Original Article
Original Article

Affiliation:
Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece
Email: aggelkal@chem.auth.gr
ORCID: https://orcid.org/0000-0002-0882-7534
Affiliation:
Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece
ORCID: https://orcid.org/0000-0003-1272-0606
Affiliation:
Department of Chemistry, University of Ioannina, 45110 Ioannina, Greece
Explor Foods Foodomics. 2024;2:767–787 DOI: https://doi.org/10.37349/eff.2024.00061
Received: June 28, 2024 Accepted: November 08, 2024 Published: November 29, 2024
Academic Editor: Claudia Zoani, C.R. Casaccia, Italy
The article belongs to the special issue Metrological Aspects in the Analysis of Nutrients, Functional Compounds, Additives and Contaminants in Food and Feed
Aim: Fast urbanization of free land, industrial progress, and improvement of human life quality, have led to increased consumption of different chemical substances recognized as emerging contaminants (ECs) that are chemicals posing potential risks to the environment and human health, but are not yet subjected to regulatory criteria. Pharmaceuticals consumed by humans are continuously discharged into aquatic environments through urban effluents and numerous classes of them have been widely detected in surface waters. We have to highlight that pharmaceuticals, deposited in the aquatic environment end up in human body through the consumption of marine organisms. Consequently, seawater can be considered as feed. Aim of the study is the development, optimization and validation of a multi-residue analytical method concerning the detection of pharmaceuticals in seawaters.
Methods: The present study describes the development of a highly selective and sensitive analytical method using solid phase extraction (SPE) followed by UHPLC-LTQ/Orbitrap MS for the determination of 18 frequently prescribed pharmaceuticals and 3 transformation products (TPs) in seawater. In order to optimize the extraction method different cartridge types were tested.
Results: Main results showed that the appropriate type of sorbent was concluded to be Oasis HLB, which presented the highest recoveries fluctuating between 61.6% and 118.8%, with a relative standard deviation below 4% and below 5%, for intra-day and inter-day precision, respectively. Limits of detection (LODs) ranged from 0.3 ng L–1 for venlafaxine to 9.8 ng L–1 for oxolinic acid, and the limits of quantification (LOQs) ranged from 1.2 ng L–1 for carbamazepine to 26.4 ng L–1 for oxolinic acid, while in all cases, the linearity, as measured by the correlation coefficient, was greater than 0.991 and ranged within the method’s quantification limit for each chemical and 1,000 ng L–1.
Conclusions: Concerning the conclusions positive detections were found in the seawater samples for oxytetracycline, sulfadiazine, caffeine, paracetamol, and trimethoprim.
The following section refers to the emerging contaminants (ECs), their presence in the aquatic ecosystem as well as its chemical properties, the consequences in human health and their influence in the food chain. Moreover, the multi-residue analytical method is given in brief, the knowledge gaps and the future perspectives. In recent years, an increasing number of scientific studies have focused on environmental pollution. According to the European Union (EU) and the United States Environmental Protection Agency (USEPA) many pharmaceuticals and personal care products (PPCPs) are listed as priority pollutants [1–3]. These chemical substances are recognized as ECs, because of their global use, their physicochemical properties, their incomplete elimination in sewage treatment plants (STPs) and their pseudo-persistent characteristics [1, 3–17]. ECs are pharmaceuticals that are a potential threat to aquatic ecosystems, wildlife and even to human health [18–26]. As a result, long-term attention should be paid to their ability to bioaccumulate in water, sediment, or biota in marine environments, as well as to the toxicity of their transformation products (TPs) [27–29]. ECs, which are deposited in the aquatic environment may end up in the human body through the consumption of the relevant commodities. As a result, sea water can be considered as a possible contributor to food contamination [30]. ECs include several classes of pharmaceuticals such as antibiotics, non-steroidal, anti-inflammatory drugs, β-blockers, antipsychotics, cholesterol-lowering drugs, anticonvulsants, hormones, alkyl phenols, plasticisers, pesticides, PPCPs, phthalates, and artificial sweeteners which are commonly identified in surface waters close to major cities around the world [31–36]. Many PPCPs cause bacterial resistance [10, 37–42], exhibit acute toxicity [10, 43–46] or cause endocrine disruption [47–50]. Pharmaceutically active compounds (PhACs) are highly hydrophilic and have low volatility, so their bioaccumulation might be considered negligible, mainly when compared to other pollutants, such as pesticides and persistent organic compounds (POPs) [27, 51]. Due to their lipophilicity and ability to restrain to organic matter [27–29], these pollutants bioaccumulate in different organisms causing adverse effects in food chain [1, 3, 46, 52–57]. To the best of our knowledge, the fate, occurrence, and toxicity of complex mixtures of pharmaceuticals and their by-products should be further investigated [58, 59]. Furthermore, the growth of dangerous bacteria and algae in water caused by pharmaceutical products can harm aquatic life and render the water unsuitable for human use. Additionally, they can introduce new or higher amounts of different chemicals into the environment, upsetting the aquatic ecosystems’ delicate balance [60]. Certain components of PPCPs are recognized or suspected endocrine disruptors, which can impair living organisms’ hormone systems’ ability to operate normally. Endocrine-disrupting substances can have a harmful effect on an animal’s or an individual’s growth, development, and ability to reproduce.
Recent studies have shown that, approximately 200 pharmaceuticals of several classes have been confirmed in Greece, with concentrations that vary from ng/L to μg/L [16, 37, 61–65]. It is imperative to develop a sensitive and selective multi-residue analytical method in order to detect low concentrations of ECs in complex matrices, such as seawater. Liquid-liquid extraction (LLE) is not preferred to solid phase extraction (SPE), because SPE provides high sensitivity and low cost, using small amounts of solvent [66]. SPE is the most commonly used method for the extraction and pre-concentration of organic pollutants in aqueous matrices [67–70]. Thus, the present study elucidates the development and optimization of sample extraction (SPE) and LC-LIT/Orbitrap MS analysis as well as the method validation of a rigorous, rapid, sensitive, selective, low-cost, and environmentally friendly multi-residue analytical method for the determination of 18 parent pharmaceuticals and 3 TPs in seawater (Table S1), selected according to their detection frequency in surface waters and their persistence in the environment. Selected compounds were extracted by off-line SPE followed by liquid chromatography-high resolution Orbitrap mass spectrometry. HRMS provides accurate and reliable information about the identification and quantification of multiclass pharmaceuticals in seawater samples [71–74]. Finally, the proposed method was applied to real seawater samples. Positive detections were found in the seawater samples for some of the selected pharmaceutical compounds, such as oxytetracycline, sulfadiazine, caffeine, paracetamol, and trimethoprim.
