 Case Report
Case Report

Affiliation:
1Genome Analysis Laboratory, Institute of Genome Research, Vietnam Academy of Science and Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0005-5109-6115
Affiliation:
1Genome Analysis Laboratory, Institute of Genome Research, Vietnam Academy of Science and Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0009-0002-4648-4226
Affiliation:
1Genome Analysis Laboratory, Institute of Genome Research, Vietnam Academy of Science and Technology, Hanoi 10000, Vietnam
ORCID: https://orcid.org/0000-0002-0889-4534
Affiliation:
1Genome Analysis Laboratory, Institute of Genome Research, Vietnam Academy of Science and Technology, Hanoi 10000, Vietnam
2Graduate University of Science and Technology, Vietnam Academy of Science and Technology, Hanoi 10000, Vietnam
Email: nguyenhaiha@igr.ac.vn
ORCID: https://orcid.org/0000-0002-5431-5935
Explor Endocr Metab Dis. 2025;2:101423 DOI: https://doi.org/10.37349/eemd.2025.101423
Received: October 11, 2024 Accepted: January 20, 2025 Published: February 13, 2025
Academic Editor: Marco Falasca, Curtin University, Australia
Metabolic disorders are due to a deficiency of enzymes, which can severely impact health or cause serious complications without treatment. This study aimed to identify the molecular causes of an infant death who had been hospitalized with complicated health problems and metabolism syndrome. Whole-exome sequencing (WES) was used to screen pathogenic variants in the patient’s genome, followed by examination of variants segregation in her parents. The WES analysis identified two homozygous variants, c.[614C>G; 649A>G] in the HMGCL gene of the patient. These two variants co-locate within the exon 7 of the HMGCL gene, resulting in 2 amino acid substitutions, p.[T205S; M217V], in the conservative region of enzyme protein. Sanger sequencing showed that the patient’s unaffected mother and father carried one mutant allele of the HMGCL gene containing two c.[614C>G; 649A>G] variants. The HMGCL gene encodes the 3-hydroxy-3-methylglutaryl-CoA lyase enzyme, which is critical in the ketogenic pathway. The deficiency of this enzyme was reported to be a life-threatening illness in the neonatal period, and two variants detected in this study were also found in a Japanese patient with sudden, unexpected death in infancy. The frequency of these two variants in the Vietnamese in-hour database and their further functional analysis were also reported in this study. The results of this study have explored the molecular etiology that causes the severe, deadly condition of the patient and provide an understanding of the risk of disease in her family.
Metabolic disorders are medical conditions that disrupt the processes of catabolism and anabolism in the normal body’s metabolism pathway. This disruption leads to a deficiency of metabolic enzymes, accumulation of substrate toxins, and scarcity of energy-producing substances. Untreated metabolic disorders can result in severe and irreversible multiple organ damage and the development of comorbidities such as atherosclerosis, type 2 diabetes mellitus, etc. Genetic mutations, particularly those inherited from parents, can cause metabolic disorders that manifest early in life leading to inborn errors of metabolism with an incidence rate from 1 in 10,000 to 1 in 200,000 [1]. This poses specific challenges in neonates due to non-specific symptoms and rapid progression. Therefore, the timely diagnosis facilitated by newborn screening programs is paramount to preempt severe complications.
Mitochondrial enzyme 3-hydroxy-3-methylglutaryl-CoA lyase [HMG-CoA lyase; HMGCL; Online Mendelian Inheritance in Man (OMIM): #613898] is encoded by the HMGCL gene located in the short arm of chromosome 1p36.11. HMGCL takes in an essential step in ketogenesis by catalyzing an HMGC into acetyl-CoA and acetoacetate, and alternately in leucine catabolism, which is the final step for releasing stored energy [2]. HMGCL deficiency (HMGCLD; OMIM: #246450) is a rare autosomal recessive genetic disorder that occurs when the enzyme HMGCL is dysfunctional or absent due to variations in the HMGCL gene. The accumulation of leucine and HMGC, toxic substances byproducts, and malfunctioning metabolic processes create lethargy, hypotonia, hypoglycemia, and metabolic acidosis. The clinical symptoms manifest in early newborns from mild to severe with episodes of vomiting, diarrhea, dehydration, coma, and death.
