 Original Article
Original Article

Affiliation:
Laboratory of Food Chemistry and Technology, School of Chemistry, Aristotle University of Thessaloniki, 54121 Thessaloniki, Greece
Email: niknen@chem.auth.gr
ORCID: https://orcid.org/0000-0002-4624-0419
Affiliation:
Laboratory of Food Chemistry and Technology, School of Chemistry, Aristotle University of Thessaloniki, 54121 Thessaloniki, Greece
ORCID: https://orcid.org/0000-0003-1677-8290
Explor Foods Foodomics. 2024;2:326–338 DOI: https://doi.org/10.37349/eff.2024.00040
Received: February 13, 2024 Accepted: March 29, 2024 Published: July 18, 2024
Academic Editor: Luca Rastrellii, University of Salerno, Italy
The article belongs to the special issue Metrological Aspects in the Analysis of Nutrients, Functional Compounds, Additives and Contaminants in Food and Feed
Aim: A protocol relying on quantum chemical calculations to assist prioritization of phenolic compounds as antioxidants in terms of hydrogen atom donation efficiency is presented. The use of reference compounds, an important metrological issue for a future harmonization and standardization of computational approaches in research is also considered.
Methods: A density functional theory (DFT) approach, namely B3LYP/6-311G++(2d,2p)//B3LYP/6-31G in the gas-phase was used for structure optimization, frequency calculation and single point energy (SPE) calculation to obtain the bond dissociation enthalpy (BDE) value of the most active O-H of olive oil phenols. For some of them used as a test set and for comparison, BDE values were calculated using three more approaches, M05-2X in the gas-phase, Becke three-parameter Lee-Yang-Parr (B3LYP) and implicit solvent effects (n-heptane to model bulk oils) with the integral equation formalism version of the polarizable continuum model (IEF-PCM), Minnesota 05 functional with double nonlocal exchange (M05-2X) at a single step using 6-31G+(d) basis set and solvation model density (SMD) as solvation model. Phenol and Trolox were used as reference compounds for ΔBDE calculation.
Results: The proposed protocol was faster by 1.35-, 1.6-, and 8.3-fold respectively than the other three and provided almost the same activity trend application to other type of olive oil phenols indicated that prioritization based on ΔBDE values was in accordance with the limited existing experimental findings in bulk oils, and the order of activity was generally in agreement with the structure-antioxidant activity criteria.
Conclusions: Present findings highlighted the usefulness of quantum chemical calculations as a tool to screen/prioritize molecules with an established structure saving experimental effort and waste production. The expression of results relatively to phenol and Trolox BDE values, may assist comparisons among research findings and facilitate standardization. Based on the findings hydroxytyrosol and related compounds should be efficient hydrogen atom donors compared to other potent virgin olive oil phenols.
In a recent review article [1], we updated the usefulness of the introduction of quantum chemical calculations in the antioxidant activity studies of phenolic compounds as a screening tool for their prioritization. The theoretical approaches in combination with the information derived from the characterization of the compounds present in a food or natural product matrix using high throughput techniques [2, 3] modernizes further the armory of antioxidant activity assessment approaches [4] giving insight into the potential of a phenolic compound to donate electrons or hydrogen atoms [5]. Such a combination assists decisions on fractionation of extracts or the isolation of specific compounds for further testing with in vitro and in vivo protocols [6]. Thus, quantum chemical calculations can serve as a green mid-tool for the prioritization regarding antioxidant behavior before producing experimental data and reducing, thus, the cost in human resources and consumption of chemicals [1]. Nowadays, this is feasible as the improvement of hardware permits to run fast routine calculations for small (< 30 atoms) and medium size (30–50 atoms) molecules [7]. The determination of the same molecular indices that may predict the activity of a phenol can be achieved through various approaches [1, 8]. These may include semi-empirical, ab initio, mixed mode, or multilevel (different basis sets) procedures, solvation simulation with an array of models, and numerous functionals, all affecting computational time and accuracy [1, 8]. So far there is no standardized methodology [1] in literature. Exception to this rule is the Quantum Mechanics-based test for Overall Free Radical Scavenging Activity (QM-ORSA) introduced by Galano and Alvarez-Idaboy [9] regarding rate constant estimation in solution.
As it is widely accepted among all types of natural antioxidants, the phenolic ones are of particular importance in food, feed, cosmeceutical, pharmaceutical, and medical applications [10, 11]. Phenolic compounds are a diverse group of secondary plant metabolites which are exerting various biological properties, including scavenging of free radicals [12]. The scavenging via hydrogen atom transfer (HAT) is the dominant mechanism of action of phenols in edible fats and oils [13] and can be characterized via the widely accepted calculation of the bond dissociation enthalpy (BDE) value of the most reactive O-H [4, 8].