Concerning the “One Health” approach the present study gives the opportunity to highlight the accumulation of pharmaceuticals in marine species via seawater and as a consequence its presence in human body via its consumption and the consequences in the food chain. This monitoring is a very challenging and demanding task due to several factors unique to the marine environment, such as the severe dilution that is occurring, the complex hydrodynamic, and local logistical constraints. As a result, important knowledge gaps are prioritized for further investigation. Suitable passive samplers for tracking seawater levels in pharmaceuticals require more development and standardization. Approaches to non-target analysis hold promise for improving the list of compounds to be analyzed and for comprehending the destiny of the targeted molecules. It would be possible to more accurately determine the ecological risk that these compounds pose to marine ecosystems by implementing integrated monitoring through long-term ecotoxicological testing on vulnerable marine species at environmental levels [75].
A collection of 25 multiclass pharmaceuticals was chosen for the current investigation; Table S1 provides information on the target compounds, including formula, specific physicochemical parameters, and corresponding isotopically labelled internal standards (ILISs). All the selected pharmaceuticals were high-purity pharmaceutical standards (purity > 98%). Citalopram, venlafaxine, and fluoxetine hydrochloride were obtained from TCI (Zwijndrecht, Belgium). O-Desmethyl venlafaxine, norfluoxetine hydrochloride, erythromicin, carbamazepine, N-acetyl sulfamethoxazole, sulfadiazine, sulfamethazine, sulfamethizole, sulfamethoxazole, sulfamethoxypyridazine, sertraline hydrochloride, sulfapyridine, sulfaquionoxaline, sulfathiazole, caffeine, paracetamol, trimethoprim, carbamazepine, carbamazepine-10,11-epoxide, phenazone, oxytetracycline and oxolinic acid were purchased from Sigma-Aldrich (Darmstadt, Germany) and N-desmethyl sertraline hydrochloride was purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Labelled internal standards carbamazepine-d10 and fluoxetine-d5 were obtained from A2S (Saint Jean d’Illac, France). The stock standard solutions were prepared in methanol, at concentrations of approximately 2,000 mg L–1 and 1,000 mg L–1 and were stored at −20°C. By suitably diluting the stock standard solutions in methanol/water 10/90 (v/v), fresh individual and blended composite working solutions were prepared at various concentrations. A distinct mixture of ILISs was prepared in methanol at a concentration of 20 mg L−1, and thereafter diluted accordingly. Fisher Scientific (Leicester, UK) provided the methanol (MeOH) and water used as LC-MS grade solvents, while formic acid (FA) was purchased from Carlo Erba Reagents (Chaussée du Vexin, French) and hydrochloric acid (HCl) fuming 37% for analysis from Merck (Darmstadt, Germany).
Without any pharmaceutical contamination, seawater from an aquafarm in the Ionian Sea close to Ioannina was used for method development. Samples were collected and then transferred to the lab in a transportable freezer. The samples were vacuum-filtered with Millipore (0.45 μm) before analysis to eliminate any suspended solid material, and they were then stored at –20°C until extraction.
Through the use of a standard offline SPE connected to a vacuum pump, selected analytes were isolated and pre-concentrated from water samples. In order to optimize the extraction method, five different types of cartridges were tested, Oasis HLB (200 mg, 6 mL), Oasis MCX (150 mg, 6 mL), C8-CNWBond (500 mg, 6 mL), C18-CNW Bond Elut (500 mg, 6 mL) and Telos Neo PRP (polar-modified reversed phase) (200 mg, 6 mL). Another parameter tested was the pH value, pH 3 and pH 7. Water LC-MS and methanol LC-MS + 2% acetic acid 20:80 (v/v) were examined for the washing step, whereas methanol and methanol + 2% acetic acid were tested as elution solvents. Finally, the highest recoveries were obtained using Oasis HLB cartridges (200 mg, 6 mL), loading 250 mL of sample, washing with water LC-MS and elution with methanol at pH 3 adjusted with HCl 37% 1 M.
Oasis HLB cartridges were preconditioned with 5 mL of methanol and 5 mL of water LC-MS in the optimized method before extraction. Water samples (250 mL) with 5 mL of 5% Na2EDTA and a pH 3 value adjusted with HCl 1 M were passed through the SPE cartridges at a flow rate of roughly 6 mL/min using a vacuum manifold that maintained a constant pressure before the cartridge dried out. Following the percolation of the entire sample, 5 mL of water LC-MS was used to wash the cartridges. Following a 20-min vacuum drying period to eliminate any remaining water, the analytes were eluted using 2 × 5 mL of methanol, by gravity, at 1 mL/min flow rate. At 40°C, methanol extracts were dried off with a light stream of nitrogen, reconstituted in a mixture of 500 μL water/methanol 90:10 (v/v) and stored at −20°C until chromatographic analysis (Figure S1).
Accela UHPLC system (Thermo Fisher Scientific, Bremen, Germany) including Accela quaternary gradient UHPLC pump (model 1.05.0900) and Accela autosampler (model 2.1.1) was used for the LC separation. First, we tested different proportions of the reconstitution solvent, 50-50, 70-30, 80-20, and 90-10 H2O:MeOH (LC-MS). The optimal solvent was determined to be H2O:MeOH (90:10 v/v).
Water (A) and methanol (B), both containing 0.1% FA, made up the mobile phase. A reversed-phase Hypersil Gold C18 analytical column (Thermo Fisher Scientific, Göteborg, Sweden) measuring 100 mm in length, 2.1 mm in diameter, and 1.9 μm in particle size was used for the separation. The column was kept at 35°C for the duration of the analysis. The mobile phase gradient started with 95% mobile phase A and was maintained for 1 min. In the following minute, the amount of mobile phase A was reduced to 30%, reaching 0% in 5 min, and then stabilizing for an additional 2 min. Afterwards, the mobile phase was restored to 95% A and maintained for more than 3 min for re-equilibration. 5 μL was the injection volume that was set, the autosampler tray temperature was stabilized at 20°C and the mobile phase flow rate was stable at 250 μL throughout the analysis.
Thermo Fisher Scientific (Bremen, Germany), provided the hybrid LIT Orbitrap XL Fourier transform mass spectrometer, and an Ion Max Electrospray Ionization (ESI) probe was installed in the hybrid MS system’s linear ion trap. The following ESI settings were used in the positive ionization mode: tube lens, 90 V; sheath and auxiliary gas flow rates, 10 arbitrary units (au); spray voltage, 4 kV; capillary temperature, 320°C; mass range, 120–1,000 m/z; and automatic gain control (AGC) target, 4 × 105. The resolution was 60,000 in full-scan (FS) MS mode, while in the data-dependent MS2 mode (ddMS2), it was 15,000 and the normalized collision energy (NCE) was 30%. Xcalibur 2.2 software (Thermo Electron, San Jose) was used for instrument control and qualitative and quantitative evaluations.
In compliance with the Document No. SANTE/12682/2019, method validation was performed on the optimized method in accordance with the actual guidelines [76]. The method’s accuracy, linearity, precision, limits of detection (LOD) and qualification, matrix effect (ME), and process efficiency (PE) were all assessed.