HMGCLD clinical onsets within the first few months of life and can be diagnosed via newborn screening by measuring 3-hydroxy isovalerylcarnitine (C5-OH) [3]. Diagnosis can also be based on the elevated level of (i) 3-methylglutaric, 3-hydroxy-3-methylglutaric, and 3-hydroxyisovaleric acids in the urinary pattern; (ii) C5-OH in the blood acylcarnitine profile [4]. Directly measuring the enzymatic activity in the cells of patients can be a diagnosis method.
This study identified the causative variants of the metabolic syndrome resulting in the death of a 12-month-old baby and discussed managing instances involving complex metabolic syndrome in Vietnamese children.
A 12-month-old female baby was hospitalized at the Vietnam National Children’s Hospital in 2021 due to exhibiting extreme symptoms and underwent clinical examination. She was the firstborn child of a healthy couple who was reported as non-consanguineous, born and raised in the same village.
The patient was described as being hospitalized with symptoms including extreme lethargy, cyanosis, diminished muscle tone (hypotonia), arrhythmias, shortness of breath, and experiencing multiple instances of coma. With irregular heart rhythms in the heart, cyanosis, and shortness of breath, a congenital heart block diagnosis was first given. Her clinical urine biochemistry showed elevated lactic aciduria, 3-OH-butyric, and 3-methylglutaconic, lack of nutrition, and extreme hypoglycemia were presented. Transaminase activities were increased indicating liver dysfunction (aspartate transaminase: 142.4 U/L) and kidney dysfunction with high urea and creatinine (urea: 7.20 mmol/L; creatinine: 71.1 mmol/L). Infection was ruled out based on low C-reactive protein at 0.85 mg/L in Table 1. High levels of amino acids and fatty acids accumulated in the blood with leucine (371.43 µmol/L), glutamic acid (667.74 µmol/L), and valine (430.78 µmol/L). Biomarkers for metabolic disorders in infant were elevated with acetylcarnitine (C2, 84.11 µmol/L), 3-hydroxy butyrylcarnitine (C4-OH, 0.78 µmol/L), glutarylcarnitine (C5-DC, 0.87 µmol/L), hexanoylcarnitine (C6, 0.27 µmol/L), adipylcarnitine (C6-DC, 0.56 µmol/L), octanoylcarnitine (C8, 0.28 µmol/L), decanoylcarnitine (C10, 0.33 µmol/L), decenoylcarnitine (C10:1, 0.20 µmol/L) in Table 1. The abnormal plasma amino acids profile and blood acylcarnitine profile suggested an amino acid/ fatty acid metabolism disorder in the patient. Unfortunately, before receiving a proper diagnosis, the patient’s condition elevated to several episodes of coma and subsequently death.