Considering the above, in the present study a gas-phase multilevel density functional theory (DFT)/Becke three-parameter Lee-Yang-Parr (B3LYP) approach is proposed as a green tool that can serve for screening and ranking phenolic compounds in terms of their ability to donate an O-H hydrogen atom to free radicals. B3LYP was selected as it is considered the most popular functional of choice in many studies including antioxidant activity ones [8]. Calculation in the gas-phase was chosen as a simpler approach and a multilevel strategy employing a low basis set for structure optimization to reduce cost and a higher one for single point calculations to improve accuracy [5]. Taking into account metrological aspects of the computational process, focus was given on the selection and use of reference compounds. The group of phenols examined first (test group-1) were hydroxytyrosol, tyrosol, and four related compounds naturally present in virgin olive oils (Figure 1), for which a health claim has been assigned in the European Union relative to the protection of blood lipids from oxidative stress [14], whereas evidence indicate that they may induce endogenous antioxidant enzymes [15].
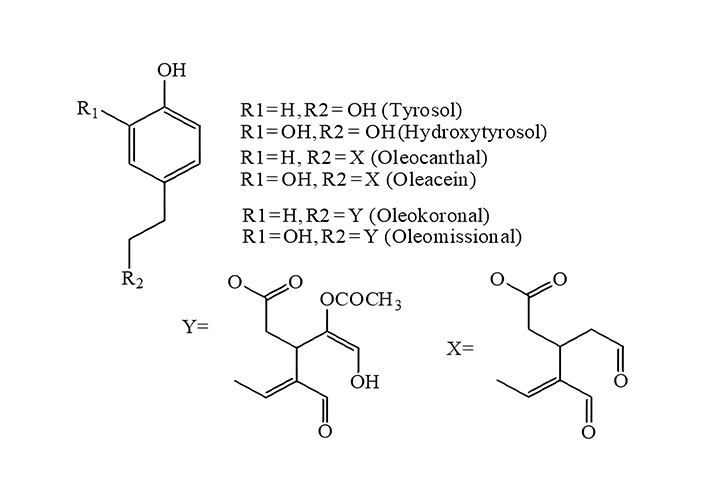
Structures of tyrosol, hydroxytyrosol, and related compounds used as the test group-1 for BDE calculation employing different quantum chemical approaches. BDE: bond dissociation enthalpy
For comparison, the BDE values for these compounds relatively to that of phenol and Trolox were also calculated using three more approaches. These were chosen taking into account some trends in computational studies of antioxidant activity. The proposed gas-phase DFT/B3LYP protocol was then applied to a second group of phenolic compounds (test group-2) widely studied for their antioxidant activity, namely hydroxybenzoic acids (Figure S1) and finally prioritization was sought among the most commonly reported compounds of phenolic nature present in virgin olive oil including hydroxycinnamic acids, secoiridoids, lignans, flavonoids, and hydroxychromans (test group-3) (Figure S2). The proposed approach is relatively fast, requires no strong theoretical background and training, nor too powerful computational resources, being, thus, suitable for experimental researchers to add it to the battery of methods used in the field of antioxidants.
All the calculations were performed by the Gaussian 16-Revision C.02 set of programs [16] using the institutional HPC (high performance computing) infrastructure “Aristotelis” employing 128 cores and 128 GB memory (https://it.auth.gr/en/hpc-en/). For the purpose of the study four different protocols were used to calculate the BDE value of the most active O-H group (C-4) of the tested phenols according to the formula:
BDE = Hr + Hh – Hp [Equation 1]
where Hr is the enthalpy of the radical generated by H-abstraction, Hh is the enthalpy of the H-atom, and Hp is the enthalpy of the parent molecule.
Protocol 1 (Prot 1): The B3LYP exchange correlation potential [17, 18] was used for geometry optimization and computation of harmonic vibrational frequencies using the 6-31G basis set [19, 20] [unrestricted B3LYP (UB3LYP) was used for the resulting radicals]. Then, single point energies (SPEs) were obtained by using the 6-311++G(2d,2p) basis set. The molecular enthalpy in the gas-phase at 298 K for each of the involved species in the calculation of BDE consisted of (U)B3LYP/6-311++G(2d,2p) calculated SPE values and (U)B3LYP/6-31G thermal contributions to enthalpy [TCE, in which the vibrational contributions include zero-point vibrational energy (ZPVE)]. The enthalpy of the H-atom at this level of theory was –0.499897 hartree.
Protocol 2 (Prot 2): The same procedure followed in Prot 1 was adopted using the Minnesota 05 functional with double nonlocal exchange (M05-2X) [21] instead of B3LYP. The enthalpy of the H-atom at this level of theory was –0.496933 hartree.
Protocol 3 (Prot 3): The same procedure followed in Prot 1 was adopted considering also solvation (n-heptane) employing the integral equation formalism version of the polarizable continuum model (IEF-PCM) [22]. The enthalpy of the H-atom at this level of theory was –0.499909 hartree.
Protocol 4 (Prot 4): Τhe M05-2X functional was used for geometry optimization, computation of harmonic vibrational frequencies, TCE and molecular enthalpy using the 6-31+G(d) basis set, whereas solvation (n-heptane) was taken into account employing the solvation model density (SMD) model [23]. The enthalpy of the H-atom at this level of theory was –0.495549 hartree.
