to the mineralocorticoid receptor (MR), enabling aldosterone to bind to the MR instead [23].
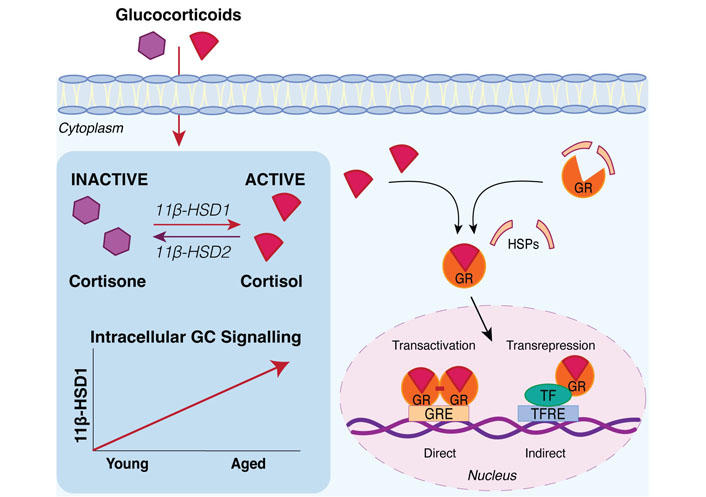
Intracellular glucocorticoid signaling increases with ageing through 11β-hydroxysteroid dehydrogenase enzymes. Tissue specific glucocorticoid (GC) levels at sites including the skeleton are elevated with ageing via the glucocorticoid activating enzyme, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) which converts inactive cortisone into active cortisol. 11β-HSD2 catalyzes the conversion of cortisol into inactive cortisone but is expressed primarily in mineralocorticoid target tissues such as the kidneys, colon, and salivary glands [22]. Active glucocorticoids exert their genomic effects by binding to the cytoplasmic glucocorticoid receptor (GR). Upon binding glucocorticoids, the GR undergoes a conformational change where it dissociates from accessory heat shock proteins (HSPs), enabling the GR-ligand complex to translocate into the nucleus [24, 25]. In the nucleus, the glucocorticoid-activated GR regulates gene expression through different mechanisms such as direct heterodimeric binding to glucocorticoid response elements (GREs) in the DNA or indirect monomeric tethering to other transcription factors bound to transcription factor response elements (TFREs) in the DNA [25]. TF: transcription factor
Active glucocorticoids exert most of their functions by binding to the cytoplasmic glucocorticoid receptor (GR), which then undergoes a conformational change where it dissociates from accessory proteins, enabling the GR-ligand complex to translocate into the nucleus with the help of chaperone proteins [24, 25]. Within the nucleus the glucocorticoid-activated GR regulates gene expression through different mechanisms such as direct heterodimeric binding to glucocorticoid response elements (GREs) in the DNA or indirect monomeric tethering to other transcription factors bound to the DNA [25]. Growing evidence suggests that the GR can also act via non-genomic mechanisms through interactions with membrane lipids and cytoplasmic proteins to elicit rapid cellular responses that occur within a few minutes and do not require changes in gene transcription [26, 27].
Ageing is associated with significant changes in the body’s 24-hour circadian rhythms, characterized by a loss of normal rhythmicity in circulating hormones, including glucocorticoids, and a decline in sleep duration and quality [4, 28–30]. Cumulative evidence indicates that disruption to circadian clocks, either by genetic deletion or environmental disturbances in the light-dark cycle, reduces lifespan and may promote the development of ageing-related diseases such as cataracts, cancer, bone loss and osteoarthritis [31–34]. Remarkably, aged rodents transplanted with a graft of fetal suprachiasmatic nucleus (i.e., a younger master clock) appear to regain robust circadian rhythms and live longer than control animals [31, 35–37].
Glucocorticoids are a central part of the circadian system in coordinating time cues from the master clock to synchronize internal clocks in peripheral organs and tissues throughout the body [38, 39]. However, with ageing the pattern of glucocorticoid secretion becomes markedly disturbed and basal hormone concentrations generally tend to increase with age [4, 5]. These changes are observed both systemically in the circulation (through impaired regulation of the HPA axis) (Figure 1) and locally in tissues via increased intracellular metabolism of glucocorticoids by the 11β-HSD isoenzymes (Figure 2). With age, glucocorticoid actions can also be affected by local changes in the expression and activity of the GR.
In young adults, circulating glucocorticoid levels peak early in the morning upon awakening with a smaller second peak in the afternoon. Thereafter, glucocorticoid concentrations gradually decline with lowest levels observed around midnight [40]. In contrast, glucocorticoid rhythmicity in the elderly is characterized by a disturbed pattern of secretion whereby the morning peak in circulating glucocorticoids occurs earlier and is reduced, with higher cortisol secretion at night [4, 5, 41, 42] (Figure 1). Clinical studies suggest that compared to young adults, the nocturnal nadir of circulating glucocorticoid levels can be up to four-fold higher in people over the age of 70 years, translating into an overall increase in mean 24-hour plasma cortisol levels in older individuals [4, 5, 41]. Of note, some studies have found no differences in nadir or mean 24-hour serum cortisol levels between younger and older adults, possibly due to differences in age stratification or fewer intervals between blood sampling [43, 44]. Since cortisol levels are also highly variable within individuals and fluctuate due to changing metabolic demands or stress, it is recommended that these factors be controlled and samples be collected over several days to improve the validity of studies examining diurnal cortisol profiles [45].