By fortifying the seawater samples at the start of the extraction and comparing the concentrations obtained after the extraction with the initial spiking level, accuracy—expressed as method recoveries—was assessed at three different concentration levels of 25 ng L−1, 100 ng L−1, and 250 ng L−1 (n = 5). Recoveries were computed using the provided equation (Equation 1):
where Cspiked is the concentration for spiked seawater samples; Creference is the concentration obtained for seawater extracts spiked after the whole process; and Cblank is the concentration of a seawater extract without any spike to confirm the absence of analytes.
Using linear least squares fit for the areas under the peak versus concentrations, a nine-point method calibration curve was constructed for each compound, allowing for the determination of the linearity of the optimized technique in a range from the limits of quantification (LOQ) to almost 100 times the LOQ. The relative standard deviation (%RSDr) of each of the five repetitions on the same day was used to calculate intra-day precision, also known as repeatability. Five days in a row, the same analysis (n = 5) was performed to determine the inter-day precision (reproducibility), which was reported as %RSDR. In a quantitative approach, the mean recoveries range from 70% to 120% with an RSD ≤ 20%, according to SANTE guidelines. If the mean recovery is not less than 30% or more than 140%, with an RSD ≤ 20%, mean recovery values outside of the range of 70–120% may be acceptable in exceptional circumstances.
Using the signal-to-noise ratio (S/N) of 3:1 and 10:1, respectively, the LOD and LOQ were calculated from the extracted ion chromatograms (XICs) at the lowest concentration obtained from the recovery test. To check for variations in the analytical signal, a non-spiked blank sample was injected each time quantification was performed. The values obtained from this sample were then deducted from the values obtained from the spiked samples. A five-point matrix-matched calibration curve in methanol/water extract 10/90 (v/v) was then created using blank seawater sample extracts and injected at the start of each sequence. Every five samples, a solvent was supplied to prevent partial blockage of the ion transfer capillary and a drop in signal intensities.
In order to assess ME, two calibration curves were created: one using seawater extract and the other using a solvent mixture. Both calibration curves were prepared within the same concentration range (2.5–1,000 ng L−1), and a blank extract was used to represent the signal of the non-spiked matrix extract. Calculations were based on the following equation (Equation 2):
According to Scordo et al. [77] (2020), a positive matrix indicating signal enhancement is present when ME > 0, while a negative ME and signal suppression are indicated by values of ME < 0. However, ME% values in the range of ± 20% to ± 50% are regarded as low, medium, and strong, respectively. To reduce the impact of the matrix on the analytes, suitable techniques must be used for values more than ± 20% [78–81]. The following equation (Equation 3) was used to calculate the %PE in order to correlate the ME with the recovered recoveries.
As the PE values are close to 100% indicate that the optimized method is accurate and effective, while recoveries are also close to 100% and ME values are low [81, 82].
First, for the optimization of SPE, five different types of cartridges, Oasis HLB (200 mg, 6 mL), Oasis MCX (150 mg, 6 mL), C8-CN WBond (500 mg, 6 mL), C18-CNW Bond Elut (500 mg, 6 mL), Telos Neo PRP (200 mg, 6 mL) and two different pH values, pH 3 (adjusted with HCl 1 M) and pH 7 (no pH adjustment needed), were tested. Prior to the extraction a conditioning step took place, 5 mL of methanol (LC-MS) followed by 5 mL of water (LC-MS) were added to the cartridges. Then, 250 mL of the sample was loaded into the cartridges at a flow rate of 6 mL/min and washed with 5 mL of water (LC-MS). Afterwards, the cartridges were dried under vacuum for approximately 20 min, and the elution step was carried out using 2 × 5 mL methanol (LC-MS) [19, 52]. The final extracts were evaporated under a gentle stream of nitrogen (N2) until dryness and reconstituted in 500 μL 90/10 water/methanol (LC-MS). In all cases, 5 mL of Na2EDTA 5% solution was added to the samples. Prior to extraction, Na2EDTA is added in order to complex with metal ions and prevent metal interferences [19, 67, 83–85]. Preliminary experiments took place at pH 7 for all cartridges and the two cartridges that gave better recoveries were further investigated at pH 3. Results showed that Oasis HLB (200 mg, 6 mL) gave recoveries from 10% to 180%, Oasis MCX (150 mg, 6 mL) 23.4–99.2% for 13 pharmaceuticals, C8-CNWBond (500 mg, 6 mL) gave recoveries for only 9 pharmaceuticals and values ranged between 4.0–59.4%, C18-CNW Bond Elut (500 mg, 6 mL) recoveries were found at values 4.3–147.0% for 15 pharmaceuticals and Telos neo PRP (200 mg, 6 mL) 10.8–138.2% at pH 7 (Figure 1). In accordance with the results analyzed above, we further examined Oasis HLB and Telos neo PRP at pH 3. Finally, better recoveries were obtained for Oasis HLB cartridges at pH 3, 65.0–110.2%, while recoveries for Telos neo PRP ranged between 11.0% and 122.7% (Figure 2).
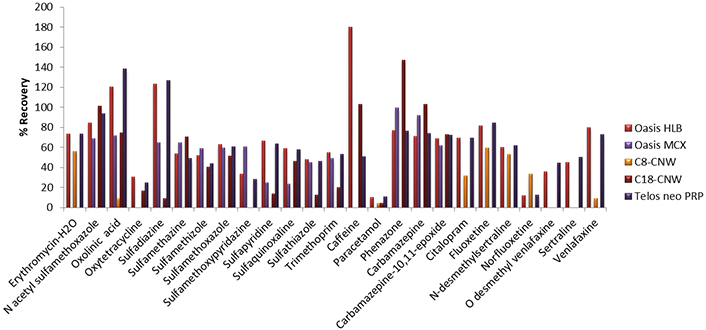
Recoveries obtained with five different cartridges (Oasis HLB, Oasis MCX, C8-CNW, C18-CNW, Telos neo PRP) at pH 7. PRP: polar-modified reversed phase
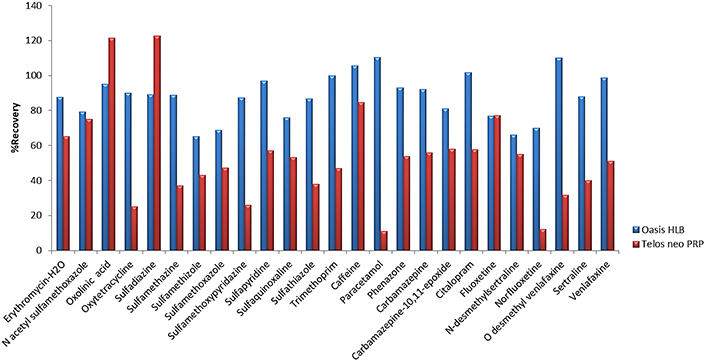
Recoveries obtained with cartridges Oasis HLB and Telos neo PRP cartridges at pH 3. PRP: polar-modified reversed phase