Patient’s serum biochemistry index
| Metabolic profiling | Index | Normal range |
|---|---|---|
| Blood biochemistry | ||
| Urea | 7.20 mmol/L | 1.3–5.9 mmol/L |
| Creatinine | 71.1 mmol/L | 10–33 mmol/L |
| Aspartate transaminase (AST) | 142.4 U/L | 19–61 U/L |
| C-reactive protein (CRP) | 0.85 mg/L | < 6.0 mg/L |
| Mass spectrometry-based newborn screening (µmol/L) | ||
| Glutamic acid (Glu) | 667.74 | 137–542 |
| Leucine (Leu) | 371.43 | 46–241 |
| Lysine (Lys) | 568.72 | 116–405 |
| Phenylalanine (Phe) | 215.52 | 19–75 |
| Proline (Pro) | 271.92 | 39–221 |
| Tyrosine (Tyr) | 236.82 | 14–109 |
| Valine (Val) | 430.78 | 36–208 |
| Free carnitine (C0) | 32.73 | 8.5–59 |
| Acetylcarnitine (C2) | 84.11 | 4.5–52 |
| 3-hydroxy butyrylcarnitine (C4-OH) | 0.78 | 0.0–0.33 |
| Glutarylcarnitine (C5-DC) | 0.87 | 0.0–0.25 |
| Hexanoylcarnitine (C6) | 0.27 | 0.0–0.18 |
| Adipylcarnitine (C6-DC) | 0.56 | 0.0–0.25 |
| Octanoylcarnitine (C8) | 0.28 | 0.0–0.21 |
| Decanoylcarnitine (C10) | 0.33 | 0.01–0.26 |
| Decenoylcarnitine (C10:1) | 0.20 | 0.0–0.18 |
After the patient’s death, her DNA sample was transferred to the Institute of Genome Research for genetic analysis, as authorized by a consent document signed by her parents. Whole-exome sequencing (WES) was performed on the patient’s DNA, followed by screening of metabolic-related genes. The screening process identified two homozygous variants, c.[614C>G; 649A>G] resultes in p.[T205S; M217V] at exon 7 of the HMGCL gene (NM_000191.3), which encodes the HMGCL enzyme. Mutations in the HMGCL gene are known to cause HMGCLD, an autosomal recessive metabolic disorder [5]. These variants were first reported in a retrospective genetic screening of neonatal sudden unexpected death in Japan [6].
Table 2 summarizes predictions from in silico tools, including sorting intolerant from tolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2), and Protein Variation Effect Analyzer (PROVEAN), indicating that the c.[614C>G; 649A>G] variants are damaging, deleterious, disease-causing, or probably pathogenic [7–9]. Chromatograms showing homozygous peaks for c.[614C>G; 649A>G] in the proband sample confirmed the WES screening results. Each parent was identified as a carrier of one mutant HMGCL allele, with compound heterozygous variants c.[614C>G; 649A>G] located on the same DNA strand (Figure 1A). Figure 1B illustrates the segregation of these variants in the family tree, demonstrating genotypes and phenotypes consistent with an autosomal recessive inheritance pattern.
Genetic variant screening by WES
| Gene | Variant change | Zygosity | Region | In silico prediction | |||
|---|---|---|---|---|---|---|---|
| cDNA | Amino acid | SIFT | PolyPhen-2 | PROVEAN | |||
| HMGCL | c.649A>G | p.M217V | HOM | Exon 7 | D | D | D |
| c.614C>G | p.T205S | HOM | Exon 7 | D | D | D | |
D: damaging/deleterious/disease-causing; HOM: homozygous; PolyPhen-2: Polymorphism Phenotyping v2; PROVEAN: Protein Variation Effect Analyzer; SIFT: sorting intolerant from tolerant; WES: whole-exome sequencing
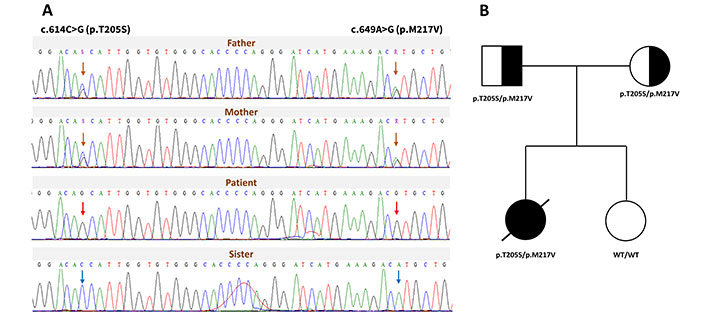
Molecular genomic analysis of the family pedigree. (A) Sanger sequencing results show the segregation of two HMGCL variants within the family. The orange arrow marks heterozygous variants in both parents, the red arrow indicates homozygous variants in the patient, and the blue arrow shows the WT alleles in the sister. (B) The family tree depicts the segregation of the two HMGCL variants. The black arrow indicates the affected proband, marked with a crossed-black symbol to denote the deceased status. Black-and-white symbols represent carriers, while white symbols represent unaffected individuals. HMGCLD: 3-hydroxy-3-methylglutaryl-CoA lyase deficiency; WT: wild-type
The replaces threonine with serine at position 205 positioned in the conservative motif of pocket side area and p.M217V is also evolutionary conservative among different species navigated via MULTIZ alignment (UCSC Genome Browser) in Figure 2A [10]. The three-dimensional model of human HMGCL structure and residues was observed Protein Data Bank (PDB, entry: 2CW6) by PyMOL Molecular Graphics System version 2.5.5 (Figure 2B). Threonine at position 205, located conterminous with β6-strands, forms a hydrogen bond with G203 and interacts with the metal-binding residue H235. The appearance of T205S probably affects the pocket dynamics of the enzyme HMGCL in binding with the ligands (Figure S1).