There is good clinical evidence that the increase in circulating glucocorticoid levels with age occurs from a blunted and delayed inhibition of ACTH secretion within the HPA feedback loop [12–14]. This age-related impairment in the sensitivity of the HPA axis to circulating glucocorticoids concentrations could be due to factors such as reduced expression of the GR in various regions of the brain or neuronal degeneration. Studies in animals have shown that GR levels are decreased with ageing in the hippocampus, which is a key brain region known to exert a negative feedback response on the HPA axis in conditions of stress [46–49]. Supporting this, Murphy et al. [50] found that chaperone proteins responsible for GR nuclear translocation are also decreased in the hippocampus of aged rats, manifesting in deficient binding of the GR to GRE regions in the DNA. In addition to playing a key role in the stress response, the hippocampus is also particularly vulnerable to the effects of increased stress. In 1986, Sapolsky et al. [51] proposed the “Glucocorticoid Cascade Hypothesis” in which elevated glucocorticoid levels from impaired negative feedback during ageing may cause damage and atrophy to the hippocampus and in turn worsen the inhibitory regulation of glucocorticoid release. Moreover, this damage to the hippocampus would reduce the expression of GRs, resulting in decreased HPA sensitivity to glucocorticoids that could potentiate damage to the hippocampus in a feed-forward cycle [51]. While elegant, the Glucocorticoid Cascade Hypothesis was found to be inconsistent with other observations and was later adapted as the “Glucocorticoid Vulnerability Hypothesis” which recognizes that elevated glucocorticoid levels can make the hippocampus vulnerable to damage but are not the only determinant for neuronal loss in the aged hippocampus [52]. In addition to the hippocampus, decreased GR expression has also been observed in the prefrontal cortex, amygdala, and hypothalamus of aged compared to young animals [46, 48], which could contribute to reduced sensitivity of the HPA axis with ageing, although some studies do not support these findings [53, 54]. Although the SCN (master clock) does not express the GR, its degeneration and decreased sensitivity to light cues with ageing have also been linked to reduced amplitudes in daily circadian rhythms [55, 56] and could contribute to the dampening of glucocorticoid rhythms with age, considering light is the major cortisol zeitgeber (time cue) [6]. In summary, there are multiple factors that may contribute to ageing-related changes in the sensitivity of the HPA axis that drives increases in circulating glucocorticoid levels. These include reduced expression of the GR or neuronal degeneration in various regions of the brain such as the hippocampus. Additionally, increased inflammation during ageing could also be involved.
The term ‘Inflammaging’ first described by Franceschi et al. [57] refers to a state of low-grade chronic inflammation that develops with age. Inflammaging involves a steady increase in circulating cytokines including interleukin-6 (IL-6), IL-1 and tumour necrosis factor α (TNF-α) that indicate changes in innate and adaptive immune function and can predict morbidity, frailty and mortality in elderly people [58, 59]. In response to inflammation and increased cytokine levels, the HPA axis is activated to release glucocorticoids into the circulation [60]. At the tissue level, inflammatory cytokines can also stimulate 11β-HSD1 to increase local glucocorticoid concentrations [61, 62]. This mechanism has been described as ‘anti-inflammaging’, whereby the increase in glucocorticoid levels, attempts to counteract the inflammaging process in an adaptive defence response to inhibit immune activity [63]. However, chronically elevated endogenous glucocorticoid levels may also have negative implications and can lead to the development of prolonged immunosuppression that increases the risk of infection [64–66]. Additionally, glucocorticoid resistance may occur in which glucocorticoids are no longer able to exert their anti-inflammatory effects on target tissues due to alterations in the expression and function of the glucocorticoid receptor [63, 67].
On the other hand, there is some evidence that suggests increased glucocorticoid levels in older adults can induce significant changes in innate and adaptive immune responses and drive inflammaging. For instance, higher levels of perceived stress characterised by increased serum cortisol levels is associated with upregulation of inflammatory biomarkers, oxidative stress and immunosenescence [68]. Notably, elderly caregivers exhibiting greater distress with increased salivary cortisol levels, display blunted lymphocyte proliferation, lower IL-2 levels and reduced lymphocyte sensitivity to glucocorticoids compared to age-matched non-stressed caregivers [69]. In a different study, elderly caregivers were found to have increased IL-6 levels as high as four-fold compared to non-caregivers [70]. Other studies have also shown that distress can increase an individual’s risk of acquiring acute respiratory infections [65, 66] as well as inhibit the immune antibody response to vaccine administration [71]. Together these studies provide evidence that increased glucocorticoid levels from chronic distress can alter cell-mediated immunity and may accelerate premature ageing of the immune response [69]. As such, it is unclear whether the age-related rise in glucocorticoid levels is simply a consequence of inflammaging, or invertedly plays an active role in driving chronic inflammation with age.