Prot 1 was applied to calculate the BDE value of the most active O-H group of the phenolic compounds belonging to all test groups of phenols (1–3), whereas Prot 2–4 were applied for the same purpose only to the compounds of test group-1.
The optimized structures of the test group-1 olive oil phenols in the gas-phase at 6-31G are given in Figure 2. The structures were based on preliminary work and literature search described in the recent review article by Nenadis et al. [1]. The compounds bear an intramolecular hydrogen bond in the catechol group, which is favored for stability purposes, whereas the side chain is located away from the aromatic ring plane for all of the tested compounds [24]. In the same Figure, the structure of phenol and Trolox, the two proposed reference compounds are included as well. In the case of Trolox, the chroman ring is distorted. More specifically the dihedral angle formed by oxygen and the three carbons of the ring is almost 60°. The -OH of the acid is oriented towards the oxygen of the chroman ring to form a hydrogen bond.
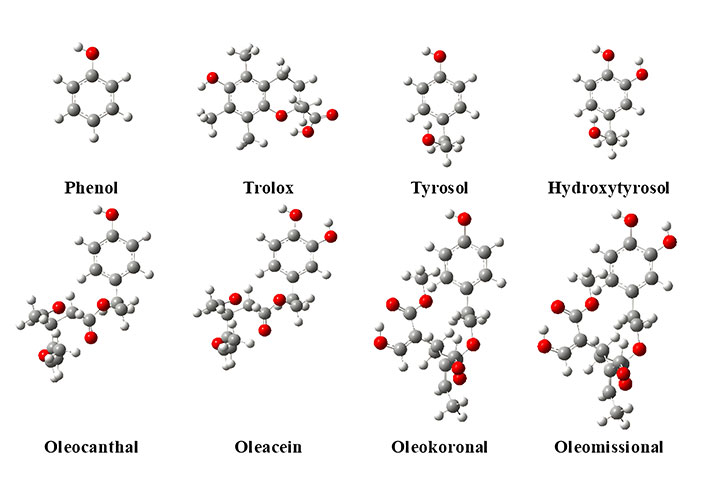
Optimized structures of reference compounds and test group-1 phenols in the gas-phase at B3LYP/6-31G level of theory
In Table 1, certain bond length, angle, and dihedral angle values are given selectively for hydroxytyrosol after optimization in the gas-phase at 6-31G level of theory. In the same Table the relative difference in the corresponding values obtained after optimization using the possible combinations of two different basis sets, DFT functionals, in the gas or liquid phase (employing IEF-PCM or SMD models) are given to examine similarities and discrepancies (for atom numbering see Figure S1).
Selected bond length, bond angle, and dihedral angle values for hydroxytyrosol after optimization with B3LYP or M05-2X, at 6-31G or 6-31G+(d) in the gas or in the liquid (n-heptane) phase employing IEF-PCM or SMD models
| Selected bonds, angles, and dihedral angles of hydroxytyrosol structure | Gas-phase | Liquid phase | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| A* | B** | C** | D** | E** | F** | G** | H** | I** | J** | K** | L** | |
| R11(5,8) | 1.3843 | –0.02 | –0.01 | –0.02 | 0.00 | –0.02 | 0.00 | –0.02 | 0.00 | –0.02 | –0.01 | –0.02 |
| R12(6,7) | 1.4015 | –0.02 | –0.01 | –0.03 | 0.00 | –0.02 | 0.00 | –0.02 | –0.01 | –0.02 | –0.01 | –0.03 |
| R13(7,15) | 0.9740 | –0.01 | –0.01 | –0.01 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | –0.01 |
| R14(8,16) | 0.9801 | –0.01 | –0.01 | –0.01 | 0.00 | –0.01 | 0.00 | –0.01 | 0.00 | –0.01 | 0.00 | –0.01 |
| R18(10,11) | 1.4538 | –0.03 | –0.01 | –0.04 | 0.00 | –0.03 | 0.00 | –0.03 | –0.01 | –0.03 | –0.01 | –0.03 |
| R21(11,21) | 0.9798 | –0.01 | –0.01 | –0.01 | 0.00 | –0.01 | 0.00 | –0.01 | –0.01 | –0.01 | 0.00 | –0.01 |
| A14(4,5,8) | 119.983 | –0.26 | 0.26 | 0.12 | –0.02 | –0.32 | 0.00 | –0.31 | 0.23 | –0.31 | 0.27 | 0.06 |
| A17(1,6,7) | 125.377 | –0.51 | –0.08 | –0.53 | 0.06 | –0.47 | 0.00 | –0.52 | 0.01 | –0.52 | –0.07 | –0.50 |
| A18(5,6,7) | 114.206 | 0.87 | –0.09 | 0.69 | 0.06 | 0.93 | 0.07 | 0.97 | –0.05 | 0.97 | –0.02 | 0.77 |
