
Abstract
Aim:
Cynometra cauliflora (namnam) belongs to the family Fabaceae and is native to eastern Peninsular Malaysia. It grows well with an annual rainfall of 1,500–2,000 mm. Even though a considerable amount of research has been carried out with C. caulifora, there is a dearth of information about biomolecules that may pave the way for drug discoveries and food supplements, which is a gap addressed in this study.
Methods:
The study presented in this paper has identified several antimicrobial, antioxidant, and anti-inflammatory substances, and an in silico approach was used to understand the behaviors of kaempferol-3-O-rhamnoside (K-3-Rh) and β-sitosterol acetate against Alzheimer’s disease (AD). The molecular dynamics (MD) simulations were performed with the selected protein ligand complex of two natural molecules and the synthetic ligand to analyze the dynamic behaviors and binding free energy throughout the 100 ns simulation time. Further, both natural molecules that were investigated comply with Lipinski’s drug-likeness rules.
Results:
The docking scores of both K-3-Rh and sitosterol were found to be compatible with the synthetic AD drug molecules [donepezil analogue (H0L)] used as a reference in the study. Hence, the phytochemicals of Cynometra caulifora showed comparatively similar potency against acetylcholinesterase (AChE).
Conclusions:
Overall, the potential binding affinity from molecular docking and static thermodynamics features from MD simulation suggest that K-3-Rh and β-sitosterol acetate could be considered as a potential therapeutic lead to inhibit AChE leading for AD treatment.
Keywords
Flavonoids, Fabaceae, acetylcholinesterase, microorganisms, drug, biomolecules, molecular docking, molecular dynamicsIntroduction
The tropical plant Cynometra cauliflora belonging to the family Fabaceae commonly grows in Asia, Africa, America, and Australia. It is an indigenous fruit tree to eastern Peninsular Malaysia [1–6]. It is widely cultivated in the northern part of Malaysia [6]. Additionally, this plant grows in Indonesia, Thailand, India, and Sri Lanka [4, 7–9]. This tree grows well in areas with a rainfall of 1,500–2,000 mm, a temperature of 22°–35°C, and is in favor of dry periods and wind resistance [5, 9]. In native languages, this tropical tree is known as ‘‘namnam” or ‘‘katak puru-puru”. The tree gets its local names due to its shrubby fruit and its appearance of the skin. Besides, there are also some vernacular names used in other regions (Table 1) [4, 9].
| Country | Vernacular names |
|---|---|
| Malaysia | Katong-Katong (Sabah) Puki Anjing Salah Nama Buah Katak Puru Namnam (Peninsular) |
| Indonesia | Namu-Namu Nam-Nam Ka-Namu-Namu (Manado, Sulawesi) Lamuta Ulias (Ambon) Nam-Na (Java) Nam-Nam Kapi Anjing Pukih (Sundanese) |
| Thailand | Amphawa (Central) Nang-Ai (Bangkok) Hima (Pattani) |
| Sri Lanka | Namnam (Sinhalese) |
The edible part of the plant is its fruit, which can be eaten raw, made into salad, juices, pickled with sugar, and fried in batter (Figure 1). The unripe fruit is sour/acidic while the ripened fruit is sweet and sour, and is sometimes used as a spice pound together with chili [1, 6, 9, 10]. This plant could also be used as an ornamental plant in the home garden or as a bonsai [9]. As far as its medicinal applications are concerned, records on folk medicine have revealed fruits to have been used as a treatment for loss of appetite [1, 6]. The seed oil is used as a curing agent especially for skin disease treatment for diabetes [1, 2, 6, 7, 9, 11], and hyperlipidemia/high blood pressure as a traditional medicine [2, 4].

C. cauliflora. (A) A healthy grown tree; (B) stem and fruits; (C) ripe fruit; (D) ripe fruit and its seed; (E) leaves; (F) leaves with asymmetrical oblique bases
Many edible and non-edible parts of this plant are rich in compounds that can be used for medicinal purposes. However, several countries have reported the use of edible parts, especially fruits in underutilized form even though many ventures use C. cauliflora fruit for value-added products [7]. Since fruits, leaves, and other parts of the C. cauliflora tree have not been given much attention compared to the other tropical fruits, some studies have started to investigate bioactive compounds extracted using C. cauliflora and their applications. For instance, studies revealed that C. cauliflora has phytochemicals such as phenols and flavonoids including other bioactive molecules demonstrated as having antioxidant ability, antibacterial, antiviral, anti-human immunodeficiency virus (HIV), and cytotoxicity activities [5, 10, 12]. Based on the health benefits of the C. cauliflora tree and the identification of nutritive qualities, this plant has potential usages as a resource bank [7]. In addition, some of the secondary metabolites present in this plant are considered a good source for the discovery of new pharmaceuticals [2]. As many new drugs have been reported to contain negative side effects, these natural resources will help implement new medicinal explorations as an alternative to synthetic drugs [2, 5, 10, 12]. Many studies have been conducted in Malaysia, while other countries have not done so despite having abundant growth of this plant [7].
The World Health Organization (WHO) has reported that the majority (80%) of people from developing countries continue the use of traditional medicine as a primary form of healthcare [4]. However, many studies have revealed that C. cauliflora is an underutilized tropical fruit plant [3, 13, 14]. Therefore, many researchers have focused their studies on these types of underutilized medicinal plants. Nonetheless, there is no comprehensive information composed together as most of the results of these studies are scattered and most of such details are available in ancient manuscripts and in modern times, in the native languages of the countries where this plant has an ethnopharmacological presence.
Alzheimer’s disease (AD) is considered a progressive neurodegenerative disorder characterized by the deterioration of memory and cognitive functions affecting the brain and it is responsible for the most common type of dementia [15]. AD has been identified as the one of leading causes of death, disability, and morbidity for the older population in the world [16]. Among the validated drugs as a treatment for AD, Acetylcholinesterase (AChE) inhibitors have shown potential therapeutic properties [17–19]. AChE is a monomer that belongs to the serine hydrolase enzyme category and it plays a vital role in terminating the cholinergic impulse transmission process at neuromuscular junctions and cholinergic brain synapses. The hydrolyzing of acetylcholine (ACh) to acetate and choline is the major biological function of AChE [20, 21]. The protein structure (Figure 2) consists of 14 α helices and 12 stranded β sheets. Remarkably, human AChE has a deep and narrow gorge-like special feature which is almost 20 Å long halfway, which runs into the protein structure. The base of the enzyme is lined with aromatic amino acids with many sub-sites [20].
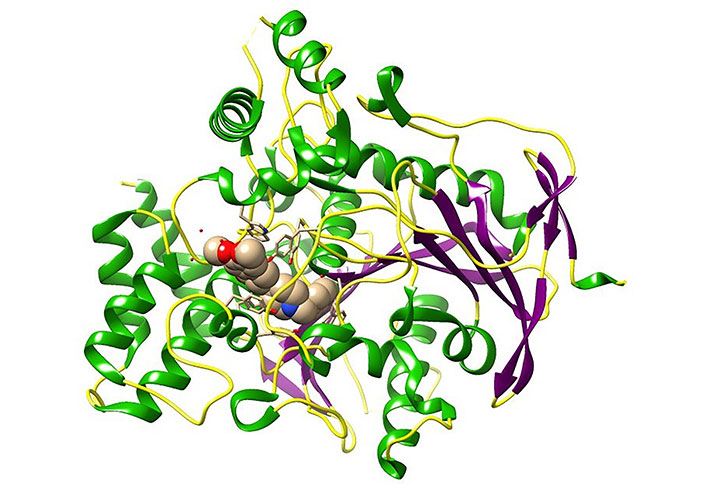
Three-dimensional (3D) crystal structure of AChE bound donepezil analogue (H0L) molecules [protein data bank (PDB) ID: 7D9O; the ligand is in spheres, green: helix, purple: sheets, yellow: coils]. The image was created using Chimera software 1.14 in conjunction with the pertinent PDB files. This visual representation illustrates the binding pocket of the AChE. H0L is the synonym given to (2R)-2-[[4-fluoranyl-1-[(4-fluorophenyl)methyl]piperidin-4-yl]methyl]-5,6-dimethoxy-2,3-dihydroinden-1-one
The ability of the inhibitory action of AChE reduces the hydrolysis of ACh into acetate and choline. Meanwhile, it consequently increases the ACh levels at the synaptic cleft stimulating cholinergic receptors which promote memory function [18]. Currently, there are only a few synthetic drugs, acting as AChE inhibitors such as donepezil, galantamine, rivastigmine, and tacrine, recommended for the treatment of symptoms related to moderate to severe AD. However, these drugs have been recognized as imparting adverse side effects including nausea, vomiting, loss of appetite, headache, constipation, and dizziness [22].