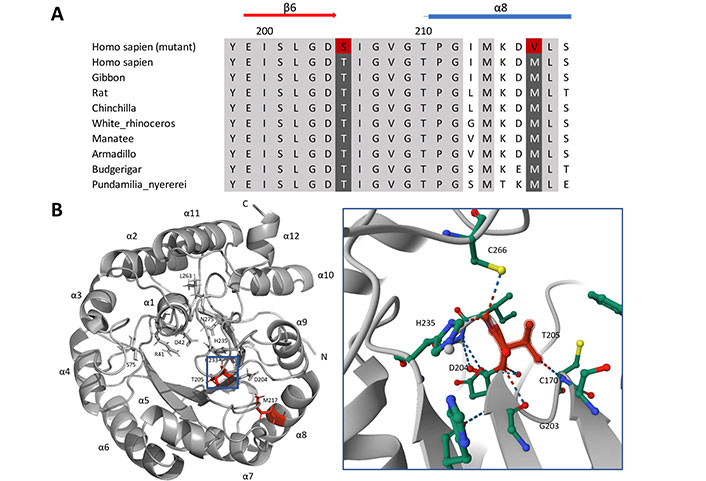
Human HMGCL T205S and M217V via multiple sequence alignment and ribbon diagram, surface presentation. (A) Structure of HMGCL β-strands in red arrow and α-helices present in blue cylinders. Highly conservation regions were in gray/back color and the alternative amino acids were in red. (B) Three-dimensional structure of HMGCL, obtained using PyMOL Molecular Graphics System version 2.2.5. Left, the amino acids T205 and M217 were labeled in red within the monomer structure of HMGCL with β-strands and α-helices (numbered) present in gray with COOH, NH2-terminal. Essential amino acids of HMGCL protein are positioned surrounding the active side (R41, D42, S75, D204, H233, H235, L263, and N275). Right, the neighbors’ interaction of T205 within 5 Å. The residue of interest was highlighted in red with hydrogen bond interactions shown in the dashed line. HMGCL: 3-hydroxy-3-methylglutaryl-CoA lyase
HMGCLD is a severe metabolic disease that often appears in newborns with serious disruption in the synthesis pathway of ketogenesis and leucine degradation for fatting energy. This results in lethargy, hypoglycemia, metabolic acidosis, cyanosis, and episodes of vomiting, diarrhea, and dehydration in newborns. There are a few uncommon symptoms that some patient expresses in dilated cardiomyopathy with arrhythmia or hepatomegaly [11]. The mortality rate of HMGCLD is reported to be around 16.1%, with a median of 9.5 months [12].
This study reported the HMGCLD disorder present in a 12-month-old patient with lethargy, cyanosis, hypotonia, arrhythmias, and shortness of breath, which later develops into coma and death. The observed biochemical profile, characterized by excessive accumulation of the amino acid leucine, and an abnormal blood acylcarnitine profile, indicates disruptions in the metabolic pathway. The WES analysis from the patient’s genome detected two homozygous variants c.[614C>G; 649A>G] co-located in the exon 7 region of the HMGCL gene. Both c.614C>G and c.649A>G are rare variants, absent from the 1,000 Genomes Project population, and predicted as damaging variants. The p.T205S substitution occurs within a conserved motif on the pocket side area, and both alterations are evolutionarily conserved across various species. The exon 7 has the second-highest incidence of mutations, mainly missense alterations affecting the functional domain of the HMGCL enzyme (Figure 3, Figure S2).