| A19(6,7,15) | 112.713 | –2.09 | 0.94 | –2.01 | 0.21 | –1.97 | 0.08 | –2.07 | 1.12 | –2.07 | 1.01 | –1.88 |
| A20(5,8,16) | 109.469 | –0.93 | 0.81 | –0.90 | 0.01 | –0.83 | 0.12 | –0.74 | 0.83 | –0.74 | 0.91 | –0.80 |
| A21(3,9,10) | 112.673 | 0.22 | –1.51 | –1.65 | 0.07 | 0.36 | 0.25 | 0.55 | –1.51 | 0.55 | –1.30 | –1.65 |
| A27(9,10,11) | 112.378 | 0.71 | –1.06 | –0.49 | 0.04 | 0.70 | 0.24 | 0.91 | –1.05 | 0.91 | –0.87 | –0.53 |
| A33(10,11,21) | 109.143 | –0.95 | 0.87 | –1.13 | –0.01 | –0.96 | –0.05 | –0.99 | 0.79 | –0.99 | 0.77 | –1.15 |
| D17(2,3,9,10) | –84.5004 | –10.72 | –2.03 | –11.18 | –0.86 | –13.04 | –0.72 | –12.04 | –4.05 | –12.04 | –3.07 | –13.15 |
| D35(3,9,10,11) | 62.2989 | 0.28 | –1.34 | –1.65 | 0.43 | 0.77 | 0.85 | 1.07 | –1.02 | 1.07 | –0.67 | –1.37 |
| D44(9,10,11,21) | –62.5456 | 3.53 | –0.23 | 4.49 | 0.70 | 3.58 | 0.45 | 3.20 | 1.81 | 3.20 | 0.57 | 5.21 |
The characters in the first column: R: bonds; A: angles; D: dihedrals. The characters in the header: A: B3LYP/6-31G; B: M05-2X/6-31G; C: B3LYP/6-31+G(d); D: M05-2X/6-31+G(d); E: B3LYP/6-31G (IEF-PCM); F: B3LYP/6-31+G(d) (IEF-PCM); G: M05-2X/6-31G (IEF-PCM); H: M05-2X/6-31+G(d) (IEF-PCM); I: B3LYP/6-31G (SMD); J: B3LYP/6-31+G(d) (SMD); K: M05-2X/6-31G (SMD); L: M05-2X/6-31+G(d) (SMD). * Bond length in angstrom (Å), angle, and dihedral angle in degrees; ** relative difference from the values of column A
The difference in the bond length values was maximum 0.03 Å, that is less than 1.5%. Regarding the bond angle values in most cases the variability was within 1° with few exceptions where values differed by 1.88° to 2.09°, which accounted for a 2% deviation.
Higher differences were evidenced for the dihedral angle (D17) that is the one defined by the side chain and the plane of the aromatic ring. The range of values differed from almost 1° up to 13°. Regardless of the gas or liquid phase model used or even the selected functional, the variability was negligible when the basis set was kept the same. By introducing the diffusion and polarization function to the basis set, the value of the dihedral increased by more than 10°. As a consequence, a small decrease in dipole moment (0.2 D) of hydroxytyrosol was observed and an increase in its polarizability by approx. 20 a.u. (from 100 a.u. to 120 a.u.). At this point it should be highlighted that for the optimized structure of hydroxytyrosol published by Dávalos et al. [24] employing M05-2X at 6-311++G(d,p) in the gas-phase the value of D17, D35, and D44 were 87.5°, 60.8°, and 59.2°, differing by ~3°, 1.2°, and 3° respectively to the corresponding ones obtained in the present study with 6-31G.
The calculation of the BDE values of the most active -OH (the one at C-4) obtained via employing the four selected protocols described in detail in the materials and methods section and expressed relatively to the BDE value of phenol (ΔBDEPH) are given in Figure 3. The relative expression was adopted to rank the compounds and compare findings avoiding discrepancies caused by possible methodological effects on calculated absolute BDE values, as e.g., for Prot 1 the absolute BDE value for phenol in the gas-phase (84.3 kcal/mol) deviated by almost 4.5 kcal/mol from the experimental one [25].