The study of dynamic behaviors of protein ligand complexes provides a deep understanding of molecular-level interactions of AChE [23]. This helps to develop new drugs using novel approaches to drug designing. Furthermore, no in silico studies have been reported on the use of these molecules (selected in this study) against neurodegenerative disorders, especially against AD. Based on this perception, this current paper attempts to prepare a comprehensive appraisal of this underutilized medicinal plant properties, health benefits, and its potential usage with bioactive compounds with in silico analyses to understand kaempferol-3-O-rhamnoside (K-3-Rh) and β-sitosterol acetate (a derivative of β-sitosterol) (Table 2) which are present in the C. cauliflora extracts, with the molecular-level interaction between the receptor and ligand, giving an insight of the therapeutic potential of these two phytochemicals leading to the development of new drugs as lead molecules. Additionally, this comprehensive information would be helpful for future studies as reference material, as increasing trends occur in studies on local tropical medicinal plants in developing countries for drug designing with new technologies.
Physicochemical and drug-likeness properties of identified phytochemicals of C. cauliflora
| Property | Results (SwissADME and Molinspiration) | ||||
|---|---|---|---|---|---|
| Procyanidin tetramer | Vitexin | β-Sitosterol acetate | Catechin | K-3-Rh | |
| Molecular formula | C60H50O24 | C21H20O10 | C31H52O2 | C15H14O6 | C21H20O10 |
| Molecular weight | 1,155.02 g/mol | 432.38 g/mol | 456.74 g/mol | 290.27 g/mol | 432.38 g/mol |
| Hydrogen bond (H-bond) donors | 20 | 7 | 0 | 5 | 6 |
| H-bond acceptors | 24 | 10 | 2 | 6 | 10 |
| Rotatable bonds | 7 | 3 | 8 | 1 | 3 |
| Log P (partition coefficient, predicted value) | 2.11 or 5.0 | –0.02 or 0.52 | 7.63 or 8.94 | 0.85 or 1.37 | 0.60 or 1.13 |
| Lipinski’s rule of five | No | Yes | Yes | Yes | Yes |
| Ghose filter | No | Yes | No | Yes | Yes |
| Veber’s rule | No | No | Yes | Yes | No |
| Egan | No | No | No | Yes | No |
| Muegge filter | No | No | No | Yes | No |
| BBB permanent | No | No | No | No | No |
| CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4 inhibitors | No | No | No | No | No |
| Bioavailability score | 0.17 | 0.55 | 0.55 | 0.55 | 0.55 |
BBB: blood brain barrier; CYP1A2: cytochrome P450 1A2
Materials and methods
Phytochemical types, their activities, and health benefits with medicinal properties
The literature review which follows hereafter was conducted to identify the presence of phytochemical types to build a comprehensive justification for the molecular docking study. The literature review was conducted based on the keywords “Cynometra cauliflora”, “Namnam”, “phytochemicals”, “bioactive compounds”, “health benefits” and “medicinal properties” using Google Scholar, PubMed, and Web of Science. All the relevant literature (30 articles) was used to obtain all the required details. Based on the chosen articles, a list of phytochemicals identified from different parts of C. cauliflora and their bioactivities against microorganisms were evaluated. These bioactive phytochemicals were used to determine the most suitable compounds for conducting molecular docking.
Structural arrangement of protein and processing of the phytochemical types with synthetic molecules
The 3D crystal structure of AChE protein bound to H0L, shown in Figure 2 (PDB ID: 7D9O, resolution: 2.45 Å, R-value: 0.188) was retrieved from the PDB. This structure was used as the initial structure for the study. The chain A of 7D9O was isolated and the ligand was removed to obtain apoprotein, using University of California San Francisco (UCSF) Chimera 1.81 software and prepared for molecular docking simulation. Other non-stranded molecules and crystalline water molecules from the retrieved crystal structure using the same software. The apoprotein was subjected to short molecular dynamics (MD) run of 4 ns to remove constraints of the crystal structure.
Several compounds from the C. cauliflora plant were identified from the literature review mentioned above and verified by the phytochemical databases [24, 25]. Lipinski’s rule was used to identify the drug-likeness of the compounds selected for the study [24, 25]. The 3D structures of the five molecules that exist in C. cauliflora were taken from the PubChem website (https://pubchemdocs.ncbi.nlm.nih.gov). The promising phytochemicals: procyanidin tetramer, β-sitosterol, K-3-Rh, catechin, and vitexin were used for physicochemical and drug-likeness property analysis and results are shown in Table 2. The molecular docking simulations were carried out with only four molecules which satisfied Lipinski’s rule (Table 2). Prior to the molecular docking simulation study, the geometry optimization of four selected molecules was performed using the density functional theory (DFT) method at the B3LYP level, employing the 6-31G (d,p) basis set in GAUSSIAN 09 C.01 [26]. Additionally, the 3D structure of the synthetic ligand, H0L [(2R)-2-[[4-fluoranyl-1-[(4-fluorophenyl)methyl]piperidin-4-yl]methyl]-5,6-dimethoxy-2,3-dihydroinden-1-one)], was modeled as the reference molecule using Avogadro software 1.93.0, applying the same conditions that were used for the geometry optimization of phytochemicals in GAUSSIAN 09 C.01 [26]. A study reported that H0L has been recognised as a highly potent, next-generation drug candidate for AD, minimizing the adverse effect of donepezil which is one of the best-known drugs [27].
Molecular docking and MD simulation
The docking software, Dock 6.9 was used to search for potential binding modes [28]. The optimized ligand structures were docked (flexible docking method) into the active site of the structure of AChE. The residues of active sites were identified using the ligand bound crystal structure of protein and further verification was made using the CASTp server (http://sts.bioe.uic.edu/castp/). The binding affinity of the protein and the ligand was ranked based on the resulting grid scores. In general, a lower grid score implies higher binding strength between the receptor and the ligand. H0L was considered as the reference ligand for the docking study. The MD simulations were done for the docked complexes with the highest binding affinity and favorable pose with the binding pocket of the receptor. All MD simulations were conducted using the GROningen MAchine for Chemical Simulations (GROMACS) 2018.1 [29–31]. Protein topologies were generated by using GROningen MOlecular Simulation (GROMOS) 54A7 united atom force field [32]. The force field parameters for the plant molecules that were found in the C. cauliflora plant and the synthetic molecule (H0L) were generated using the Automated Topology Builder (ATB) server [33]. The simulation box was defined as having a volume of 10.57 × 10.57 × 10.57 nm3. The docked complexes were placed at the center of the simulation box and then solvated with simple point charge (SPC) water molecules [33]. Further, Na+ ions were added appropriately, in order to keep the whole system neutral. In the end, the final systems contained nearly 100,800 atoms. Moreover, the electrostatic interactions were calculated by a particle mesh Ewald (PME) summation scheme keeping a short-range cutoff in 1.2 nm [34]. The isothermal-isobaric ensemble conditions were maintained during all the simulations. Both the temperature and pressure were kept constant at 300 K and 1 bar according to Berendsen’s weak coupling algorithm [35]. The bonds of the molecules are maintained on fixed bond lengths at their equilibrium distances using the linear constraint solver (LINCS) algorithm [36]. In addition, the steepest descent algorithm was used to minimize the energy of the system with 2,000 steps and then the entire system was equilibrated with a 500 ps long MD simulation. Then MD simulation was conducted with a 2-femtosecond time step at 100 ns. Further, MD trajectory configurations were stored at every 2 ps intervals. Finally, the entire simulation protocol mentioned above was used for the apoprotein alone to study its overall stability. The trajectory information was analyzed using GROMACS utilities, UCSF Chimera, Pymol 2.3.0, and Rasmol 2.7.1 software [37]. The ligand and the protein non-covalent interactions were evaluated by the LigPlot + v.14.5 software and ProteinPlus web server [38, 39].