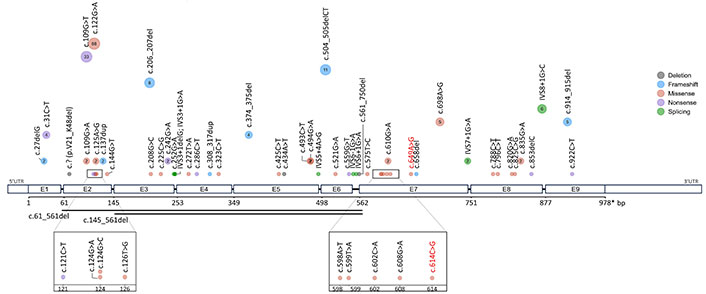
Schematic of mutation spectrum throughout the HMGCL gene. The dots represent a distinct mutation, with dot size and inner number indicating the corresponding count of mutation encounters. The smallest dot signifies a single encounter of the mutation, while larger dots contain a numerical annotation representing the number of mutation encounters. The two horizontal lines mark a large segment deletion mutation. Reported variants c.614C>G and c.649A>G were highlighted in red. Mutation types were distinguished via color legend
The HMGCL’s active site is located at the C-terminal end of the β-barrel structure, which is crucial to the enzyme’s function [13]. The HMGC ligand binding across a cavity located at the C-terminal end of human enzyme HMGCL for acetoacetate catalyzation (Figure S1). The p.T205S is likely to disturb H235 active cation ligands and interfere with residue G203 is reported mutated form and affects enzyme activity and cavity entrance [14]. Residues G203 and D204 are situated closely and interact with T205S via hydrogen bonds on β6-strands of the triosephosphate isomerase (TIM) barrel structure (Figure 2B). The alteration from threonine to serine probably diminishes methyl group (-CH3) interaction and affects the remaining two sensitive residues. T205 was spotted in a significant functional domain of HMGCL protein, and shown to have hydrogen bonds interact with HMGCL ligand (2.7 Å) indicating a significant role in the catalysis process of enzymes [13].
To date, approximately 58 mutations of the HMGCL gene have been reported in the Leiden Open Variation Database (LOVD) and Human Gene Mutation Database (HGMD). The distribution and prevalence of HGMD mutations are illustrated in Figure 3. The missense mutations constitute the most frequent alteration of the HMGCL gene with 50%, followed by frameshifts, nonsense, and splicing mutation with equal 13.79%, and deletion accounts for 8.62% detail (Figure S2A). Exons 2 and 7 exhibited the highest distribution of mutations, predominantly consisting of missense alterations, likely associated with the functional domain of the enzyme HMGCL (Figure 3, Figure S2B).
Two heterozygous c.[608G>A; 610G>A] in cis of each healthy parent transmitted to proband in our WES results. However, the heterozygous compound c.[608G>A; 610G>A] in Japanese cases was reported to be fatal in infant patients [6]. The difference in pathogenicity manifestations could resolve in the cis/trans configuration of variants impact [15]. In Oshima et al. [6] retrospective sudden unexpected death case detected compound heterozygous c.[608G>A; 610G>A], no definite diagnosis and biochemical test was made, though there was metabolic disease speculation. With that limitation, these variants are maintained as variant of uncertain significance in the ClinVar database. In this report, we provide a case with a suitable genetic pattern, additional in silico residues interaction, and HMGCL’s mutations comprehensive analysis support evidence for a definite classification. Based on American College of Medical Genetics and Genomics (ACMG) standards and recommendations for variant interpretation and the additional shreds of evidence of c.[608G>A; 610G>A], the two variants could be classified as likely pathogenic [15].