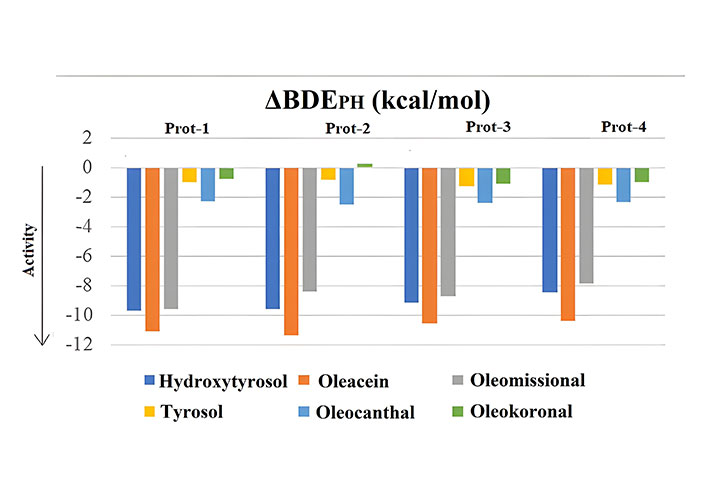
ΔBDEPH values for the test group-1 phenols using different computational approaches (Prot 1–4)
The trend in all approaches (Prot 1–4) was rather the same, although some quantitative differences were evidenced. This was not surprising as solvation is not expected to have a large effect on the hydrogen atom donation of phenols [26]. Thus, the order of activity obtained was oleacein > hydroxytyryrosol ≥ oleomissional > oleocanthal > tyrosol > oleokoronal. However, the time required to obtain the results employing each protocol was different. Using, as an example, the largest of the compounds (oleomissional) of the test group-1, the time required to calculate the BDE value was lower by Prot 1 by 1.35-, 1.6-, and 8.3-fold respectively compared to Prot 2–4. Taking into account phenol as a reference, it is evident that the compounds oleokoronal and tyrosol, were expected to present some weak or even zero hydrogen atom donating efficiency considering that the ΔBDEPH was ~1 kcal/mol. Oleocanthal, though monophenol, without any other substituents in the aromatic ring, except for the side chain, was expected to exert higher hydrogen atom donation potential (ΔBDEPH = –2.5 kcal/mol). Clearly, the compounds bearing a catechol moiety were expected to be efficient as the corresponding ΔBDEPH values were –8 kcal/mol to –11 kcal/mol, verifying that a catechol moiety is a requirement for efficient radical scavenging [13]. In this way it can be assumed that all of the 6 tested compounds may contribute to the oxidative stability of virgin olive oil and to its biological activity [14, 15], but those bearing a catechol moiety should confer the most.
Taking into account Trolox activity (Figure 4), it was evident that the monophenolic compounds were predicted to be poor antioxidants. The trend was the same using all of the 4 approaches.
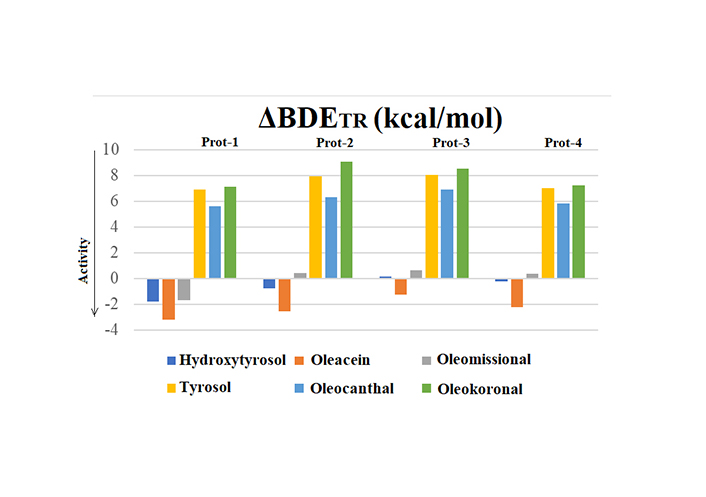
ΔBDETR values for the test group-1 phenols using different computational approaches (1–4)
Regarding the catecholic compounds although the trend was almost similar, there were differences concerning the effectiveness of the compounds compared to Trolox. Oleacein was predicted to be more efficient than Trolox as the ΔBDETR was –1 kcal/mol to –3 kcal/mol under all procedures followed. Hydroxytyrosol was found to be more potent than Trolox using Prot 1, slightly better using Prot 2 and rather of similar activity when liquid phase was taken into account (B3LYP/IEFPCM or M05-2X/SMD). Nevertheless, as the compounds are catecholic ones, and in case HAT is the dominant mechanism, a second hydrogen atom donation may follow from -OH at C-3 [13], as depicted in Figure 5 for hydroxytyrosol. Consequently, the three catecholic compounds should all be more efficient hydrogen atom donors than Trolox.
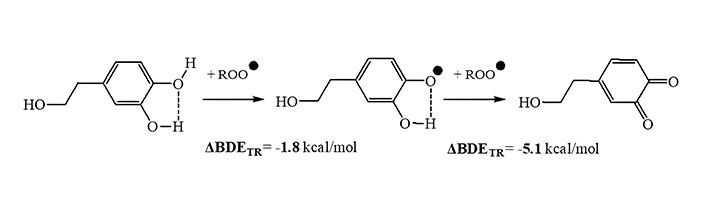
Stepwise hydrogen atom donation to free radicals from hydroxytyrosol at B3LYP/6-311G++(2d,2p)//B3LYP/6-31G level of theory in the gas-phase
In this way the total ΔBDETR value will become even higher if we consider the sum of ΔBDEs from the two steps (–6.9 kcal/mol). Therefore, the importance of the catechol moiety for efficient antioxidant capacity through the HAT mechanism becomes more evident.
The information on the activity of the selected compounds, especially under conditions favouring HAT such as in bulk oils is rather limited. Furthermore, to our knowledge Trolox has not been used in experiments involving bulk oil oxidation [27–31] to verify theoretical findings. Taking into account literature, and particularly the prioritization of certain olive oil phenols reported by Papadopoulos and Boskou [27] under accelerated conditions of oxidation of stripped olive oil (63°C) via hydroperoxide determination, and protection factor (PF) calculation, the high difference in the PF values for tyrosol (1.3) and hydroxytyrosol (9.5) is in accordance with their high difference in the corresponding ΔBDE values (Figures 3 and 4).