Molecular mechanics Poisson-Boltzmann surface area analysis
The molecular mechanics Poisson-Boltzmann surface area (MM-PBSA) approach was used to estimate the binding free energies of selected protein ligand complexes [40]. The binding free energy (ΔGbind) between a protein and a ligand was calculated using the formula given below:
ΔGbind = ΔGcomplex − (ΔGprotein + ΔGligand)
Where ΔGcomplex is the total free energy of the protein ligand complex and ΔGprotein and ΔGligand are the total free energies of the separated protein and ligand in a solvent, respectively. The bootstrap analysis was performed to obtain the standard error associated with energy terms and the energy contribution per residue in AChE to the binding energy. The entropy change was assumed to be the same for all the systems investigated.
Results
Phytochemical types
The literature review has revealed that the C. cauliflora plant can be considered a rich source of bioactive compounds, many phytochemicals were found that would facilitate the future development of value-added products with phytomedicinal mechanisms of action, as well as in the development of functional food products. Most of the chemical compounds found in the literature for C. cauliflora are listed in Table 3 with their bioactivity.
List of the phytochemicals found in the C. cauliflora plant which have known functional properties
| Parts of the tree | Extracted compound(s) | Category of bioactive compounds | Function | Reference |
|---|---|---|---|---|
| Leaves | K-3-Rh (C21H20O10) | Flavonoids | Active lipase inhibitor | [41] |
| Leaves | Procyanidin tetramer Catechin Procyanidin trimer Taxifolin pentoside Vitexin Isovitexin Kaempferol hexoside Quercetin pentoside Quercetin hexoside Kaempferol coumaroyl-hexoside Quercetin hexoside Isorhamnetin hexoside Apigenin-6-C-glucoside-8-C-glucoside Procyanidin hexamer | Flavonoids | Antioxidant activity AChE inhibitory activity Tyrosinase inhibitory activity α-glucosidase inhibitory activity | [1] |
| Leaves | Taxifolin 3-O-arabinofuranoside Apigenin 8-C-glucoside (vitexin) Apigenin 6-C-glucoside (isovitexin) Acacetin 7-O-β-glucoside | Flavonoids | Antioxidant activity AChE inhibitory activity Butyrylcholinesterase (BChE) inhibitory activity α-glucosidase inhibition activities | [2] |
| Leaves | Vitexin (apigenin 8-C-glucoside) | Apigenin flavone glucoside | - | [11] |
Phytol (C20H40O) Vitamin E (C29H50O2) β-Sitosterol (C29H50O) | Phenol | - | ||
| Leaves | Amorphigenin Capensine Epigallocathecin gallate Isorhamnetin Nobiletin Xanthotoxin | Flavonoids | - | [42] |
Apigenin Cyanidin Fraxetin Malvidin Naringenin Oenin | α-glucosidase inhibition activities | |||
| Leaves | α-Thujene α-Pinene Ethyl 2-methyl pentanoate (E)-2-Methyl-3-octene-5-yne β-Pinene Myrcene δ-3-Carene α-Terpinene p-Cymene Limonene (Z)-β-Ocimene γ-Terpinene Terpinolene Linalool α-Terpineol neo-Dihydrocarveol 6-Camphenol acetate cis-Carvone oxide trans-Dihydro-α-terpinyl acetate p-Vinyl guaiacol Verbanol acetate Aromadendrene α-Himachalene β-Chamigrene α-Bulnesene trans-Cadina-1,4-diene Monoterpenes Oxygenated monoterpenes Sesquiterpenes Non-terpene derivatives | Essential oil (EO) | - | [6] |
| Fruits | K-3-Rh (C21H20O10) | Flavonoids | Active lipase inhibitor | [41] |
| Fruits | Limonene cis-Thujone trans-Pulegol β-Cubebene β-Elemene α-Guaiene cis-β-Farnesene Prezizaene 9-epi-(E)-Caryophyllene Sesquisabinene Ishwarane β-Chamigrene Germacrene D α-Muurolene β-Bisabolene α-Bulnesene γ-Cadinene Cubebol (E)-γ-Bisabolene γ-Cuprenene trans-Cadina-1,4-diene Selina-3,7(11)-diene Elemol β-Calacorene Longipinanol Globulol Longiborneol trans-β-Elemenone 1,10-di-epi-Cubenol Eremoligenol γ-Eudesmol α-Acorenol Hinesol α-Muurolol Agarospirol Valerianol Allohimachalol epi-β-Bisabolol Occidenol Longiborneol acetate 11-αH-Himachal-4-en-1-β-ol Nootkatol Cryptomerione Curcumenol (2E,6E)-Farnesol (E)-Nuciferol (Z)-Lanceol α-Chenopodiol Occidol acetate Linoleic acid Monoterpenes Oxygenated monoterpenes Sesquiterpenes Oxygenated sesquiterpenes Non-terpene derivatives | EO | - | [6] |
| Twigs | Apigenin Luteolin | Flavonoids | N/A | [43] |
| Twigs | (Z)-β-Ocimene Santolina alcohol Dihydromyrcenol Linalool trans-Sabinene hydrate cis-Verbenol Octanoic acid cis-4-Caranone Fragranol Geraniol (E)-cis-Jasmonol Decanoic acid Dodecanoic acid Squamulosone α-Eudesmol acetate Occidol acetate Linoleic acid Monoterpenes Oxygenated monoterpenes Oxygenated sesquiterpenes Non-terpene derivatives | EO | - | [6] |
| Heartwood | Apigenin (4’,5,7-trihydroxyflavone) | Flavone | Antioxidant activity | [12] |
-: no data; N/A: not applicable
Molecular docking results and MD simulations analysis
According to the docking results (Table 4), among all the plant-based compounds used in this study, only K-3-Rh and β-sitosterol showed the highest binding affinities of −44.5 kJ mol–1 and −44.9 kJ mol–1 respectively to AChE. However, the H0L showed a greater binding affinity of –55.8 kJ mol–1. Procyanidin tetramer showed a similar binding affinity towards the AChE, despite the failure of Lipinski’s rules. The binding affinities shown as grid score represent the approximate binding energy of the selected ligand as the summation of both the electrostatic and van der Waals energies [28, 44]. Furthermore, the H-bonds formation between the ligand and receptor indicates that both AChE-K-3-Rh and AChE-sitosterol formed stable complexes with 2 H-bonds and 1 H-bond respectively.
Docking results of selected compounds with AChE
| Molecule | Grid score/kJ mol–1 |
|---|---|
| K-3-Rh | –44.5 |
| Procyanidin tetramer | –42.7 |
| Catechin | –40.8 |
| Vitexin | –46.1 |
| β-Sitosterol acetate | –44.9 |
| H0L (reference) | –55.8 |
The validity of docking parameters incorporated with the docking procedure was confirmed by the root mean square deviation (RMSD) value between the two structures which was 1.053 Å (Figure 3). The upper limit value of the accepted RMSD value is 2.0 Å for a similar pose [45]. Thus, the best docking poses to the ligand binding pocket (LBP) with the most negative grid score were selected for the MD simulation study to investigate the stability of the protein ligand complexes in an aqueous environment. As seen in Table 4, the phytochemicals extracted, K-3-Rh and sitosterol acetate, were selected for the MD simulation study. In addition, apoprotein and H0L-protein complex were also subjected to MD simulation using the same conditions as described in the subsequent section.