While it is evident the interactions between the immune and endocrine systems are bimodal, several questions remain. 1) What is the timing of onset of inflammaging, neurodegeneration and increased glucocorticoid levels with age? 2) How does inflammaging affect diurnal rhythms in glucocorticoid secretion? 3) How do disturbances in glucocorticoid diurnal rhythmicity influence immune activity with ageing? Further studies that measure 24-hour diurnal rhythms in cortisol and inflammatory biomarkers over time will help unravel the complex interactions between immune and endocrine systems during ageing and may provide some important insights into the development of ageing-related pathologies.
Glucocorticoid levels in peripheral tissues such as the skeleton have been shown to increase with ageing, via the action of the glucocorticoid activating enzyme, 11β‐hydroxysteroid dehydrogenase type 1 (11β-HSD1) (Figure 2). Cooper et al. [72] showed in primary human osteoblast cultures that 11β-HSD1 reductase activity was positively correlated with the age of the bone tissue donor. Studies in aged mice have confirmed that 11β-HSD1 gene expression is increased with ageing at skeletal sites of the vertebrae and tibia compared to younger animals [3, 73]. Muscle, skin, adipose and hippocampal tissue also show elevated 11β-HSD1 expression with ageing [74–77]. Increases in cellular 11β-HSD1 levels and activity with age could be driven by higher circulating glucocorticoid concentrations since dexamethasone and cortisol treatment have been shown to enhance 11β-HSD1 actions in primary human osteoblast cultures [72]. In this way, 11β-HSD1 acts to increase glucocorticoid activation in the presence of elevated glucocorticoid levels [72]. Alternatively, local ageing-related changes in cells such as increased expression of inflammatory pathways and cytokines can also increase 11β-HSD1 levels in tissues independently of disturbances in circulating glucocorticoids [62, 78].
It is well established that physiological levels of endogenous glucocorticoids are essential for normal development and maintaining skeletal bone mass and mineral homeostasis [79–83]. However, mild chronic glucocorticoid excess, as it may occur in chronic stress or as part of the ageing process in older adults, can have detrimental effects on a range of systems including impairments in cognitive function, loss of muscle and/or bone mass, visceral obesity, and hypertension, among others [84–86]. Chronically elevated glucocorticoid levels can also affect the normal stress response in the elderly, making them more vulnerable to stressful stimuli [87]. Current findings from studies utilizing animal models that disrupt glucocorticoid signaling in mice have provided valuable knowledge into how elevated glucocorticoid concentrations with age can drive the development of ageing-related skeletal diseases including osteoporosis, sarcopenia and osteoarthritis. These studies and their findings are summarized in Table 1 and Figure 3.
Phenotypes of 11β-HSD1/2 and glucocorticoid receptor genetically modified mouse models in ageing related skeletal diseases
| Genetic model | Cre promoter | Cells targeted | Age/Sex studied | Phenotype | Reference |
|---|---|---|---|---|---|
| Osteoporosis | |||||
| 11β-HSD2 transgenic (C57BL/6) | Osteocalcin gene 2 (OG2) | Mature osteoblasts and osteocytes | 21 months males and females | Vertebrae: higher BMD, cancellous BV, BFR and greater compression strength.Femur: higher BMD and bending strength.Osteoblast and osteocyte apoptosis reduced.Protected from age-related deteriorations in bone vasculature. | [73] |
| 11β-HSD2 transgenic (CD-1) | 2.3kb Pro-a1(I) collagen (Col1a1) | Osteoblasts and osteocytes | 12 and 18 months males and females | Vertebrae: females displayed higher trabecular thickness, but no change in BV/TV in either gender.18-month-old mice protected from fat accumulation, and insulin and leptin resistance. | [91] |
| GR KO (C57BL/6) | Osterix (Osx) | Osteoblast progenitor cells (plus hypertrophic chondrocytes) | 21 months females | Femur: reduced femoral cortical bone area and trabecular bone mass.Serum markers of bone formation (PINP) and resorption (TRAcP5b) reduced. | [98] |
| Sarcopenia | |||||
| 11β-HSD1 KO (C57BL/6) | Myogenic differentiation 1 (MyoD) | Myogenic progenitor cells | 22 months males | Increased muscle mass and strength. | [108] |
| 11β-HSD1 over-expressing(C57BL/6) | Global | All cells | 2 months males | Glucocorticoid overexpression induced muscle atrophy. | [108] |