For verification of the above observations made for the prioritization among the six compounds of test group-1, calculations were also carried out for another set of compounds (test group-2) consisted of a series of hydroxy-benzoic acids (Figure S2). The ΔBDE values of the most active -OH (the one at C-4) using both phenol and Trolox as references are given in Table 2.
ΔBDE values of test group-2 of phenols at B3LYP/6-311G++(2d,2p)//B3LYP/6-31G level of theory in the gas-phase
| No. | Phenolic compounds | *ΔBDEPH | **ΔBDETR |
|---|---|---|---|
| 1 | 4-Hydroxy-benzoic acid | 3.8 | 11.7 |
| 2 | 3-Hydroxy-benzoic acid | 3.6 | 11.5 |
| 3 | 4-Hydroxy-3-methoxy-benzoic acid | 2.5 | 10.4 |
| 4 | 4-Hydroxy-3,5-dimethoxy-benzoic acid | –3.2 | 4.7 |
| 5 | 3,4-Dihydroxy-benzoic acid | –5.6 | 2.3 |
| 6 | 2,5-Dihydroxy-benzoic acid | –2.7 | 5.2 |
| 7 | 3,4,5,-Trihydroxy-benzoic acid |
*BDE phenol: 84.3 (kcal/mol); **BDE Trolox: 76.4 (kcal/mol). No.: number of phenolic compound (for structures see Figure S2). The compounds were grouped into those that are expected to be inactive (ΔBDEPH values in bold), those that are expected to be less efficient (ΔBDEPH and TR values non-highlighted) or better hydrogen atom donors than Trolox (ΔBDEPH and TR values highlighted in green)
The prioritization of the acids was found to be in accordance with the number and position in the aromatic ring of the -OH groups. The most potent one—as expected—was found to be the trihydroxy benzoic acid, better known as gallic acid. Indeed, it was the only one with a BDE value lower than that of Trolox. The 3,4-dihydroxy one (protocatechuic acid), followed, whereas the 2,5-dihydroxy one was predicted as less active. Finally, those bearing a single -OH group should be expected to be rather inactive due to their higher BDE values than that of phenol (positive ΔBDEPH). Introduction of a methoxy group seems to little affect the activity and the presence of a second one is required for substantial activity. The presence of two -OH at C-2 and C-5 was shown to favour less the activity than when being located at C-3 and C-4. The prioritization of compounds 1 to 5 in terms of ΔBDEPH was in accordance with that found by Bountagkidou et al. [28] upon oxidation of purified olive oil at 120°C using the Rancimat apparatus. Indeed, the compound 3 hardly offered protection, whereas 1 and 2 presented a rather prooxidant activity as evidenced in terms of calculated PF values. The same trend for 1, and 3-5 has been reported by Papadopoulos and Boskou [27] at milder conditions of autoxidation (63°C). The fact that gallic acid (7) is expected to be more efficient than all of the other hydroxybenzoic acids was in line with the findings of Cuvelier et al. [29] who tested various phenols under accelerated conditions using methyl linoleate as an oxidizable matrix (110°C). A discrepancy has been observed regarding the predicted activity of 6, compared to that of 5 and 4 with the experimental findings [efficient quantity (EQ)] of the same authors who reported that 6 was more potent as its EQ value (6.8) was lower than that of 5 (7.5) and even more than that of 4 (61). Such a discrepancy was rather unexpected considering that the hydrogen atom of the -OH at C-2 should be rather blocked due to the formation of an intramolecular hydrogen bond with the carboxylic group attached at C-1 as in the case of salicylic acid [32]. Furthermore, the other -OH at -C-5, is a substituent in m-position, which should not favour hydrogen atom donation [33]. This is an issue that probably needs further theoretical insight into the molecular properties of the particular compound, considering that other researchers have found in sunflower oil under accelerated oxidation conditions (120°C) the better efficiency of 6 compared to 5 and 4 [31].
The gas-phase approach was finally employed to screen the hydrogen atom donating efficiency of a diverse group of phenols identified in the polar fraction of virgin olive oil [1, 34–37]. The group included phenols, beyond those of Figure 1, and namely hydroxycinnamic acids, secoiridoids, lignans, flavonoids, and hydroxychromans (Figure S3). The ΔBDE values using both references are given in Table 3.