In the current study, all selected pharmaceuticals were detected and quantified with FS mode in positive ionization mode as [M + H]+ adduct ions, along with precise mass spectra obtained at 60,000 FWHM as profile data. The calculated accurate mass, which should match the theoretical accurate mass within a mass tolerance of ± 10 ppm, and the retention time, which needs to match the analyte in the sample within a tolerance of ± 30 sec in the same analytical batch, were used to identify them. MS fragmentation (at least one fragment ion) was used for a more precise identification. First, a mix of standard solutions (50 μg L−1) of the pharmaceuticals analyzed was injected directly into the mass analyzer in FS mode to find the precursor ion, the most abundant fragments and the necessary collision energy (Table 1). A preliminary test to determine the most suitable column was carried out by testing Thermo Scientific, diphenyl (50 mm × 2.1 mm; 2.6 μm) and hypersil gold C18 (100 mm × 2.1 mm; 1.9 μm) from Thermo Scientific. Peak shape and sensitivity were good in both cases, but a slightly better separation of the standards was achieved on the hypersil gold C18 column due to more specific interactions possible between the stationary phase and the analytes. In the present study, two different mobile phases were tested to optimize the separation and detection conditions. It is known that the addition of FA or ammonium formate to the mobile phase is a common practice in positive mode to improve ionization: (1) water and methanol both acidified with 0.1% FA and (2) water and methanol with 0.1% FA and 5 mM ammonium formate. Τhe first combination was selected because of its applicability and effectiveness in multi-residue methods covering a broad spectrum of ECs giving better signal for most of the analytes. Another parameter in the MS is the flow rate. 400 μL min−1 and 250 μL min−1 were tested and the optimal value was found to be 250 μL min−1. Column temperature was also evaluated at 27°C and 35°C, with the latter being the best option. In addition, different solvent compositions (water/methanol 50:50 v/v, methanol/water 10:90 v/v, methanol/water, 10:90 v/v, methanol/water acidified with 0.1% FA, water/methanol 70:30 v/v, and methanol/water 20:80 v/v) were tested as reconstitution solvents for the final extract. Better and sharper peaks were obtained with 10:90 (v/v) methanol/water acidified with 0.1% FA. The high resolution allowed for the separation of peaks that were not fully separated in the chromatographic dimension. Regarding MS conditions, AGC target, tube lens, sheath gas, auxiliary gas and spray voltage were studied as a function of the obtained analytical signal. For AGC target, among the two tested values (4 × 105 and 106), 4 × 105 was selected as the best. Tube lens values of 90 V and 110 V were tested, and 90 V significantly improved the signal. Sheath gas tested values were 38 and 10, with the second one found to be the best, while the auxiliary gas flow studied values of 15, 10 and 10 were selected. Spray voltage values of 3.6 kV and 4 kV were tested and the last one was found to be the most appropriate. As shown in Table 1, at a resolution of 60,000, the mass error was always less than 0.5 ppm. The fact that accurate mass measurements are not influenced by the complex matrix components proves that LTQ Orbitrap MS contributes to the separation of compounds among potential interferences.
Ultrahigh-performance liquid chromatography-LTQ Orbitrap MS parameters
| Pharmaceuticals | IS* | Elemental composition [M + H]+ | tR (min) | Theoritical mass [M + H] | Experimental mass | Mass accuracy (Δppm) | DBE | Fragment ions (NCE 30eV)** |
|---|---|---|---|---|---|---|---|---|
| Antibiotics | ||||||||
| Erythromycin-H2O | CBZ d-10 | C37H66NO12+ | 4.41 | 716,4580 | 716,4598 | 1.767 | 5.5 | 558.3627/540.3530/522.3413 |
| N-Acetyl sulfamethoxazole (M) | CBZ d-10 | C12H14N3O4S+ | 3.96 | 296,0700 | 296,0700 | 0.091 | 7.5 | 188.0822/136.0758/236.0492/198.0222/194.0385/190.0977/202.0975 |
| Oxolinic acid | CBZ d-10 | C13H12NO5+ | 4.11 | 262,0710 | 262,0712 | 1.225 | 8.5 | 244.0607/262.0711 |
| Oxytetracycline*** | CBZ d-10 | C22H25N2O9+ | 3.74 | 461,1555 | 461,1558 | 0.788 | 11.5 | 443.1447/426.1180 |
| Sulfadiazine | CBZ d-10 | C19H11N4O2S+ | 3.46 | 251,0597 | 251,0599 | –1.525 | 7.5 | 156.0114/158.0017/108.0441 |
| Sulfamethazine | CBZ d-10 | C12H15N4O2S+ | 3.74 | 279,0901 | 279,0914 | 1.351 | 7.5 | 186.0335/204.0440/156.0116/124.0869 |
| Sulfamethizole | CBZ d-10 | C9H11N4O2S2+ | 3.71 | 271,0318 | 271,0322 | 1.500 | 6.5 | 156.0117/108.0443 |
| Sulfamethoxazole | CBZ d-10 | C10H12N3O3S+ | 3.84 | 254,0594 | 254,0597 | 0.793 | 6.5 | 156.0115/188.0822/147.0792/190.0977/160.0870/194.0387/108.0442 |
| Sulfamethoxypyridazine | CBZ d-10 | C11H13N4O3S+ | 3.76 | 281,0703 | 281,0706 | 1.290 | 7.5 | 156.0115/188.0127/126.0662/108.0442 |
| Sulfapyridine | CBZ d-10 | C11H12N3O2S+ | 3.59 | 250,0645 | 250,0645 | –0.095 | 7.5 | 156.0115/184.0872/108.0442 |
| Sulfaquinoxaline | CBZ d-10 | C14H13N4O2S+ | 4.09 | 301,0754 | 301,0753 | –0.076 | 10.5 | 156.0115/108.0440 |
| Sulfathiazole | CBZ d-10 | C9H10N3O2S2+ | 3.52 | 256,0209 | 256,0208 | –0.213 | 6.5 | 156.0115/108.0442 |
| Trimethoprim | CBZ d-10 | C14H19N4O3+ | 3.64 | 291,1462 | 291,1450 | –1.329 | 7.5 | 230.1163/261.0983/123.0663 |
| Stimulant | ||||||||
| Caffeine | CBZ d-10 | C8H11N4O2+ | 3.76 | 195,0877 | 195,0879 | 1.783 | 5.5 | 138.0662/133.0858/89.0592 |
| Analgesics | ||||||||
| Paracetamol | CBZ d-10 | C8H10NO2+ | 3.44 | 152,0706 | 152,0705 | –0.691 | 4.5 | 110.0598/134.0601 |
| Phenazone | CBZ d-10 | C11H13N2O+ | 3.91 | 189,1022 | 189,1024 | 0.637 | 6.5 | 161.1073/131.0728/144.0807/146.0838/172.0757/174.0787/130.0649 |
| Anticonvulsants | ||||||||
| Carbamazepine | CBZ d-10 | C15H13N2O+ | 4.51 | 237,1022 | 237,1024 | 0.550 | 10.5 | 194.0965/220.0753 |
| Carbamazepine-10.11-epoxide (M) | CBZ d-10 | C15H13N2O2+ | 4.20 | 253,0972 | 253,0969 | –0.135 | 10.5 | 236.0704/210.0912 |
| Antidepressants | ||||||||
| Citalopram | CBZ d-10 | C20H22FN2O+ | 4.09 | 325,1711 | 325,1710 | –0.240 | 10.5 | 262.1027/280.1131/307.1606/109.0445 |
| Fluoxetine | FXT d-5 | C17H19F3NO+ | 4.54 | 310,1413 | 310,1417 | 1.272 | 7.5 | 148,1123 |
| N-Desmethyl sertraline (M) | CBZ d-10 | C16H16Cl2N+ | 4.75 | 292,0654 | 292,0652 | –0.519 | 8.5 | 275,0000 |
| Norfluoxetine (M) | FXT d-5 | C16H17F3NO+ | 4.54 | 296,1257 | 296,1262 | 1.637 | 7.5 | 134,0000 |
| O-Desmethyl venlafaxine | CBZ d-10 | C16H26NO2+ | 3.81 | 264,1958 | 264,1961 | 0.433 | 4.5 | 246.1857/107.0490 |
| Sertraline | CBZ d-10 | C17H18Cl2N+ | 4.68 | 306,0810 | 306,0809 | –0.397 | 8.5 | 275.0388/129.0689 |
| Venlafaxine | CBZ d-10 | C17H28NO2+ | 4.06 | 278,2115 | 278,2114 | –0.344 | 4.5 | 260.2011/121.0646 |
IS*: internal standards (CBZ d-10: carbamazepine d-10/FXT d-5: fluoxetine d-5); NCE**: normalized collision energy; oxytetracycline*** NCE: 20 eV
The final optimized parameters were as follows: Oasis HLB (200 mg, 6 mL) cartridges, conditioning of sorbent with methanol and water, both LC-MS analysis, wash with water and as elution solvent was used methanol. Then, 250 mL of seawater was filtered, followed by pH adjustment with HCl 37% 1 M at pH 3 and 5 mL of Na2EDTA 5% was added to the sample. Elution extracts were evaporated under a gentle stream of nitrogen until dry and reconstituted with 90:10 water/methanol. The analytical method was validated for its identification and quantification capabilities. All validation performance characteristics are shown in Table 2.