| GR KO (C57BL/6) | Skeletal muscle actin (ACTA1) | Striated myofibers | 20 weeks old males and females | Increased muscle mass.Diurnal variation and fasting-dependent temporal elevation of plasma alanine levels reduced.Lipolysis increased in adipose tissue. | [109] |
| GR KO(FVB/Balb/c) | Muscle creatine kinase (MCK) | Striated and cardiac myofibers | 3–4 months females | Increased muscle mass.Protected from glucocorticoid-induced muscle atrophy.Attenuated expression of atrophy related genes (MuRF1 and MAFbx, 4E-BP1, and MT2) induced by nutritional deprivation.No effect on muscle atrophy induced by denervation. | [110] |
| GR KO (C57BL/6) | Myosin light chain 1F (Mlc1f) | Striated myofibers | 12 weeks old males | Attenuated muscle atrophy. Reduced expression of proteolysis-associated genes induced by reduced food intake. | [112] |
| Osteoarthritis | |||||
| 11β-HSD2 transgenic (CD-1) | 2.3kb Pro-a1(I) collagen (Col1a1) | Osteoblasts and osteocytes | 26 and 38 weeks old males | In young mice, no change in cartilage loss, subchondral bone sclerosis and osteophyte formation.In aged mice, cartilage loss, subchondral bone sclerosis, and osteophyte size were reduced. No changes in synovial inflammation. | [3] |
| GR KO (C57BL/6) | Pro-a1(II) collagen (Col2a1)-tamoxifen inducible Cre (Cre-ERT2) | Chondrocytes | 24, 26, 30, and 38 weeks old males | Less severe cartilage loss (all timepoints) and reduced synovial inflammation (at 24 weeks).Decreased chondrocyte and synoviocyte hypoxia inducible factor (HIF)-2α expression.Reduced chondrocyte senescence and catabolic signaling.No change in subchondral bone sclerosis and osteophytes. | [128] |
GR: glucocorticoid receptor; 11β-HSD1/2: 11β-hydroxysteroid dehydrogenase types 1 and 2; KO: knockout; Cre: cre recombinase; BMD: bone mineral density; BV: bone volume; BFR: bone formation rate; BV/TV: bone volume/tissue volume; Cre-ERT2: cre fused with a mutated ligand-binding domain of estrogen receptor that is tamoxifen inducible; PINP: procollagen type I N-propeptide; TRAcP5b: tartrat-resistant acid phosphatase 5b; MuRF1: muscle RING-finger protein-1; MAFbx: muscle atrophy F-box; 4E-BP1: eukaryotic translation initiation factor 4E-binding protein 1; MT2: metallothionein 2; HIF-2α: hypoxia inducible factor 2α; kb: kiloba
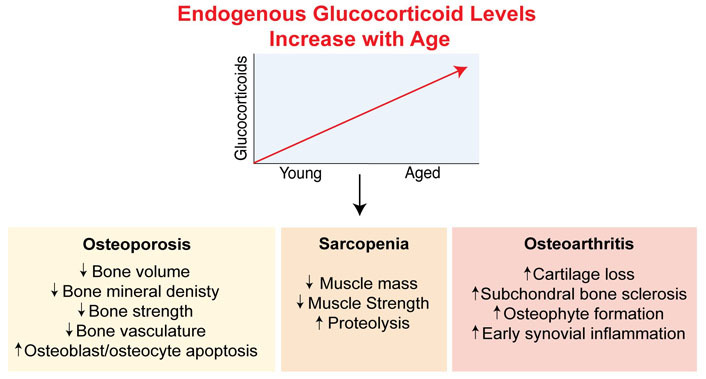
Elevated glucocorticoid concentrations with age have detrimental effects on skeletal diseases. Both circulating and local glucocorticoid levels are increased with ageing. Increased systemic glucocorticoid levels are driven by disturbances in the feedback of the hypothalamic-pituitary-axis due to reduced expression of the glucocorticoid receptor and neuronal degeneration in the brain, as well as inflammaging. Increases in intracellular glucocorticoid levels are elevated with ageing via the glucocorticoid activating enzyme, 11β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) which converts inactive cortisone into active cortisol in tissues. These changes in glucocorticoids have been found to play a role in driving the progression of skeletal diseases including osteoporosis, sarcopenia and osteoarthritis
Osteoporosis is the most common bone disease globally, affecting approximately 1 in 3 postmenopausal women and 1 in 5 men over the age of 60 years [88]. With rapid population ageing, the incidence of osteoporosis is expected to markedly increase and place a significant burden on public healthcare systems. Osteoporosis is characterized by a reduction in bone mass and a deterioration in bone microarchitecture that compromises bone strength and increases the risk of fractures, particularly as people age [89]. Similar to humans, mice also exhibit trabecular and cortical bone loss and reduced bone density with age, making them suitable models to investigate the pathophysiology of osteoporosis. In the majority of murine strains, bone mass peaks at 4–6 months of age and then decreases thereafter [90–92].
Ageing, chronic stress and long-term treatment with glucocorticoids have similar detrimental effects on bone quality and volume and are therefore considered causes of osteoporosis. Increased endogenous salivary cortisol levels at circadian nadir are associated with reduced lumbar bone mineral density (BMD) in healthy 70-year-old subjects [85]. This observation is supported by studies demonstrating serum cortisol levels are positivity associated with the rate of bone loss at the lumbar spine and hip in healthy older adults, suggesting elevated cortisol secretion in older adults may contribute to the development of osteoporosis [93, 94].