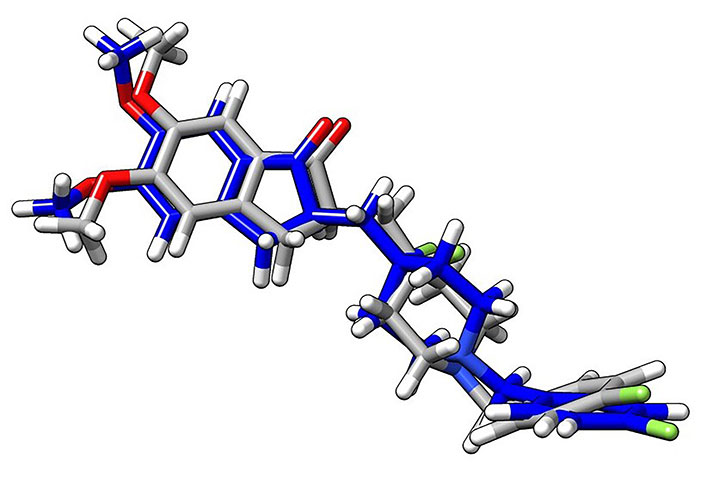
Superimposition arrangement of docked H0L with the same from the crystal structure of the protein ligand complex (gray: crystal structure; blue: docked structure of the ligand, H0L; white: H atoms; red: oxygen atoms; green: fluorine atoms). The superimposition of the crystal structure and the docked structure is used to validate the docking simulation procedures employed in the research. In this figure, both the crystal structure and the docked structures showed a good superimposition within the expected limits, as demonstrated in the relevant reference [45]
MD simulations analysis was performed, with a 100 ns long simulation time, to find the stability of protein ligand complexes in an aqueous environment and for the dynamic conformational changes analysis. The complexes of AChE-H0L, AChE-K-3-Rh, AChE-sitosterol, and the apoprotein tertiary structure were subjected to MD simulation in an aqueous medium. The Lennard-Jones short-range (LJ-SR) and Coulombic short-range (Coul-SR) potentials were used to calculate the average interaction energies of each complex (AChE-ligand). The plotted results, as shown in Figure 4, revealed that the ligand shows a linear Lennard-Jones (LJ) interaction at around −100 kJ mol−1 to −280 kJ mol−1 while Coul-SR has a range of 0 to –50 kJ mol−1 or +50 kJ mol−1. Moreover, the energy analysis, performed using g_energy tools, indicated that LJ-SR interactions play a significant contribution to the stability of ligand-receptor complex than that of coulombic interaction.
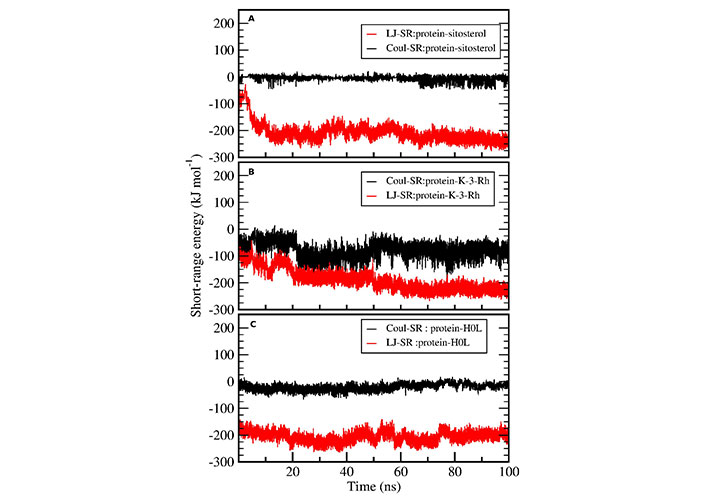
Protein ligand interaction energy analysis. Coul-SR (black) and LJ-SR (red) of ligands interacting with AChE
RMSD calculations were used to assess and compare the dynamic stability of all four systems including apoprotein during the 100 ns simulation period. The RMSD plot shows the magnitude of the deviation of the receptor structure as a function of simulation time with respect to the initial receptor structure. All three systems exhibited reasonable stability in an aqueous system (Figure 5). Further, there were fewer conformational changes for all complexes including the apoprotein. However, the AChE-K-3-Rh complex showed almost similar RMSD behavior compared with the AChE-H0L complex. The results evidently denoted that all the small molecules bind well with the AChE binding site.
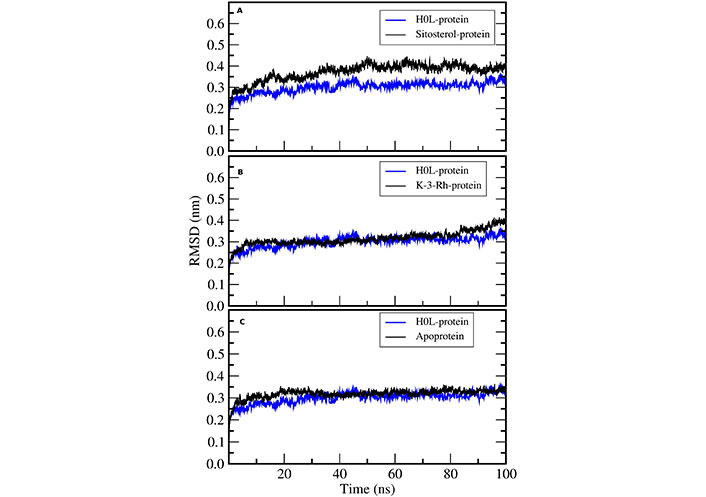
RMSD of H0L-protein complex with (A) sitosterol-protein, (B) K-3-Rh-protein, and (C) apoprotein
Radius of gyration (Rg) of AChE was calculated to investigate the compactness of the receptor protein in all three systems. This exhibits the overall spread of atoms relative to the center of the mass of the protein that can be used to analyze the conformational stability of the protein. The constant and low value of Rg revealed the better compactness of the molecule. In addition, the structural changes and compactness of AChE also showed the same after binding to all the ligands. The Rg plot shown in Figure 6 inferred that the two plant molecules-AChE complexes were stable in terms of protein conformational stability and uniform compactness throughout the simulation. The results showed that the Rg values of both K-3-Rh and sitosterol were around 2.35 nm at the beginning of the simulation. However, the results further indicated that K-3-Rh and sitosterol systems reached a lower value of Rg, which was almost the same as that of H0L, after 90 ns. As the fluctuation difference of other complexes in comparison with H0L was less than 0.1 nm, all three complexes and apo systems maintained reasonable stability in the aqueous medium.
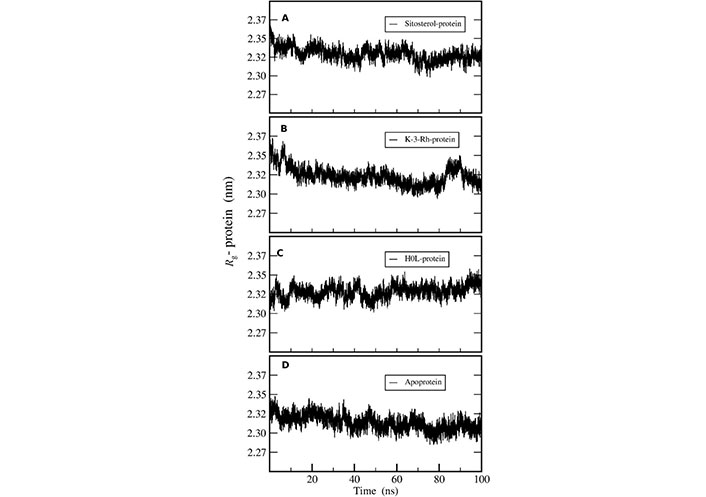
Rg of AChE-ligand complexes of (A) Sitosterol-protein, (B) K-3-Rh-protein, (C) H0L-protein, and (D) apoprotein
The residue-wise root mean square fluctuation (RMSF) of AChE was calculated for all the protein ligand systems and apoprotein systems during the course of the MD simulation process. All systems performed a similar fluctuation pattern throughout the entire 100 ns of the MD simulation (Figure 7). The residues fluctuated around 400 and 500 and these regions corresponded to the surface lined resides. The active residues that bound to ligands showed narrow fluctuation amplitude in RMSF plots. Moreover, it is noted that the RMSF results were the same for three ligand-receptor complexes except for minor variations in the binding sites.
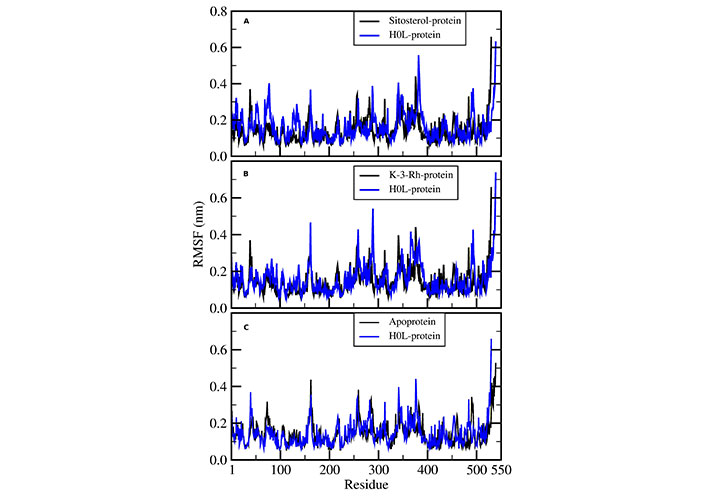
RMSF of AChE-H0L complex with (A) sitosterol-protein, (B) K-3-Rh-protein, and (C) apoprotein
Solvent accessible surface area (SASA) analysis was conducted to understand the solvent accessibility of AChE-ligand complexes using MD trajectories. The SASA plots indicate the bimolecular surface area accessible to solvent molecules. The relative expansion was indicated by an increase in SASA values (Figure 8). Among the protein ligand complexes, the H0L system clearly indicated a constant SASA value throughout the 100 ns simulation. Both K-3-Rh and sitosterol complexes showed a significant decrease in the SASA value during the first 20 ns of the MD simulation. However, the average SASA value range for both natural molecule systems was 235–250 nm2, which was slightly greater than that of the H0L system. Further, all systems reached a value which is around 235 nm2, giving an insight that natural molecular systems can also mimic the action of synthetic drug molecules against AChE.