Method validation parameters: linearity, linear range, limits of detection (LODs), limits of quantification (LOQs), mean recoveries (%) and relative standard deviations (RSDs%, n = 5) of three different spiked levels (n = 5) studied for intra-day and inter-day, matrix effect (ME%) and process efficiency (PE%) for pharmaceutical residues in seawater
| Pharmaceuticals | Linearity (r2) | Linear range(ng L–1) | LOD(ng L–1) | LOQ(ng L–1) | 25 ng L–1 | 100 ng L–1 | 250 ng L–1 | 25 ng L–1 | 100 ng L–1 | 250 ng L–1 | ME% | PE% | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Rec (%) | RSDr% | Rec (%) | RSDr% | Rec (%) | RSDr% | Rec (%) | RSDR% | Rec(%) | RSDR% | Rec (%) | RSDR% | |||||||
| Antibiotics | ||||||||||||||||||
| Erythromycin-H2O | 0.9977 | 25–250 | 6.0 | 18.5 | 76.2 | 1.1 | 87.6 | 1.3 | 87.0 | 0.9 | 76.0 | 1.7 | 87.4 | 1.1 | 86.8 | 1.1 | 14.9 | 100.7 |
| N-Acetyl sulfamethoxazole | 0.9989 | 25–250 | 1.8 | 5.3 | 76.4 | 0.8 | 79.2 | 1.7 | 75.6 | 1.3 | 74.0 | 1.4 | 76.8 | 2.1 | 78.2 | 1.9 | 0.6 | 79.6 |
| Oxolinic acid | 0.9976 | 25–250 | 9.8 | 26.4 | 85.2 | 0.9 | 85.0 | 1.2 | 88.6 | 1.3 | 78.6 | 1.5 | 2.0 | 86.9 | ||||
| Oxytetracycline | 0.9996 | 12.5–125 | 5.0 | 16.7 | 81.4 | 1.2 | 90.0 | 0.8 | 86.2 | 1.3 | 77.4 | 1.6 | 86.0 | 1.3 | 82.2 | 1.5 | 9.2 | 98.3 |
| Sulfadiazine | 0.9995 | 25–250 | 2.5 | 8.5 | 86.6 | 1.1 | 89.0 | 1.2 | 76.6 | 1.1 | 82.4 | 1.2 | 94.8 | 2.4 | 82.4 | 1.2 | –18.8 | 72.2 |
| Sulfamethazine | 0.9996 | 25–250 | 2.1 | 6.0 | 81.4 | 1.1 | 88.8 | 1.3 | 86.0 | 1.4 | 78.2 | 2.3 | 85.4 | 2.5 | 82.6 | 1.4 | 5.7 | 93.9 |
| Sulfamethizole | 0.9994 | 25–250 | 2.5 | 6.6 | 61.6 | 1.3 | 65.0 | 1.7 | 62.4 | 1.3 | 62.0 | 1.8 | 65.4 | 2.0 | 62.8 | 2.2 | 5.6 | 68.6 |
| Sulfamethoxazole | 1.0000 | 25–250 | 1.1 | 3.0 | 92.0 | 1.7 | 98.6 | 1.9 | 92.6 | 1.8 | 98.2 | 1.9 | 95.0 | 2.9 | 90.8 | 1.8 | –1.8 | 96.8 |
| Sulfamethoxypyridazine | 0.9997 | 25–250 | 3.4 | 10.3 | 79.4 | 1.2 | 87.2 | 0.8 | 84.2 | 1.2 | 77.4 | 1.4 | 85.0 | 2.9 | 82.0 | 2.1 | 3.1 | 89.9 |
| Sulfapyridine | 0.9995 | 25–250 | 3.0 | 10.0 | 90.2 | 1.1 | 96.8 | 1.5 | 96.2 | 0.9 | 74.8 | 1.4 | 76.6 | 1.5 | 76.0 | 0.9 | 0.9 | 97.7 |
| Sulfaquinoxaline | 0.9907 | 25–250 | 3.3 | 10.0 | 71.2 | 1.0 | 75.8 | 1.7 | 76.2 | 1.1 | 70.4 | 1.3 | 70.0 | 2.4 | 70.4 | 2.4 | 6.0 | 80.3 |
| Sulfathiazole | 0.9970 | 25–250 | 3.0 | 9.3 | 86.8 | 1.1 | 86.6 | 1.5 | 87.0 | 1.5 | 88.4 | 1.2 | 88.2 | 1.9 | 88.6 | 1.7 | –3.1 | 84.5 |
| Trimethoprim | 0.9975 | 50–250 | 5.0 | 18.0 | 98.8 | 0.6 | 101.2 | 1.6 | 98.2 | 2.2 | 92.0 | 1.3 | 93.0 | 3.1 | 95.0 | 4.2 | –0.9 | 100.2 |
| Stimulant | ||||||||||||||||||
| Caffeine | 0.9998 | 50–250 | 3.3 | 10.6 | 85.2 | 1.0 | 105.6 | 0.5 | 110.8 | 0.7 | 95.0 | 1.1 | 115.4 | 1.3 | 118.8 | 1.0 | –0.2 | 105.4 |
| Analgesics | ||||||||||||||||||
| Paracetamol | 0.9962 | 25–250 | 3.6 | 9.8 | 93.4 | 1.3 | 110.2 | 1.0 | 116.2 | 0.5 | 81.8 | 1.4 | 98.6 | 1.5 | 104.6 | 1.1 | –15.8 | 92.8 |
| Phenazone | 0.9998 | 25–250 | 2.5 | 8.3 | 81.6 | 1.1 | 93.0 | 0.9 | 80.8 | 1.0 | 85.4 | 2.1 | 96.8 | 1.7 | 84.4 | 1.0 | –14.4 | 79.6 |
| Anticonvulsants | ||||||||||||||||||
| Carbamazepine | 1.0000 | 25–250 | 0.5 | 1.2 | 92.6 | 1.5 | 92.0 | 1.3 | 87.8 | 0.8 | 91.8 | 1.6 | 91.2 | 1.3 | 87.0 | 1.5 | –0.9 | 91.2 |
| Carbamazepine-10.11-epoxide (M) | 0.9949 | 12.5–125 | 1.5 | 4.5 | 80.0 | 1.6 | 83.2 | 3.9 | 84.0 | 1.2 | 77.4 | 1.3 | 80.4 | 2.2 | 83.2 | 4.8 | –8.3 | 76.3 |
| Antidepressants | ||||||||||||||||||
| Citalopram | 0.9998 | 25–250 | 0.7 | 2.1 | 62.6 | 1.8 | 81.6 | 1.0 | 96.0 | 1.0 | 62.0 | 1.6 | 88.6 | 1.5 | 82.8 | 1.6 | 18.1 | 96.4 |
| Fluoxetine | 0.9944 | 12.5–250 | 1.4 | 4.1 | 84.6 | 0.6 | 86.8 | 1.1 | 82.0 | 1.2 | 85.6 | 1.1 | 87.8 | 2.2 | 83.0 | 1.5 | 14.8 | 99.6 |
| N-Desmethyl sertraline (M) | 0.9942 | 50–500 | 3.1 | 8.5 | 62.0 | 1.8 | 65.4 | 2.6 | 68.8 | 0.9 | 61.8 | 3.2 | 64.0 | 2.6 | 64.4 | 1.8 | –0.9 | 67.8 |
| Norfluoxetine (M) | 0.9925 | 100–500 | 2.2 | 6.0 | 64.6 | 1.7 | 69.8 | 1.2 | 71.6 | 1.6 | 66.6 | 1.7 | 64.8 | 2.0 | 66.4 | 1.7 | –14.1 | 60.0 |