Studies in mice support these clinical findings and suggest that elevated endogenous glucocorticoid levels drive bone loss during ageing. To date, two studies have utilized the 11β-HSD2 transgenic mouse model to investigate the effects of disrupted glucocorticoid signaling specifically on bone quality in aged mice. In this mouse model, the glucocorticoid inactivating enzyme 11β-HSD2 is overexpressed in specific types of bone cells to reduce the intracellular bioavailability of glucocorticoids prior to binding to the GR. In a study by Weinstein et al. [73], 11β-HSD2 was overexpressed in mature osteoblasts and osteocytes using the osteocalcin gene 2 (OG2) promoter, to determine the role of glucocorticoids specifically in bone forming cells during skeletal ageing in male and female C57BL/6 mice. In wild-type mice, ageing was associated with an increase in the adrenal production of glucocorticoids and the expression of 11β-HSD1 in bone. Between 16 to 21 months of age, wild-type mice displayed a significant reduction in BMD. Importantly, OG2-11β-HSD2 mice were protected from the adverse effects of ageing on bone compared to wild-type animals. By the age of 21 months, OG2-11β-HSD2 mice had higher vertebral BMD, cancellous bone volume, bone formation rate and greater compression strength compared to wild-type age-matched controls. Similarly at the femur, BMD and bending strength were higher in OG2-11β-HSD2 mice compared to wild-type mice. The prevalence of osteoblast and osteocyte apoptosis was reduced by 47–57% in OG2-11β-HSD2 mice compared to wild-type 21-month-old controls, indicating that protecting osteoblasts and osteocytes from excessive endogenous glucocorticoids ameliorates the adverse effects of ageing on bone. These 21-month-old OG2-11β-HSD2 mice were also found to be protected from age-related deteriorations in bone vasculature compared to wild-type animals. Remarkably, bone vasculature in the 21-month-old OG2-11β-HSD2 transgenic mice was virtually restored to that of the 8-month-old animals [73].
The effects of endogenous glucocorticoids on ageing have also been studied by Henneicke et al. [91] who utilized mice in which 11β-HSD2 was overexpressed in osteoblasts and osteocytes using the 2.3kb Pro-a1(I) collagen (Col1a1) promoter in a CD-1 background strain (Col2.3-11β-HSD2). In male and female mice, peak bone mass occurred at 3 months of age and then steadily declined over time. Notably, at 12 and 18 months of age, female Col2.3-11β-HSD2 mice displayed higher vertebral trabecular thickness compared to wild-type age-matched controls. However, no differences were observed in vertebral BV/TV between transgenic and wild-type mice of either sex, suggesting glucocorticoid signaling has a mild effect vertebral trabecular architecture in aged female mice. Such discrepancies in skeletal phenotypes between the studies of Weinstein et al. [73] and Henneicke et al. [91] could be related to differences in the developmental phenotypes of these two Cre reporter strains. Weinstein and colleagues [73] previously reported that 5-month-old OG2-11β-HSD2 mice exhibit similar skeletal BMD and strength compared to age matched wild-type animals. This data indicates that glucocorticoid signaling in mature osteoblasts and osteocytes does not affect normal development or turnover in young mice [95]. Conversely, Col2.3-11β-HSD2 mice exhibit lower bone mass, delayed bone formation and reduced bone strength and stiffness compared to wild-type littermates, indicating that glucocorticoid signaling in early derived osteoblast cells is critical for bone development [91, 96, 97]. Thus, differences in the accrual of bone mass during skeletal maturation between these two mouse models may affect ageing-related bone loss in older mice. Additionally, other factors may also explain the varied results in these two studies including strain variation, or the age at which mice were studied (18 versus 21 months old).
On the other hand, a recent study by Pierce et al. [98] has reported that the GR is important for maintaining bone health in 21-month-old female C57BL/6 mice. In this study, the GR was deleted in mice using an Osterix (Osx) Cre that specifically targets osteoblast progenitor cells, as well as hypertrophic chondrocytes. Osx-GR knockout mice display reductions in femoral cortical bone area, and trabecular bone mass at 21 months of age compared to wild-type control animals. Serum markers of bone formation (PINP) and resorption (TRAcP5b) were reduced in Osx-GR knockout compared to wild-type mice. These data suggest that loss of the GR in osteoblast progenitor cells has detrimental effects on bone mass. This finding has also been observed in younger mice whereby the GR was deleted specifically in osteo-progenitor cells using either a Runx2 or Osx Cre promoter [82, 83]. Similarly, as mentioned Col2.3-11β-HSD2 mice display a lower bone mass phenotype compared to wild-type littermates at 3 and 7 weeks of age [91, 96, 97]. Collectively, the findings of Weinstein et al. [73], Henneicke et al. [91] and Pierce et al. [98] indicate that the effects of glucocorticoids on bone are bimodal and dependent on their concentrations. When GR signaling is completely removed, the normal beneficial effect of glucocorticoids on bone formation is absent, resulting in lower bone mass. Conversely, when there is excessive glucocorticoid signaling such as in circumstances of ageing, this can also lead to bone loss, indicating too little or too much of glucocorticoid signaling has detrimental effects on the skeleton.