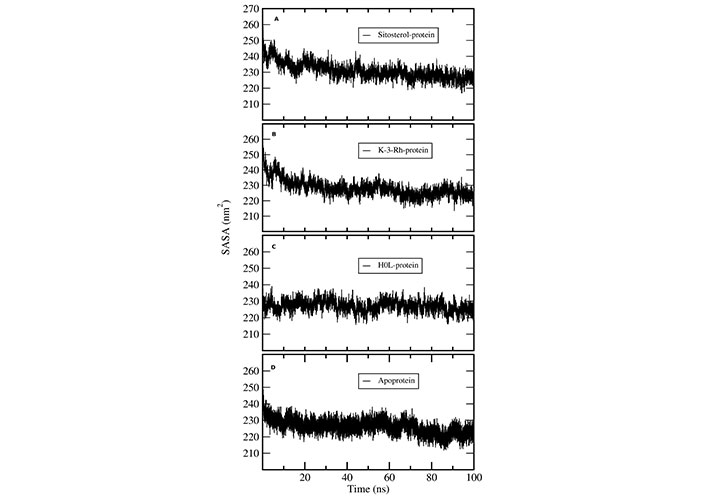
SASA plots of AChE-ligand complexes. (A) sitosterol-protein; (B) K-3-Rh-protein; (C) H0L-protein; (D) apoprotein
The MM-PBSA method, utilizing the GROMACS tool g_mmpbsa, was employed to estimate the binding affinity of the ligands to AChE. The determination of free energy, using MM-PBSA, provides a comprehensive and detailed description of the binding affinities between the receptor and the ligand compared to molecular docking and other forms of energy calculations, such as LJ-SR, LJ long-range (LJ-LR), and Coul-SR potential calculations.
Binding free energy calculations, as shown in Table 5, included potential energy in a vacuum (van der Waals and electrostatic energy), polar solvation energy, and non-polar solvation energy. According to the results presented in Table 5, the analysis indicates that all three molecules bind spontaneously to the AChE protein, and the order of binding affinity varies as follows: sitosterol approximately equals H0L > K-3-Rh. The primary component of the binding energy in all three cases was the van der Waals interaction energy.
Binding energy component and binding free energy for ligands from MM-PBSA (all energy terms are in kJ mol−1; using bootstrap analysis)
| System | Van der Waals energy | Electrostati energy | Polar solvation energy | Non-polar solvation energy | Binding free energy |
|---|---|---|---|---|---|
| H0L | –218.15 ± 1.79 | –10.71 ± 0.61 | 72.34 ± 1.79 | –21.10 ± 0.15 | –177.72 ± 1.69 |
| K-3-Rh | –199.62 ± 4.09 | –37.49 ± 1.34 | 115.22 ± 3.08 | –19.15 ± 0.27 | –140.86 ± 2.54 |
| Sitosterol | –219.42 ± 3.36 | –2.51 ± 0.48 | 59.10 ± 1.91 | –20.76 ± 0.29 | –183.68 ± 3.01 |
However, it was seen that electrostatic energy contributes more than the non-polar solvation energy for H0L and sitosterol, while electrostatic energy is a significant contributor than non-polar solvation energy for K-3-Rh. Among all molecules used for the study, the synthetic ligand H0L, and natural ligand, sitosterol showed the highest binding affinity. The larger negative van der Waals energy and relatively lower positive polar solvation energy can lead to higher stability of both plant molecules towards AChE.
H-bond analysis showed the presence of a number of H-bonds between the ligand and the active site of the receptor. It provides details for interacting amino acid residues with the ligand viz, K-3-Rh, and sitosterol. The presence of H-bonds was calculated using g_hbond tools and the results, shown in Figure 9, clearly indicate the existence of H-bonds between AChE and three ligands present in the three respective simulations. Furthermore, the Ligplot analysis of protein ligand complexes was carried out to obtain 2D diagrams of H-bonds arrangement of the protein-K-3-Rh and protein-sitosterol complexes using LigPlot + v.2.2.2 software, and resulting figures are shown in Figures 10 and 11 respectively. In Figures 10 and 11, the interaction between ligands and the amino acid residues of LBP of both K-3-Rh and sitosterol are given in Figure 10A and C, Figure 11A and C respectively. Furthermore, As shown in Figure 12, there are H-bond interactions between H0L and LBD at 100ns (Figure 12B) and 0 ns (Figure 12D) respectively. The interaction of active residues and H0L can be clearly seen in Figure 12A and B.
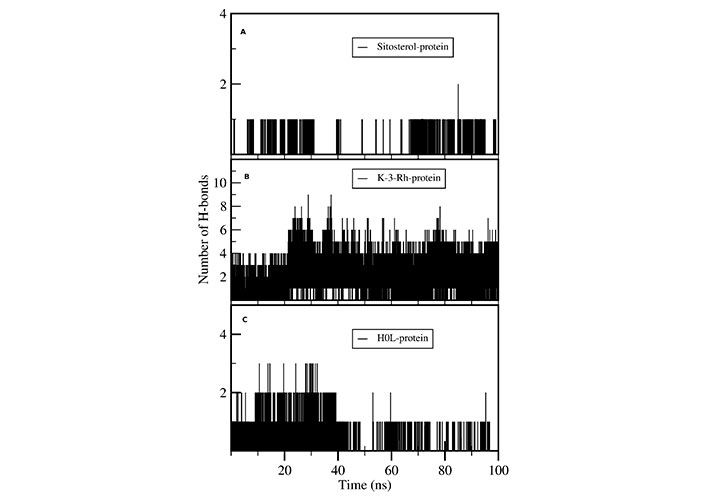
Number of intermolecular H-bonds formed between active site residues of the protein with (A) sitosterol, (B) K-3-Rh, and (C) H0L as a function of the simulation time
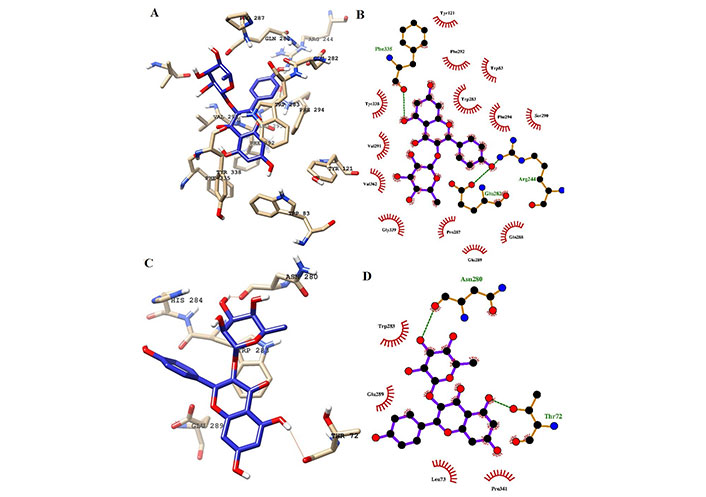
Interaction of ligands with the key residues at the binding pocket of AChE. (A) Ligand K-3-Rh at the binding pocket of AChE at 100 ns; (B) Ligplot depiction of H-bond and hydrophobic interactions between K-3-Rh and AChE at 100 ns; (C) ligand K-3-Rh at the binding pocket of AChE at 0 ns; (D) Ligplot depiction of H-bond and hydrophobic interactions between K-3-Rh and AChE at 0 ns. Ball and stick: the ligands and protein side chains; green dotted line: H-bonds; spoked arcs: residues making non-bonded contacts