| O-Desmethyl venlafaxine | 0.9960 | 25–250 | 0.8 | 2.0 | 77.0 | 1.4 | 110.0 | 1.3 | 101.0 | 1.0 | 74.4 | 1.9 | 103.0 | 1.3 | 93.8 | 1.4 | 8.7 | 119.6 |
| Sertraline | 0.9987 | 2.5–500 | 1.3 | 4.2 | 85.6 | 1.4 | 86.4 | 1.8 | 87.8 | 1.7 | 82.1 | 1.1 | 85.0 | 2.3 | 87.8 | 1.7 | 0.7 | 82.7 |
| Venlafaxine | 0.9981 | 25–250 | 0.3 | 1.5 | 68.0 | 1.2 | 98.8 | 1.3 | 91.0 | 1.1 | 69.2 | 1.2 | 100.0 | 1.1 | 92.4 | 1.5 | 4.9 | 103.7 |
RSDr repeatability relative standard deviation; RSDR reproducibility relative standard deviation
Three spiking levels (25, 100, and 250 ng L–1) were selected to conduct the recovery experiments. A nine-point matrix-matched calibration curve was used to calculate concentrations in the spiked samples compared with the initial spiking level. Recoveries in all cases, at the intermediate spiking level ranged from 64.8% for norfluoxetine to 115.4% for caffeine, while recoveries for the higher level were found to be between 62.4% for sulfamethizole to 118.8% for caffeine and for the lower level 61.6% for sulfamethizole and 98.2% for sulfamethoxazole. Method precision was calculated as the standard deviation ranging from 0.5% for caffeine and paracetamol to 1.9% for carbamazepine-10,11-epoxide and from 0.9% for sulfapyridine to 4.8% for carbamazepine-10,11-epoxide for intra-day and inter-day, respectively.
Linearity was checked using spiked seawater samples in order to construct a nine-point calibration curve, in a range from LOQ to 1000 ng L–1. The developed method exhibited excellent linearity (r2 > 0.991) for all compounds. The linear range and correlation coefficients for each compound are listed in Table 2.
Method precision was assessed in terms of repeatability (intra-day) and reproducibility (inter-day) at three spiking levels of 25, 100, and 250 ng L–1 (Table 2). This was measured as the standard deviation of the recovery percentages of the spiked samples obtained on the same day (n = 5) and five different days. Low relative standard deviations are crucial because of sensitivity variations in the equipment, even within the same sequence, which may result in variable results.
LOD and quantification were found in nanograms per gram as described previously. LODs ranged from 0.3 ng L–1 for venlafaxine to 9.8 ng L–1 for oxolinic acid, while LOQs from 1.2 ng L–1 for carbamazepine to 26.4 ng L–1 for oxolinic acid. It is noteworthy that in high-resolution MS detectors, the chromatogram may not provide information about the background noise, making the calculation of the S/N difficult. When background noise is not present in the chromatogram, the limits are estimated by injecting decreasing concentrations corresponding to a peak signal value around 104, as recommended elsewhere [33, 86].
The ME was studied as a validation parameter to assess the potential interferences of the matrix components in the ionization. The residual matrix components contained in complex solid matrices typically interfere in the ionization, affect the analytical signal measurements and hence cause errors, leading to inaccurate results. The use of matrix-matched calibration is highly recommended and isotope standards constitute an additional way to minimize ME. Matrix compounds can be eluted at the same retention time as the target ones. A matrix-matched calibration curve from pristine samples was prepared and the slope was compared with the slope of a curve in a solvent mixture. As a result, the ME was calculated using Equation 2. As shown in Table 2 and Figure 3A ME values found to be low, between –18.8% for sulfadiazine to 18.1% for citalopram, indicating a slight ME (lower than ± 20%) [81]. The overall method efficiency was expressed as the PE, correlating the MEs with the recoveries obtained. PE values were calculated using Equation 3. Results revealed that the PE was over 60% for the majority of the compounds analyzed. The highest values were calculated for norfluoxetine, while the lowest were found for caffeine and O-desmethyl venlafaxine (Table 2 and Figure 3B).