Intriguingly, aside from the skeletal phenotypes, mice with disrupted glucocorticoid signaling in bone cells also exhibit changes in whole body metabolism. In Col2.3-11β-HSD2 mice, Henneicke et al. [91] found that disruption of glucocorticoid levels in bone forming cells protected 18-month-old mice from fat accumulation, as well as insulin and leptin resistance, compared to aged-matched wild-type controls. This data indicates that skeletal endogenous glucocorticoid signaling is also important in the development of obesity and metabolic disease during normal ageing. This concept is supported by in vitro studies showing the increased expression of 11β-HSD1 and thus higher intracellular cortisol levels in human mesenchymal cells can redirect differentiation towards the adipocyte rather than osteoblast lineage [99]. This effect could explain decreases in bone formation and quality, favoring the development of osteoporosis with age. On the other hand, while Pierce et al. [98] found that body mass was reduced in 21-month-old Osx-GR knockout mice, whole body percent fat mass was increased compared to wild-type animals. Further, metabolic rate was higher in Osx-GR knockout mice, with a trend toward lower fasting blood glucose levels at 6 months of age compared to wild-type mice. Given the high prevalence of metabolic disease in the ageing population, further studies are warranted in investigating how increased skeletal endogenous glucocorticoid signaling affects whole body metabolism during ageing.
Sarcopenia is a progressive skeletal muscle disorder involving the accelerated loss of muscle mass, strength and function [100]. As with bone mass, muscle mass declines substantially after 50 years of age [101]. Sarcopenia is primarily driven by ageing-related changes in muscle metabolism including a reduction in the number of myofibers and satellite cells, the infiltration of fat and fibrous tissue, and reduced muscle innervation that results in decreased protein turnover [101]. These changes can also be driven by multiple factors such as lack of physical activity, malnutrition or inadequate protein intake, chronic diseases, systemic inflammation, glucocorticoid exposure and hormonal changes [101]. The health implications of sarcopenia are often severe in older adults resulting in increased falls, disability, frailty and mortality [100]. As a result of population ageing, sarcopenia is expected to place a significant economic burden on health care systems. Given there is no approved therapeutic drug to treat Sarcopenia, research focusing on understanding how sarcopenia progresses with age will assist advances in diagnosis and therapies [100].
Several clinical studies have found cortisol levels are associated with sarcopenia. In a cohort from the Longitudinal Ageing Study in Amsterdam, older persons with high salivary cortisol levels had an increased risk of reduced grip strength [102]. In a recent Mendelian Randomization study, increased morning plasma cortisol concentrations were associated with reduced lean mass and grip strength in women but not men [103]. Similar findings have been reported by other groups suggesting that subtle increases in cortisol have adverse effects on skeletal muscle metabolism in older women [104, 105]. At the tissue level, 11β-HSD1 expression has been found to be significantly increased in skeletal muscle in women over 60 years of age compared to those aged between 20 to 40 years [74]. Furthermore, 11β-HSD1 expression in skeletal muscle is associated with reduced grip strength and increased percentage of body fat in both men and women [74]. In support of this, Schluessel et al. [106] showed that 11β-HSD1 expression was negatively correlated with muscle mass and type-2 fiber diameter in patients above the age of 60 years. Interestingly, 11β-HSD1 but not GR mRNA levels were found to be significantly associated with muscle strength, suggesting that increased cortisol levels by 11β-HSD1 within muscle is a key component of sarcopenia [107].
To determine the role of endogenous glucocorticoids in sarcopenia, several studies have utilized muscle specific 11β-HSD1 and GR genetically modified mouse models. Recently, Lv et al. [108] found that blocking 11β-HSD1 expression in the muscle using a myogenic differentiation 1 (MyoD)-Cre, resulted in increased muscle mass and strength compared to wild-type mice at 22 months of age. Additionally, the authors demonstrated that overexpression of 11β-HSD1 lead to muscle atrophy in young male mice at 2 months of age, confirming that increased glucocorticoid concentrations in muscle can induce sarcopenia. Importantly, it was found that caloric restriction in aged 22-week-old male mice, reduced 11β-HSD1 expression in muscle, and improved grip strength and muscle weight compared to age-matched controls without caloric restriction [108]. This suggests that caloric restriction (with adequate nutrition) could delay sarcopenia through glucocorticoid signaling in elderly individuals.