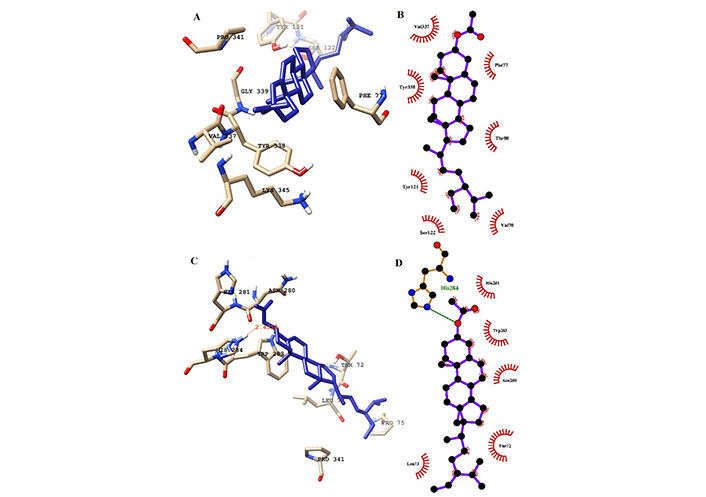
Interaction of ligands (sitosterol) with the key residues at the binding pocket of AChE. (A) Ligplot depiction of H-bond and hydrophobic interactions between sitosterol and AChE at 100 ns; (B) ligand sitosterol at the binding pocket of AChE at 100 ns; (C) Ligplot depiction of H-bond and hydrophobic interactions between sitosterol and AChE its native state from the docking experiment; (D) ligand sitosterol at the binding pocket of AChE at its native state from the docking experiment at 0 ns. Ball and stick: the ligands and protein side chains; green dotted line: H-bonds; spoked arcs: residues making non-bonded contacts
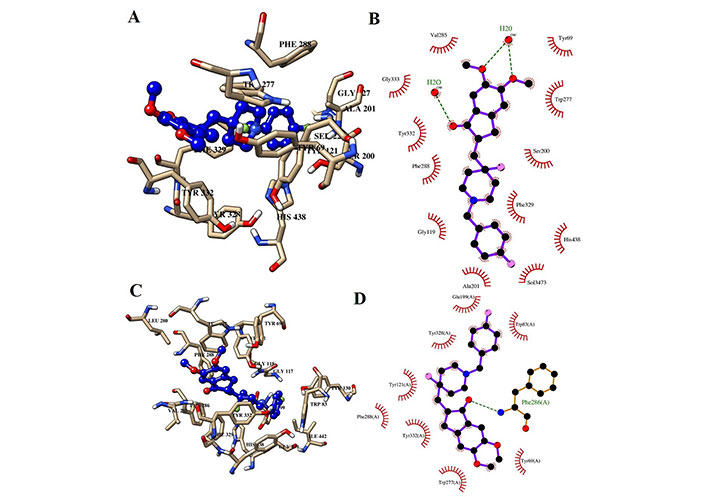
Interaction of H0L with the key residues at the binding pocket of AChE. (A) Ligand H0L at the binding pocket of AChE at 100 ns; (B) Ligplot depiction of H-bond and hydrophobic interactions between H0L and AChE at 100 ns; (C) ligand H0L at the binding pocket of AChE at 0 ns; (D) Ligplot depiction of H-bond and hydrophobic interactions between H0L and AChE at 0 ns. Ball and stick: the ligands and protein side chains; green dotted line: H-bonds; spoked arcs: residues making non-bonded contacts
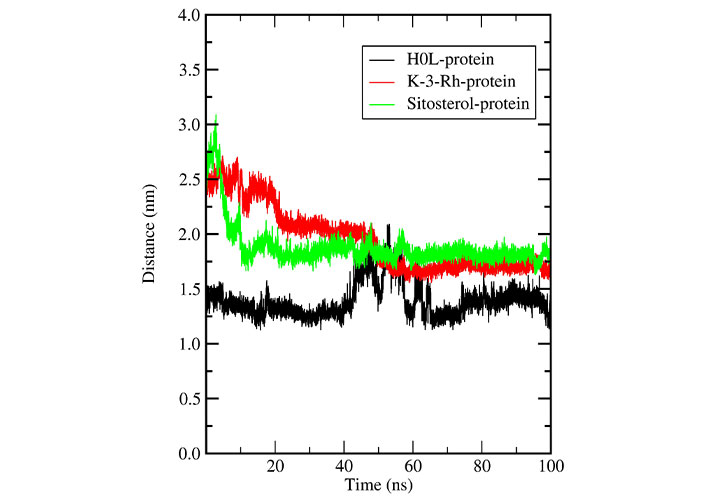
Further, the distance between the centers of mass (c.o.m) of the protein (AChE) and the c.o.m of the ligand was calculated using the g_distance tool and results are shown in Figure 13. According to Figures 10 and 11, K-3- formed a significant number of H-bonds during the whole simulation process making a strong stable interaction with LBP of AChE. This behavior cannot be seen in H0L and sitosterol. However, there was a strong interaction between H0L and AChE in the first half of the whole simulation time for maintaining 1 H-bonds to 3 H-bonds. Moreover, in the account of chemical structural features seen, the H0L, the reference molecule, has similarities to both K-3-Rh as well as sitosterol. As depicted in Figure 14, it can clearly be seen that both H0L and stigmasterol molecules are planer. In K-3-Rh, the H-bond formation was more significant than that of H0L with the receptor. This may facilitate the inhibitory activity of K-3-Rh against AChE.
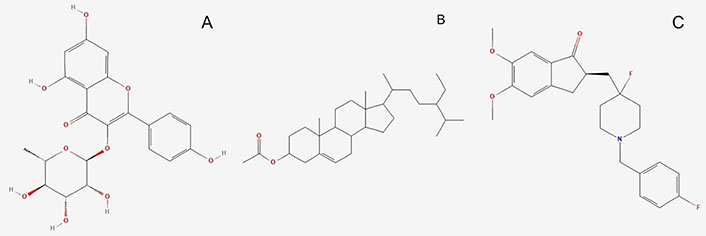
Molecules used for the investigation. (A) K-3-Rh (PubChem Identifier: CID 5316673 URL: https://pubchem.ncbi.nlm.nih.gov/compound/5316673#section=2D-Structure); (B) β-sitosterol acetate (sitosterol) (PubChem Identifier: SID 348285530 URL: https://pubchem.ncbi.nlm.nih.gov/substance/348285530#section=2D-Structure); (C) H0L (PubChem Identifier: CID 156583276 URL: https://pubchem.ncbi.nlm.nih.gov/compound/156583276#section=Structures)
In a detailed analysis of H-bonds formations, initially, the K-3-Rh-AChE complex had 2 H-bonds with Thr72 and Asn280. However, these residues were not seen in the snapshot taken at 100 ns, but Arg244, Glu283, and Phe335 formed 3 H-bonds with K-3-Rh ligand. The residue involved in H-bond formation in the stigmasterol-AChE complex was only His284 at 0 ns. Surprisingly, at the end of the simulation time, the stigmasterol-AChE complex possessed only hydrophobic interactions (Figure 9B) which indicates that the stable complex formation is possible in the presence of non-bonded interactions with the residues of the receptor. After 40 ns of simulation time, the average distance between the c.o.m of ligand and the c.o.m of protein was increased and normalized from 60 ns. Furthermore, it is clearly indicated that all three ligands occupy LBP maintaining close contact with the active residues of the binding pocket with an average distance of 1–2 nm (Figure 10). It was observed that K-3-Rh molecules maintained a stronger H-bond with active residues than H0L.