ΔBDE values of diverse virgin olive oil phenols calculated at B3LYP/6-311G++(2d,2p)//B3LYP/6-31G level of theory in the gas-phase
| Phenol | *ΔBDEPH | **ΔBDETR | Phenol | *ΔBDEPH | **ΔBDETR |
|---|---|---|---|---|---|
| 1. 4-Hydroxy-phenylacetic acid | 1.1 | 9.0 | 13. Luteolin | ||
| 2. Apigenin | 1.0 | 8.9 | 14. 3,4-Dihydroxy-phenylacetic acid | ||
| 3. Oleocanthalic acid | 0.5 | 8.4 | 15. Oleaceininc acid | ||
| 4. Hydroxy-pinoresinol | –0.3 | 7.6 | 16. 1-(3’-Methoxy-4’-hydroxy) phenyl-6,7-dihydroxy-chroman | ||
| 5. Acetoxy-pinoresinol | –0.4 | 7.5 | 17. Taxifolin | ||
| 6. 4-Hydroxy-cinnamic acid (p-coumaric acid) | –0.5 | 7.4 | 18. 1-Phenyl-6,7-dihydroxy-chroman | ||
| 7. Pinoresinol | –0.7 | 7.2 | 19. Hydroxytyrosol-linolenate | ||
| 8. Ligstroside aglycone monoaldehyde | –0.8 | 7.1 | 20. Hydroxytyrosol-oleate | ||
| 9. Ligstroside aglycone | –0.8 | 7.1 | 21. 3,4-Dihydroxy-cinnamic acid (caffeic acid) | ||
| 10. Tyrosol acetate | –1.2 | 6.7 | 22. Hydroxytyrosol acetate | ||
| 11. 4-Hydroxy-3-methoxy-cinnamic acid (ferulic acid) | –1.2 | 6.7 | 23. Oleuropein aglycone | ||
| 12. 2-Hydroxy-cinnamic acid (o-coumaric acid) | –1.7 | 6.2 | 24. Oleuropein aglycone monoaldehyde |
*BDE phenol: 84.3 (kcal/mol); **BDE Trolox: 76.4 (kcal/mol). The compounds are grouped into those that are expected to be inactive (ΔBDEPH values in bold), those that are expected to be less efficient (ΔBDEPH and TR values non-highlighted) or better hydrogen atom donors than Trolox (ΔBDEPH and TR values highlighted in green)
As evident from the Table 3 those phenols bearing a single phenolic moiety in the aromatic ring (1–3, 8–10, 12) or a methoxy group at C-3 (4–7, 11) were predicted to be poor hydrogen atom donors or even inactive granted that the a ΔBDEPH values were in the range –1.7 kcal/mol up to 1.0 kcal/mol. The compounds bearing a catechol moiety (13–24) were predicted as almost equally efficient or better than Trolox. Considering the variety of structures, it is evident that the crucial structural feature is the presence of a catechol moiety rather than the characteristics of the side chain.
The proposed gas-phase protocol (Prot 1), as well as the one considering the solvation employing the most common used model (IEF-PCM) has been extensively used by our group to elucidate the structure-antioxidant activity relationship of phenolic compounds and shed light on experimental findings, e.g., [26, 32]. Prot 3 is a modification of 1, via replacing B3LYP with M05-2X functional, which is considered advanced [7, 8], whereas Prot 4, comprises the combination of M05-2X, the solvation using the physically complete model SMD [8], and the basis set 6-31G+(d) to cover the minimum requirements proposed by Galano and Alvarez-Idaboy [9] in antioxidant activity studies. The 6-31G selected in our proposal is on purpose a low basis set, although even lower ones could have been selected. As shown in our recent review article [1] the use of 6-31G provides fast results considering that optimization is coupled to frequency calculation that is a time-consuming step. According to Ramachandran et al. [38] acceptable geometry can be obtained even with low basis sets such as 3-21G, whereas more extended ones describe better the energetics of molecular systems. Bearing this in mind, 6-31G could be a practical choice granted that the higher basis set 6-311G++(2d,2p) is proposed in literature for SPE calculations. To its usefulness adds also the fact that the particular basis set provided relatively accurate geometry for the aurone maritimetin and p-hydroxybenzoic acid as evidenced in our past studies [26, 32] and the fact that experts in the field have used it before our group in the highly cited work of Leopoldini et al. [5] to optimize geometries of phenols including, tyrosol and hydroxytyrosol prior to SPE calculations at the B3LYP/6-311++G(3df,2p) level of theory.
Taking into account that the absolute values are affected by the methodology employed, in the present work we introduce two reference compounds for result expression. The first one is phenol, that is expected to be rather inactive, a reference introduced already since many years in the benchmark work of Wright et al. [25]. The second one is Trolox, which is the most common reference in vitro antioxidant activity assays, and is recommended by appropriate bodies for standardization purposes [39]. In addition, it is the reference compound proposed by Galano and Alvarez-Idaboy [9] in their protocol. Our proposal, except for a short of data normalization, may facilitate the grouping of compounds into those that are expected to be inactive or poor, of rather mediocre (lower than Trolox) or very efficient (better than Trolox) hydrogen atom donors.