Due to the infrequency of HMGCLD in Asia and the lack of evidence of case occurrence in Vietnam, our patient was unable to be diagnosed at the molecular level before her death. The mother’s second pregnancy was advised to have a prenatal diagnosis or newborn test targeting the HMGCL gene. Fortunately, their newborn baby received two wide-type alleles of HMGCL gene from the parent. As the first case report of HMGCLD in Vietnam, we would like to emphasize the importance of diagnosing the disease to prevent neonatal mortality. HMGCLD is a rare metabolic disorder reported in some European and Asian countries since 2000 [6, 16–19]. Notably, the in-house database screening shows the presence of the HMGCL allele with two variants, c.[608G>A; 610G>A], in 3 individuals as heterozygous states, suggesting the existence of this allele in the population. Although the patient’s parents in this study are a non-consanguinity married couple within three generations, they were born and grew up in the same village. It is a possibility that the first mutations appeared very early in their small geographical population. This raises a fundamental question on the solutions of population genomic management methods by region that need to be implemented.
HMGCLD can be diagnosed through newborn screening, organic acid analysis, blood acylcarnitine profiling, and genetic mutation analysis. In our case, although the patient exhibited clinical symptoms alongside abnormal plasma amino acid and blood acylcarnitine profiles suggestive of an inborn error of metabolism, the rapid progression of the condition precluded a definitive diagnosis. Early diagnosis and treatment of HMGCLD are critical to preventing severe health issues and improving the quality of life of the patients, as patients diagnosed and treated in early life demonstrate high survival rates. Management typically involves a strict low-protein, low-leucine diet combined with carnitine or carbohydrate supplementation to mitigate metabolic complications [20]. For our case, genetic testing using WES technology offers the most powerful molecular diagnostic criteria, which significantly support the initial clinical suspicion. In rare diseases, WES data can be retrospectively analyzed and continuously updated, facilitating long-term diagnostic consolidation.
Our case report has some limitations. Firstly, a definitive diagnosis of HMGCLD is lacking due to the absence of confirmatory urine organic acid analysis. The patient presented with clinical features indicative of inborn errors of metabolism, and genetic testing suggested HMGCLD; therefore, the combination of genotypes and phenotypes will be conformational diagnosis in this case. Secondly, the pathogenicity of variants c.[608G>A; 610G>A] in this study was demonstrated solely through in silico analysis, and further functional examination is needed to confirm their pathogenic impact.
C5-OH: 3-hydroxy isovalerylcarnitine
HGMD: Human Gene Mutation Database
HMGC: 3-hydroxy-3-methylglutaryl-CoA
HMGCL: 3-hydroxy-3-methylglutaryl-CoA lyase
HMGCLD: 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
OMIM: Online Mendelian Inheritance in Man
WES: whole-exome sequencing
The supplementary figures and other supplementary material for this article are available at: https://www.explorationpub.com/uploads/Article/file/101423_sup_1.pdf.
We thank the patients and family members who participated in this study.
HTNH: Formal analysis, Writing—original draft, Writing—review & editing. KLN: Methodology, Writing—original draft. PNV: Resources, Writing—original draft, Writing—review & editing. HHN: Conceptualization, Supervision, Writing—review & editing. All authors have read and agreed to the published version of the manuscript.
The authors declare no conflicts of interest.
The study was approved by the Institutional Review Board of the Institute of Genome Research (IGR IRB), Vietnam Academy of Science and Technology (No: 4-2024/NCHG-HĐĐĐ).
Informed consent to participate in the study was obtained from the patient’s parents.
Informed consent to publication in the study was obtained from the patient’s parents.
The datasets generated for this study can be found in the Human Gene Mutation Database (HGMD), Leiden Open Variation Database (LOVD) v.3.0, and Protein Data Bank (PDB id: 2CW6). All datasets analyzed for this study are included in the manuscript and the supplementary files.
Not applicable.
© The Author(s) 2025.
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.
Copyright: © The Author(s) 2025. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3288
Download: 46
Times Cited: 0