Discussion
Phytochemical types and their activities
Plant-based natural products have been recognized as an alternative for modern medicinal uses, because of their chemical constituents. There are emerging trends in underutilized medicinal plants such as C. cauliflora based on their secondary metabolites and their diversity. A few studies have revealed that leaves, fruits, and seeds of C. cauliflora are rich sources of bioactive compounds such as α-glucosidase, AChE, and tyrosinase, which have strong antioxidant activities [1, 2, 6, 10]. Nevertheless, many studies have reported extracts of leaves, wood, twigs, bark, and seeds rich with phenols, terpenoids, flavonoids, tannins, quinones, cardiac glycosides and saponins (saponins steroids/triterpenoids) [1, 2, 6, 11, 12, 46–49]. Further, it has been reported that terpenoids are only found in the leaves and fruits, and to date, it has also been reported that cardiac glycosides are found in both mature and young leaves including the bark except stem [1, 2]. The study conducted by Seyedan et al. [11] reported that among the phytoconstituents, saponin was the main component. Moreover, studies have shown the antibacterial activity of leaf extracts [49, 50]. The phytochemical studies conducted on the fruits have reported the presence of triterpenoids, flavonoids, tannins, and saponins which showed antioxidant activities [12, 46]. Nonetheless, methanolic extracts of leaves and fruits have proven ferric reducing antioxidant power (FRAP). Furthermore, it has been reported that methanolic extracts have more capacity than aqueous extracts [46, 51]. Besides, for the first time, in C. cauliflora, it has been identified that four types of flavonoid compounds have been present, namely, apigenin 6-C-glucoside (isovitexin), apigenin 8-C-glucoside (vitexin), taxifolin 3-O-arabinofuranoside, and acacetin 7-O-β-glucoside [51]. In addition, all these compounds have cholinesterase, antioxidant, and α-glucosidase inhibition activities [2].
As natural products have been identified to be suitable for a diverse range of applications including medicinal uses, it has been revealed that constituents of natural products play vital roles in scavenging free radicals thereby, reducing cell damage and inhibiting lipid peroxidation [13]. In addition, it has the ability to act as a natural antioxidant which reduces the risk of aging, inflammation, cardiovascular disease, and cancer. It has been identified that natural products as anticancer compounds and their bioactive constituents are responsible for the cytotoxic properties [6, 13]. Foods rich in antioxidants are considered a good source to act against and prevent many chronic diseases associated with oxidative stress [1, 4, 46, 52]. Therefore, many studies have been conducted to analyze the bioactive properties of C. cauliflora, to understand its activities with health and nutritional benefits [46]. There are some reasons for the extra effects on the activity of the antioxidant properties of natural product extracts. Those are due to the presence of the active groups and their chemical structures. For instance, some bioactive compounds such as simple phenols may not have antioxidant properties due to their chemical conformations. The lack of correlation between bioactive compounds and antioxidant molecular variation is responsible for this [53].
The free radical scavenging effect of methanol, aqueous extract, and EOs of C. cauliflora have shown by using many assays such as 2,2-diphenyl-1-picryl-hydrazyl-hydrate (DPPH) free radical method, total phenolic content (TPC), total flavonoid content (TFC), FRAP assay, oxygen radical absorbance capacity (ORAC), and ferrous ion chelating activities [1, 5, 6, 12, 53]. Nitric oxide (NO) production inhibition of C. cauliflora extracts has also been reported with the involvement of xanthine oxidase inhibitory, non-phenolic antioxidants [53]. Nevertheless, it has been reported the antioxidant activity of plant EOs has been present due to the higher presence of oxygenated monoterpenes (α-terpineol and linalool) [53]. Mainly, the terpene family represents the major class of compounds in EOs in plants and it has a low molecular weight (Table 3). Besides, the oil extracts of C. cauliflora have shown free radical scavenging activities from twigs, fruits, and leaves. It has been reported that twig oil scavenging activity is higher than the activities of fruit and leaf oils [6]. The reason for this higher scavenging activity of EOs is due to the C. cauliflora fruit being rich in sesquiterpene alcohols. Moreover, leaf oil is mainly rich in monoterpenes and it is not highly active with less antioxidant activity and also less activity against pathogens [6].
Further, the anti-hyaluronidase activity of C. cauliflora extracts has been reported by Perera et al. [53] due to those extracts acting as inhibitors of hyaluronidase. This particular enzyme plays a vital role as a mediator of inflammation conditions, as well as several physiological and pathological conditions. Moreover, C. cauliflora extracts showed this ability due to the presence of polyphenolics and flavonoids. Further, many natural plant extracts have revealed hyaluronidase inhibition due to the presence of phenolic acids and flavonoids [53, 54]. Additionally, the xanthine oxidase inhibitory activity of C. cauliflora extracts has also been reported and it shows xanthine oxidase enzyme catalyzes inhibition with the interference of no phenolic antioxidants [53].
The cytotoxicity activity of C. cauliflora has been evaluated and all studies have found that the plant extracts are non-toxic. This property has been further verified by the NO production inhibitory assay [6, 13, 53, 55].
The antimicrobial activities of C. cauliflora extracts have been analyzed in different ways and are shown in summary in Table 6. The EOs extracted from the leaf, twig, and fruit have revealed a wide range of effects on bacteria (Table 6). It has been further reported that fruit oil shows positive responses only against gram-positive bacteria such as Staphylococcus aureus and methicillin-resistant S. aureus (MRSA) [6]. However, oil extracts from leaves did not show any response against microorganisms. Further, minimum inhibitory concentration (MIC) and minimum bactericidal concentration (MBC)/minimum fungicidal concentration (MFC) assay studies have proved the response of twig oil against microorganisms and it depends on the concentration of the EOs used [6]. The presence of triterpenoids, tannins, flavonoids, quinones, and saponins is responsible for strong activity against antimicrobial effects [50]. Those compounds have strong inhibitory power on microorganisms. Additionally, some studies have reported antimicrobial effects against Porphyromonas gingivalis, Staphylococcus epidermidis, S. aureus, and Pseudomonas aeuruginosa bacteria. However, the methanolic extract has reported less antimicrobial activity on Escherichia coli [47, 50]. This is due to the different activities and their bioactive molecule involvement in different and diverse mechanisms of microorganisms. For instance, flavonoids are polyphenolic compounds that are low molecular mass bioactive molecules [50, 53] and are considered strong antibacterial compounds as they have the ability to make complex compounds with the involvements of extracellular proteins. These properties also support the damage to the cell membranes of the bacteria. Moreover, flavonoids are able to inhibit cytochrome c reductase enzymes that use oxygen inside the bacteria cell and inhibit internal oxygen usage. In addition, flavonoids have the ability to inhibit the formation of DNA and RNA in bacteria and further, their effects on the cell wall permeability of the bacteria cells [50]. Additionally, antiviral activities of C. cauliflora extracts have been reported due to the presence of terpenoids and tannins, while they inhibit the microorganism metabolism and enzymes [6, 48]. Nonetheless, Sumarlin et al. [47] reported that a combination of two materials such as flavonoids, vitamin C, β-carotene, and total phenolic with methanolic extracts of C. cauliflora have stronger effects as antioxidants and antibacterial sources. Further, they have reported that the use of C. cauliflora extracts in a single form or combination would help enhance antioxidants and antibacterial activities [47]. Therefore, these findings are crucial for the use of C. cauliflora extracts as the source for the production of functional foods [47].
Types of phytochemicals extracted from different parts of C. cauliflora and its bioactivities against microorganisms
| Part of the tree | Extracted compound(s) | Character of bioactive compounds | Target organism(s) | Reference |
|---|---|---|---|---|
| Leaves | Methanolic extract | Virus | Dengue virus type 2 (DENV-2) | [48] |
| Leaves | Methanolic extract | Gram-negative bacteria | Porphyromonas gingivalis | [50] |
| Leaves | Methanolic extract | Gram-positive bacteria | S. aureus | [49] |
| MRSA | ||||
| Streptococcus mutans | ||||
| Streptococcus pyogenes | ||||
| Gram-negative bacteria | Shigella sonnei | |||
| Klebsiella pneumoniae | ||||
| Vibrio cholerae | ||||
| E. coli | ||||
| Leaves | Methanolic extract | Virus | Herpes simplex virus type-1 (HSV-1) | [14] |
| Fruit | Chloroform extract | Fungus | Candida albicans | [13] |
| Candida parapsilosis | ||||
| Cryptococcus neoformans | ||||
| Ethyl acetate extract | Fungus | C. albicans | ||
| C. parapsilosis | ||||
| C. neoformans | ||||
| Trichophyton interdigitale | ||||
| Ethanol | Fungus | C. albicans | ||
| C. neoformans | ||||
| Methanolic extract | ||||
| Water | ||||
| Fruit oil | EOs | Gram-positive bacteria | S. aureus | [6] |
| MRSA | ||||
| Twigs | EOs | Gram-positive bacteria | Bacillus cereus | [6] |
| Bacillus subtilis | ||||
| S. aureus | ||||
| MRSA | ||||
| Gram-negative bacteria | Proteus mirabilis | |||
| E. coli | ||||
| K. pneumoniae | ||||
| Yeast | Candida utilis | |||
| C. albicans |
Health benefits and medicinal properties
Studies have reported that C. cauliflora leaves are used traditionally as a treatment for diabetes and hyperlipidemia, while the fruits are used as a cure for appetite loss [9, 11, 47, 48, 56]. The study conducted using the C. cauliflora leaf extracts by Sumarlin et al. [47] reported its anti-diabetic potential, especially for type 2 diabetes mellitus. Also, leaf methanolic extracts showed inhibition activity against AChE, α-glucosidase, and tyrosinase enzymes, whereas seed oil is used for curing skin diseases [1, 6, 7, 9, 11, 41]. Traditionally, other parts of the plant such as the stem, bark, and root are reported to have medicinal properties for cancer and blood diseases [11, 41]. Additionally, some traditional approaches have been reported such as its peel being boiled in water to cure swelling and to treat diabetes, and the root being boiled in water to cure malaria [56].