The role of the glucocorticoid receptor in sarcopenia has also been examined in other studies. Deletion of the GR in muscle using either a skeletal muscle actin alpha 1 (ACTA1) or muscle creatine kinase (MCK) Cre promoter, results in increased muscle mass in mice [109, 110]. This phenotype is in keeping with MyoD-Cre 11β-HSD1 knockout mice [108]. Interestingly, Shimizu et al. [109] found that the diurnal variation and fasting-dependent temporal elevation of plasma alanine levels were almost diminished and lipolysis increased in adipose of MCK-GRKO mice, suggesting that the GR in muscle may also play a physiological role in systemic energy delivery to organs such as the liver and adipose tissues. A recent study by the same group reported that muscle GR signaling plays a crucial role in accelerating glucocorticoid-induced obesity through the induction of hyperinsulinemia [111]. Studies investigating the effects of sarcopenia during caloric restriction have also found that GR signaling is required for muscle atrophy and increases expression of proteolysis-associated genes induced by reduced food intake [110, 112]. Together these studies in 11β-HSD1 and GR knockout mice indicate that increased endogenous glucocorticoid signaling with age plays an essential role in sarcopenia.
It is also worth noting that muscle atrophy is likely to reduce skeletal loading and contribute to bone loss with ageing [113]. Conversely, endocrine and paracrine factors secreted from bone cells (termed osteokines) have been shown to also exert effects on skeletal muscle, highlighting the importance in understanding tissue-crosstalk mechanisms in bone loss and muscle atrophy during ageing [114]. While the bone and muscle specific glucocorticoid knockout models discussed in this review indicate that elevated glucocorticoid levels have direct detrimental effects on both tissues, there is the possibility that glucocorticoid signaling may also have indirect actions through inter-tissue communication networks. Further studies are warranted in understanding how the relationship between muscle and bone is impacted by ageing and elevated glucocorticoid signaling [113]. Such studies may uncover bimodal therapeutic approaches that target both muscle and bone in improving overall musculoskeletal health in the elderly.
Osteoarthritis (OA) is a chronic degenerative joint disease involving the irreversible loss of cartilage, formation of abnormal bone and secondary synovial inflammation. Osteoarthritis is the most common form of joint disorders, affecting over 600 million people worldwide in 2020. The risk of developing OA increases with age, with approximately 40% of the population aged 70 years or older being affected [115, 116]. Karlson et al. [117] found women aged 70 years or older, were nine times more likely to have a hip replacement than those aged less than 55 years. Studies in mice show that ageing also affects the severity of OA induced by surgical destabilization of the medial meniscus (DMM) [3, 118]. The DMM model, developed by Glasson et al. [119] mimics a meniscal tear injury and is a commonly used technique to induce posttraumatic OA in mice. It has been shown that cartilage loss and abnormal bone formation are more severe when DMM is performed in 6 to 12-month-old mice, compared to younger animals [3, 118]. Intriguingly, microarray data from Loeser et al. [118] revealed that 493 genes displayed differential expression between young and old DMM mice, indicating that ageing affects the way in which cells respond to OA-inducing injuries. In fact, there are several ageing-related changes in the joint that contribute to the development of ‘spontaneous’ OA including cellular senescence and the formation of advance glycation end products that alter the cartilage and bone matrix, making it more susceptible to damage and injury. These findings demonstrate the importance of investigating OA in older mice as critical molecular pathways could be missed if only younger mice are studied in OA pathogenesis. Studies utilizing older mice could improve translatability to humans.
The severity of OA is also closely associated with several other risk factors including obesity, female sex, genetic predisposition, injury, and certain occupations that involve repetitive use of joints. As such, the multifactorial nature of OA and complexities surrounding what drives the progression of the disease have hindered the development of effective disease modifying treatments. As such, current treatment relies on the use of non-steroidal anti-inflammatory drugs and intra-articular glucocorticoid injection for primary pain relief, with end stage patients requiring joint replacement surgery [120, 121].
The use of therapeutic intra-articular glucocorticoid injections in the treatment of OA has been studied extensively but is not without controversy [122]. Recent studies have reported that corticosteroid injections, especially given continuously or over the long-term can increase the risk of disease progression [123–126]. Furthermore, 20–30% of patients do not respond to glucocorticoid treatment, possibly due to differences in the degree of inflammation and pain, stage of the disease or demographic characteristics such as BMI, sex, and age [127]. Given the widespread use of intra-articular glucocorticoid injections and the projected disease burden of OA in the ageing population, there is a need to establish best practice for intra-articular glucocorticoid injections including identifying which types of patients are most and least likely to benefit, to avoid adverse outcomes.
Compared to therapeutic glucocorticoids, the effects of endogenous glucocorticoids on the progression of OA have received little attention. To date there are only two studies investigating this question, both utilizing genetically modified mice to disrupt glucocorticoid signaling specifically in either bone forming cells or chondrocytes. Using the Col2.3-11β-HSD2 transgenic mouse line, Tu et al. [3] were the first to establish that endogenous glucocorticoid signaling in osteoblasts and osteocytes plays a detrimental role in the development of OA. Importantly, the authors found that this effect was related to ageing by examining posttraumatic OA pathology of both young (26-week-old) and aged (38-week-old) transgenic and wild-type male mice. In young mice, cartilage loss, subchondral bone sclerosis and osteophyte formation were comparable between Col2.3-11β-HSD2 and wild-type mice following DMM surgery. However, in aged mice, disruption of glucocorticoid signaling in osteoblasts and osteocytes, reduced cartilage loss, subchondral bone sclerosis and osteophyte size in mice with DMM-OA. Together these results indicate that the increase in glucocorticoid signaling with age in the joint contributes to cartilage loss and abnormal bone formation in OA. A limitation of this study was that only end-stage disease was assessed. Since inflammation is known to be more pronounced in the early stages of the disease after injury, this may explain why differences in synovial inflammation between groups were not observed.