The prioritization of the compounds in terms of hydrogen atom donation according to the proposed Prot 1 were in general agreement with the principles of the structure-activity relationship set both experimentally and theoretically, e.g., [33] and which were more or less supported by the limited available experimental results derived from bulk oil autoxidation [27–31]. Focusing once again on the latter, it was evident that the presence of a single -OH group did not imply antioxidant activity through hydrogen atom donation. The co-presence of a methoxy group at C-3 was not predicted to significantly improve the hydrogen atom donation, as the OH group forms an intramolecular bond with the particular substituent and thus, a second methoxy group at C-5 is required to make an impact [27–32]. The presence of a carboxyl group directly attached to the aromatic ring should have a negative influence due to the electron withdrawing properties [28, 32]. This is rather attenuated when kept in distance from the ring without an extension of conjugation (e.g., in phenylacetic acids) considering that field/inductive effect has little effect to BDE value compared to resonance one [40]. Thus, the fact that 3,4-dihydroxy phenylacetic acid was much more efficient than the benzoic acid counterpart [27] was justified theoretically. The superiority of 3,4-dihydroxycinnamic acid compared to the benzoic acid one, despite the extension of conjugation that facilitated the negative influence of the carboxylic acid through resonance effect, presents overall a positive impact in activity as shown by the ΔBDE value. Such a result was due to the stabilization of the phenoxy radical achieved through electron delocalization [33]. In the case of hydroxytyrosol and related compounds, since there is no conjugation, and O-H BDE is affected only by local phenomena [25, 40], this may rather justify the similar ΔBDE values obtained. Nonetheless, it should be stated that the differences in the side chain size and characteristics may affect lipophilicity and this could affect performance in systems such as bulk oils [13]. Even so, this was not evidenced experimentally by Gordon et al. [30] for hydroxytyrosol, its acetate and oleuropein aglycone under the oxidation of stripped olive oil triacylglycerols (60°C, peroxide and anisidine value determination), as the three compounds, which differed in their polarity according to experimental logarithm of partition coefficient (LogP) values (0.03, 0.82 and 1.13, respectively), were found to be almost equally effective (PF = 29.8, 28.2, 27.3). All in all, the presence of a catechol moiety or pyrogallol is the feature that promotes the efficient hydrogen atom donation. By the donation of a first hydrogen atom, the radical formed is stabilized through the formation of an intramolecular hydrogen bond, and this stability is expected to be larger when a second intramolecular bond (pyrogallol) can contribute. Such a contribution was clearly reflected in gas-phase BDE values described by Nenadis and Tsimidou [4] for pyrogallol. Beyond these, a second hydrogen atom can be donated as discussed above (see Figure 5).
Present findings highlight the usefulness of quantum chemical calculations as a green tool to screen/prioritize molecules with an established structure. Although the improvement in facilities, such as the availability of HPC infrastructure and appropriate software, may significantly speed up the process (in our case was almost 42-fold faster, compared to the performance of a personal computer with 3 cores and a 900 MB memory in Windows environment) adding to their use for routine analysis, the proposed approach requiring no strong theoretical background and training or powerful computational resources, may be useful for experimental researchers in the field of antioxidants. The expression of results relatively both to phenol and Trolox values, may assist literature data comparisons as the absolute values are affected by the methodology and do not facilitate standardization. Further research may be required combined with experiments to tune the ΔBDE threshold between a poor and a mediocre antioxidant, etc. Regarding the examined olive oil compounds, it was predicted that hydroxytyrosol and related compounds should be efficient hydrogen atom donors compared to other natural molecules bearing catechol/pyrogallol moieties and should contribute the most to virgin olive oil oxidative stability and biological properties, considering their usual concentration in the oil, too.
B3LYP: Becke three-parameter Lee-Yang-Parr
BDE: bond dissociation enthalpy
DFT: density functional theory
HAT: hydrogen atom transfer
HPC: high performance computing
IEF-PCM: integral equation formalism version of the polarizable continuum model
LogP: logarithm of partition coefficient
M05-2X: Minnesota 05 functional with double nonlocal exchange
PF: protection factor
SMD: solvation model density
SPEs: single point energies
TCE: thermal contributions to enthalpy
UB3LYP: unrestricted B3LYP
ZPVE: zero-point vibrational energy
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/101040_sup_1.pdf.
The authors would like to acknowledge the support provided by the IT Center of the Aristotle University of Thessaloniki (AUTh) throughout the progress of this research work.
NN: Conceptualization, Investigation, Validation, Writing—original draft, Writing—review & editing. MZT: Conceptualization, Writing—original draft, Writing—review & editing. Both authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
M.Z.T. undertook this work within the frame of the project Dissemination and further Use of Research Achievements of the Laboratory of Food Chemistry and Technology, School of Chemistry AUTH [AUTH Research Committee code 96677]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Stella A. Ordoudi, Maria Z. Tsimidou
David Heath ... Nives Ogrinc
Alexandros Nakas ... Andreana N. Assimopoulou
Gabriel Mustatea, Elena L. Ungureanu
Florinda Artuso ... Fabio Pollastrone
Sandra Gueifão ... Inês Coelho
Despoina Langari, Fani Th. Mantzouridou
Eleni Zymvrakaki ... Urania Menkissoglu-Spiroudi
Arianna Latini ... Patrizia Galeffi
Aggeliki Kalogeropoulou ... Triantafyllos Albanis
Pierpaolo Di Bitonto ... Sabina Tangaro
Maria Z. Tsimidou ... Claudia Zoani