The methanolic extracts of C. cauliflora whole fruit showed higher cytotoxic activity on human promyelocytic leukemia HL-60 cells by inhibiting cell proliferation. However, cytotoxicity towards 3T3/NIH cells was reported with less activity [56]. Cytotoxic activity of C. cauliflora extract induces cell death by apoptosis. Moreover, mice models have shown the effectiveness of C. cauliflora extracts on WEHI-3B cells by in vivo antileukemic activity of the extract [3, 55, 56]. In addition, a recent study conducted by Samling et al. [6] revealed the use of C. cauliflora leaf, twig, and fruit oils for human breast MCF-7 and MDA-MB-231 cancer cells and human breast MCF-10A normal cells to evaluate anti-proliferative activities. It has been found that twig oil shows strong cytotoxicity against MCF-7 and MCF-10A cells whereas it does not show notable cytotoxicity against MDA-MB-231 cells [6]. However, leaf and fruit oils did not report significant anti-proliferative effects. In addition, some studies have reported that fruit extract has cytotoxicity activities against human promyelocytic leukemia HL-60 cells [43]. It has also been reported that fruits contain a low antioxidant capacity and because of it have a moderate phenolic content [10, 43]. Therefore, these kinds of studies are important in order to extract cytotoxic compounds for the treatment of cancers and the production of drugs for the treatment of cancer patients.
The antibacterial and antiviral properties of C. cauliflora extracts and their vital role as phytochemicals in the inhibition of microorganism metabolisms have proven a wide range of medicinal applications in the pharmaceutical industries [6, 48]. The presence of higher amounts of terpenoids and tannins has proven a strong bioactive potential against the substrates of bacteria and fungi [48]. In addition, it shows a higher healing capacity on scars and wounds. Furthermore, antiviral properties of C. cauliflora extracts also have been reported against DENV-2 [48].
C. cauliflora aqueous extracts have been shown to lower blood glucose in diabetes rat models by inhibiting or suppressing the free radicals by alloxan, which is an organic compound that can selectively demolish insulin-producing cells in the pancreas and inhibit lipid peroxidation [4]. Further, the hypoglycemic and hypolipidemic potentials of C. cauliflora extracts have been revealed to contain antioxidant and free radical scavenging properties [4]. Nonetheless, other observations made on obese animals have revealed its ability to reduce their body weight by reducing low-density lipoprotein (LDL) cholesterol and serum total cholesterol together with serum lipase and triglycerides [11]. All these medicinal activities are reported due to the presence of bioactive compounds, mainly the presence of phenolic compounds rich in hydroxyl groups [1, 14]. Among them, the oligostilbenoid groups have been identified as the most active groups (Figure 15), and those have displayed biological activities such as antioxidant, antibacterial, antiviral, anti-HIV, and cytotoxic properties [5, 10, 12]. Usually, phenolic compounds with their active groups are able to act as single scavengers and as reducing agents [1]. In addition, the activity against HSV-1 infection has been reported with the extracts of C. cauliflora and it shows less and moderate impacts [12]. This is because of the presence of plant alkaloids in the C. cauliflora extracts and it has the ability to reduce the interactions with active compounds. Further, it has been found that the presence of antiviral compounds with non-cytotoxic dilution of the extract acts as a prophylactic agent against HSV-1 infections [14]. Consequently, potential applications of phenolic compounds rich in the fruit of C. cauliflora have revealed health benefits and nutritional benefits for humans [1].
Molecular docking and MD simulations analysis
In this study, assessing the health benefits of phytochemicals from the underutilized tropical fruit C. cauliflora against AD, a comprehensive in silico investigation was conducted. Initially, docking simulations revealed that among the plant-based compounds examined, K-3-Rh and β-sitosterol exhibited the highest binding affinities to AChE at –44.5 kJ mol–1 and –44.9 kJ mol–1, respectively. However, the synthetic reference molecule H0L displayed superior binding affinity at –55.8 kJ mol–1. Procyanidin tetramer also demonstrated notable binding affinity despite not adhering to Lipinski’s rules. These results, expressed as grid scores, represent the approximate binding energy, encompassing both electrostatic and van der Waals energies [28, 44].
Furthermore, this investigation involved MD simulations, with a focus on stability and dynamic interactions. The RMSD analysis confirmed the structural stability of ligand-receptor complexes in an aqueous environment. All three systems, including the apoprotein, exhibited minimal conformational changes, with the AChE-K-3-Rh complex closely matching the behavior of the AChE-H0L complex. Moreover, analyses of Rg and RMSF supported the overall stability and compactness of AChE in the presence of these ligands [29–31]. Additionally, SASA analysis hinted at the potential of natural molecules, like K-3-Rh and sitosterol, to mimic the action of synthetic drug molecules against AChE. Finally, the MM-PBSA method allowed us to comprehensively estimate binding affinities, showcasing the potential of plant-based compounds in AD treatment. These findings underscore the promise of phytochemicals from C. cauliflora in AD therapy and warrant further exploration of their therapeutic applications [38, 39].
Conclusion
C. cauliflora plant-based explorations on medicinal approaches have recently become popular and it is a current requirement as many people are combining traditional medicines with new cutting-edge technologies to cure diseases including chronic diseases. Therefore, this information laid the foundation for in-depth studies with in silico studies, molecular docking, and MD simulations in order to understand the behavior of the phytochemicals. Thereby, those studies will be helpful for drug discovery and in other medicinal applications including nutraceuticals and functional foods.
In summary, this study revealed that the binding of K-3-Rh and β-sitosterol acetate to inhibit AChE is significantly similar to the synthetic drug molecule of H0L which is a derivative of donepezil. These findings were verified by the extensive studies carried out using MD simulation. The MM-PBSA analysis clearly revealed that the phytochemical can bind naturally to the AChE in a similar way to how H0L binds to the receptor. The similarities of the binding mode of both natural molecules to the active site of the AChE with H0L indicate that the inhibition action of natural ligands is as effective as the synthetic drug. This insightful finding can be useful for further design of next-generation drugs of natural origin. Further, the AChE enzyme is a better starting model for structure-based drug design, modeling, docking, and MD computations in searching for AD treatments.
Abbreviations
| 3D: |
three-dimensional |
| ACh: |
acetylcholine |
| AChE: |
acetylcholinesterase |
| AD: |
Alzheimer’s disease |
| c.o.m: |
centers of mass |
| Coul-SR: |
Coulombic short-range |
| EO: |
essential oil |
| GROMACS: |
GROningen MAchine for Chemical Simulations |
| H-bond: |
hydrogen bond |
| HSV-1: |
herpes simplex virus type-1 |
| K-3-Rh: |
kaempferol-3-O-rhamnoside |
| LBP: |
ligand binding pocket |
| LJ-SR: |
Lennard-Jones short-range |
| MD: |
molecular dynamics |
| MM-PBSA: |
molecular mechanics Poisson-Boltzmann surface area |
| MRSA: |
methicillin-resistant Staphylococcus aureus |
| PDB: |
protein data bank |
| Rg: |
radius of gyration |
| RMSD: |
root mean square deviation |
| RMSF: |
root mean square fluctuation |
| SASA: |
solvent accessible surface area |
Declarations
Author contributions
JI, HKSdZ, NY, and TCB: Conceptualization, Investigation, Writing—original draft. VYW: Validation, Writing—review & editing, Supervision. All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors declare that there are no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Data from the present manuscript will be made available upon reasonable request.
Funding
This research was funded by the Research for Oracle Program of University of Rajarata, Sri Lanka. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2024.