Since chondrocytes are critical for maintaining cartilage health and are therefore involved in OA pathology, a more recent study by our group examined whether glucocorticoid signaling in chondrocytes also contributes to the progression of OA in mice [128]. In this study, the GR was specifically deleted in chondrocytes using a tamoxifen inducible Col2a1 Cre promoter (GRCol2a1CreERT2). Similar to the study of Tu et al. [3] knee OA was induced by DMM surgery in 22-week-old GRCol2a1CreERT2 and GRflox/flox male littermates. We found that deletion of the chondrocyte GR significantly attenuated cartilage loss compared to wild-type littermates. Furthermore, early after DMM injury wild-type mice displayed extensive synovial activation characterized by synovial thickening and increased IL-1β expression, which was less pronounced in GRCol2a1CreERT2 mice. These effects on the synovium were associated with increased chondrocyte and synoviocyte hypoxia inducible factor (HIF)-2α expression in a glucocorticoid dependent manner. Chondrocyte senescence and elevated catabolic signaling [reduced yes-associated protein 1 (YAP1) and increased matrix metalloprotease (MMP)-13 expression] were also increased in osteoarthritic cartilage of wild-type mice. In contrast, chondrocyte YAP1 and MMP-13 expression, as well as chondrocyte senescence were similar in GRCol2a1CreERT2 DMM mice and control animals without OA. This data indicates that glucocorticoid signaling in chondrocytes promotes synovial activation, chondrocyte senescence and cartilage degradation by upregulation of catabolic signaling through HIF-2α in murine posttraumatic OA [128]. Together with the findings of Tu et al. [3], these results indicate that inhibition of glucocorticoid signaling in cartilage and bone may present a promising way to slow OA.
Endogenous glucocorticoid physiology becomes markedly disturbed during ageing, characterized by dampened diurnal rhythmicity and elevated basal concentrations of circulating and tissue-level glucocorticoids. Studies in genetically modified mice indicate that these alterations in glucocorticoid signaling have detrimental effects on bone quality, skeletal muscle metabolism and drive the progression of osteoarthritis. These studies provide important insights that will aid in the development of novel therapeutics to target ageing-related skeletal diseases, especially for osteoarthritis and sarcopenia which currently have no disease-modifying treatment available. Future investigations into understanding how bone-muscle-joint crosstalk mechanisms are impacted by ageing and elevated glucocorticoid signaling may uncover therapeutic avenues to treat ageing-related musculoskeletal diseases in a holistic manner.
4E-BP1: eukaryotic translation initiation factor 4E-binding protein 1
11β-HSD1/2: 11β-hydroxysteroid dehydrogenase isoenzymes 1 and 2
ACTA1: actin alpha 1, skeletal muscle
ACTH: adrenocorticotropic hormone
AVP: arginine vasopressin
BMD: bone mineral density
BMI: body mass index
BV/TV: bone volume/tissue volume
Col1a1: collagen type I alpha chain 1
Col2a1: collagen type II alpha chain 1
Cre-ERT2: cre fused with a mutated ligand-binding domain of estrogen receptor that is tamoxifen inducible
CRH: corticotropin-releasing hormone
DMM: destabilization of the medial meniscus
GR: glucocorticoid receptor
GREs: glucocorticoid response elements
HIF: hypoxia inducible factor
HPA: hypothalamic-pituitary-adrenal
IL: interleukin
KO: knockout
MAFbx: muscle atrophy F-box
MCK: muscle creatine kinase
Mlc1f: myosin light chain 1F
MMP: matrix metalloprotease
MR: mineralocorticoid receptor
MT2: metallothionein 2
MuRF1: muscle RING-finger protein-1
MyoD: myogenic differentiation 1
OA: osteoarthritis
OG2: osteocalcin gene 2
Osx: osterix
PINP: procollagen type I N-propeptide
SCN: suprachiasmatic nucleus
TRAcP5b: tartrate‐resistant acid phosphatase 5b
TNF: tumour necrosis factor
YAP1: yes-associated protein 1
EM: Conceptualization, Writing—original draft, Writing—review & editing. HZ: Writing—review & editing. MJS: Writing—review & editing. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
The authors would like to acknowledge the ongoing research support by the National Health and Medical Research Council (NHMRC) of Australia through NHMRC Ideas [APP2028787] and NHMRC Investigator [APP1196062] grants. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Amar Mann ... Sudarshan Ramachandran
Pravinath Ramachandran ... Geoffrey Hackett
Carter Coggins ... Vikrant Rai