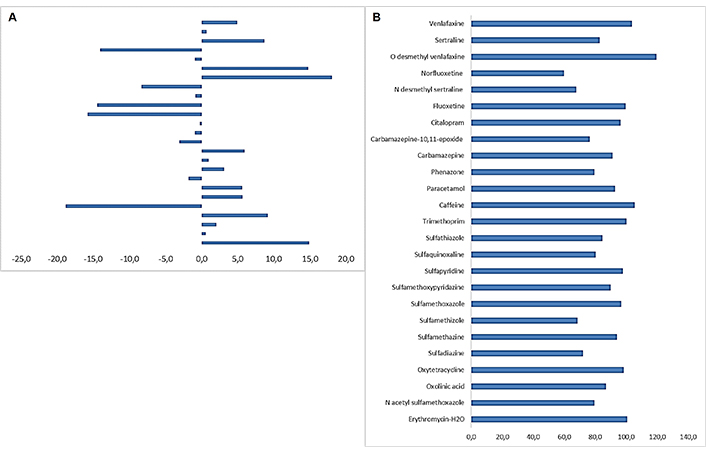
A. Matrix effects calculated from the slopes of the solvent and matrix-matched calibration curves for the pharmaceuticals studied; B. calculated process efficiency (PE%) for the selected compounds
After the development, optimization, and validation of the SPE method for the determination of pharmaceutical compounds, their application to real seawater samples from points within a fish farm located in the Epirus region (Ionian Sea) followed. First, the physicochemical characteristics of the water samples were measured as soon as they were received in the laboratory. The physicochemical characteristics of the samples are listed in Table 3. According to the results listed in Table 4, positive detections were found in the seawater samples for some of the selected pharmaceutical compounds, such as oxytetracycline, sulfadiazine, caffeine, paracetamol, and trimethoprim. In more detail, the table also shows the concentrations for each month of the annual sampling.
Physicochemical characteristics of distilled water and real fish culture samples
| Parameters | Distilled water | July 2020 | August 2020 | September 2020 | October 2020 | November 2020 | December 2020 | January 2021 | February 2021 | March 2021 | April 2021 | May 2021 | June 2021 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Conductivity (mS/cm) | 48.1 | 50.5 | 47.8 | 47.3 | 45.6 | 46.1 | 50.5 | 46.9 | 51.3 | 52.2 | 50.9 | 46.6 | 61.7 |
| TDS (mg L–1) | 80,500 | 89,500 | 85,542 | 84,250 | 77,000 | 85,000 | 91,000 | 86,500 | 84,500 | 91,500 | 84,000 | 80,500 | 90,500 |
| Temperature (oC) | 16.4 | 16.0 | 15.8 | 15.6 | 17.5 | 14.9 | 15.9 | 15.3 | 19.5 | 19.6 | 19.1 | 26.5 | 28.0 |
| Salinity (‰) | 38.5 | 41.1 | 38.9 | 38.7 | 35.3 | 38.2 | 41.2 | 38.9 | 35.9 | 39.0 | 38.4 | 38.8 | 38.8 |
| pH | 6.8 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 5.5 | 6.0 | 6.0 | 6.0 | 6.0 |
TDS: total dissolved solids
Pharmaceutical substances (ng L–1) detected in real fish farm seawater samples during annual sampling
| Compounds | July 2020 | August 2020 | September 2020 | October 2020 | November 2020 | December 2020 | January 2021 | February 2021 | March 2021 | April 2021 | May 2021 | June 2021 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Erythromycin-H2O | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Oxolinc acid | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Oxytetracycline | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | 43,817 | < LOD | < LOD | < LOD | < LOD |
| N-Acetyl sulfamethoxazole (M) | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfadiazine | < LOD | < LOD | 145.4 | < LOD | < LOD | < LOD | < LOD | 1,092 | < LOD | 411.3 | 339.3 | < LOD |
| Sulfamethazine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfamethizole | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfamethoxazole | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfamethoxypyridazine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfapyridine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfaquinoxaline | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sulfathiazole | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Trimethoprim | < LOD | < LOD | < LOD | < LOQ | < LOD | < LOD | < LOD | 32.6 | < LOD | < LOQ | 34.2 | < LOD |
| Caffeine | < LOQ | < LOD | < LOQ | < LOQ | < LOQ | < LOQ | < LOD | < LOD | < LOD | < LOD | < LOQ | < LOQ |
| Paracetamol | < LOD | < LOD | < LOD | < LOD | < LOD | < LOQ | < LOQ | < LOD | < LOD | < LOD | < LOD | < LOD |
| Phenazone | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Carbamazepine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Carbamazepine-10.11-epoxide (Μ) | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Citalopram | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Fluoxetine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| N-Desmethyl sertraline (M) | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Norfluoxetine (M) | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| O-Desmethyl venlafaxine (M) | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Sertraline | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
| Venlafaxine | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD | < LOD |
LOD: limits of detection; LOQ: limits of quantification. Positive findings are given in bold
The pharmaceuticals detected in seawater samples were mainly antibiotics and they presented the highest concentrations during winter months. Specifically, oxytetracycline given the highest concentration (43,817 ng L–1) in seawater samples in February 2021, sulfadiazine was detected in September 2020 (145.5 ng L–1), February 2021 (1,092 ng L–1), April and May 2021 (411.3 ng L–1 and 339.3 ng L–1, respectively), while trimethoprim was found in a low concentration of approximately 32.6 ng L–1 and 34.2 ng L–1 during February and May 2021, respectively. Caffeine and paracetamol were detected at levels below the LOQ values.
However, the distribution of antimicrobial resistance related to antibiotics, needs to be a major priority, in order to preserve the aquaculture environment and guarantee seafood safety.
The occurrence and fate of emerging pollutants in aquatic ecosystems play an intrinsic role in the field of environmental pollution studies. Frequently prescribed pharmaceuticals, belonging to several classes and highly consumed PPCPs are discharged into the marine environment and they are not completely removed from wastewater treatment plants (WWTPs). As a result, parent compounds and their TPs tend to bioaccumulate in aquatic biota. Therefore, there is an urgent need to develop and validate a multi-residue analytical method for the determination of ECs in seawater. Concentration levels ranged from ng/L to μg/L. Therefore, the present study illustrates an off-line SPE method, which has been optimized, validated, and applied in seawater samples from an aquafarm, near the city of Ioannina in order to determine 21 selected pharmaceuticals and their TPs. The SPE method, was developed using high-performance liquid chromatography/Orbitrap (LTQ Orbitrap) mass spectrometry. This study demonstrated the applicability of SPE extraction for organic contaminants as well as the excellent high resolution and accuracy obtained with Orbitrap HRMS. The overall analytical performance was tested in terms of accuracy (trueness and precision), linearity, LOD and quantification, ME and PE. Positive detections were found in the seawater samples for some of the selected pharmaceutical compounds, such as the following: oxytetracycline, sulfadiazine, caffeine, paracetamol, trimethoprim. In the present study, the highest concentration was found in the February sample for the antibiotic oxytetracycline (43,817 ng L–1). Table 5 presents the main characteristics of recent work on surface waters applying the SPE analytical method and analysis of the samples by mass chromatography. In most cases, liquid chromatography coupled with mass chromatography has been applied, while Cao et al. [11], used a triple quadrupole. By comparing these data with the optimized method applied in this work, we noticed that in the majority of cases the aqueous samples were acidified to a pH value of 3. Regarding the recoveries, Asghar et al. [71], found similar recovery values (62.98–105.74%) to the present work, while Chaves et al. [18], Xu et al. [43], Rivera-Jaimes et al. [14], found slightly lower recoveries. All studies found linearity > 0.99, except for Asghar et al. [71], who reported a value > 0.97, and Rivera-Jaimes et al. [14], who reported a linearity value > 0.98. Finally, regarding the limits, LODs and LOQs were found to be lower compared to the present work, except for Chaves et al. [18] and Chitescu et al. [87], who reported much higher values. Compared to the rest of the work, the results of this study appear to be satisfactory.
Analytical characteristics of recent work involving SPE extraction and LC-MS techniques for the analysis of pharmaceuticals in surface waters
| Samples matrix | Compounds | Sample pretreatment | SPE | LC-MS tecnhnique | Recovery (%) | LODs | LOQs | Linearity | References | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Cartridge | Conditioning | Elution | |||||||||
| Seawater | 25 PhACs | pH 3 (HCl) | Oasis HLB (200 mg, 6 mL) | 5 mL MeOH5 mL H2O LC-MS | 2 × 5 mL MeOH | UHPLC-LTQ/Orbitrap MS | 61.6–118.8 | 0.3–9.8 ng L–1 | 1.2–26.4 ng L–1 | 0.9907 | This study |
| Lake water | 13 PPCPs | pH 3 (HCl) | Oasis HLB (500 mg, 6 mL) | 5 mL MeOH5 mL H2O LC-MS | MeOH | UHPLC-QqQ/MS | 79.4–115.5 | - | 0.04–4 ng L–1 | - | [11] |
| Surface water | 168 PhACs | pH 3 (HCl)pH 7 (HCl or NH4OH) | Oasis HLB (200 mg, 6 mL) | 2 × 3 mL MeOH2 × 3 mL H2O (pH 3) | 2 × 2 mL MeOH | LC-MS/MS | 77–117 | < 0.3 ng L–1 | < 0.1 ng L–1 | 0.9900 | [52] |
| River water | 67 PhACs | - | Oasis HLB (200 mg, 6 mL) | 3 mL MeOH3 mL H2O | 2 × 5 mL MeOH | LC-Orbitrap-MS | - | - | - | 0.9900 | [10] |
| River water | 33 PPCPs | pH 7 | Strata-X (200 mg, 6 mL) | 4 mL MeOH4 mL H2O | 4 mL MeOH | LC-MS/MS | 47.9–114 | 1.3–7 ng L–1 | 4–200 ng L–1 | - | [18] |
| Surface water | 31 ECs | pH 3 | Chromabond HR-X (500 mg, 6 mL) | 6 mL MeOH6 mL H2O LC-MS | 2 × 5 mL MeOH | HPLC-MS/MS | 97.1–105.0 | 0.005–2.5 ng L–1 | 0.01–5.0 ng L–1 | 0.9954 | [88] |
| Surface water | 122 PhACs | pH 6–6.2 | Oasis WAX/Oasis HLB/Sep-Pak Plus AC2 | 6–10 mL MeOH6–10 mL H2O ultrapure | Different for each cartridge | UPLC-ESI-TQD-MS/MS | 50–150 | 0.0003–1.757 ng L–1 | 0.010–5.9102 ng L–1 | > 0.9950 | [43] |
| River water | 35 PhACs | - | Oasis HLB | 6 mL MeOH6 mL H2O HPLC | 6 mL MeOH | HPLC-ESI-MS/MS | 3–123 | 0.2–9.4 ng L–1 | 0.5–33 ng L–1 | > 0.9811 | [14] |
| Surface water | 29 PhACs | - | Oasis HLB (200 mg, 6 mL) | 4 mL MeOH6 mL H2O LC-MS | 6 mL 1% NH4OH in MeOH | UHPLC-HRMS | 62.98–105.74 | 0.01–2.5 ng L–1 | 0.03–8.33 ng L–1 | 0.9717 | [71] |
| River water | 67 PhACs | pH 3 (CH3COOH) | Strata-X (200 mg, 6 mL) | 6 mL MeOH6 mL H2O LC-MS | 6 mL MeOH | LC-Q-Orbitrap-HRMS | 85–115 | 0.4–10 ng L–1 | 1.5–55 ng L–1 | 0.9900 | [87] |
ECs: emerging contaminants; ESI: Electrospray Ionization; LODs: limits of detection; LOQ: limits of quantification; PhACs: pharmaceutically active compounds; PPCPs: personal care products; SPE: solid phase extraction
The analysis of real samples in recent works showed the presence of antibiotics in the water samples which is in agreement with the results of the present work. In the present work the highest concentration of oxytetracycline was found in the February sample. However, the presence of antibiotics in fish farms is perfectly reasonable due to their therapeutic use in fish samples. More specifically, Xu et al. [43], detected the antibiotics sulfadiazine and trimethoprim in surface water at concentrations of 0.91 ng L–1 and 1.63 ng L–1, respectively. Chaves et al. [18], and Tran et al. [88], detected paracetamol and caffeine at concentrations of 873.4 ng L–1 and 1,282 ng L–1 and 100.6 ng L–1 and 100.4 ng L–1, respectively. Mhuka et al. [10], detected the substances paracetamol, caffeine, trimethoprim at concentrations of 432.8, 2,979 and 85.32 ng L–1, respectively, while Gros et al. [67], detected paracetamol and trimethoprim in seawater samples at concentrations of 23 ng L–1 and 1 ng L–1. Finally, Asghar et al. [71], in the samples from the Yang and Han rivers and in the lake water from the East Lake detected the substances trimethoprim and caffeine in concentrations of 5.9/32.05 ng L–1 and 8.45/113 ng L–1, respectively.
Monitoring of pollutants in water can reveal details about the destiny of pharmaceuticals in the environment, such as the processes of transformation, partitioning, and transport, in addition to the goals of the monitoring approach. Therefore, additional research on the behavior, destiny, and toxicity of the parent chemicals and TPs is necessary for such inclusion. With the advancement and complementarity of non-target analysis, the incorporation of metabolites and degradation products into monitoring strategies is crucial and feasible, it would allow for the evaluation of their possible combined ecotoxicological consequences. The identification of priority substances would allow for the connection between the occurrence of contaminants and associated ecological responses through integrated monitoring of the environmental matrices carried out concurrently with the measurement of biomarkers of exposure and effects. It would be possible to create environmental quality standards and/or guidelines for medicines in seawater, sediments, and biota if the data regarding the ecological effects of pharmaceuticals present in the examined environment were to expand. Among the short-term steps that have been suggested to reduce the amount of pharmaceuticals that humans, animals, and ecosystems are exposed to include close monitoring of the chemicals’ effects on living things and their environmental presence. Finally, a trade-off must be made between suggested scientific methods and practical and financial limitations.
AGC: automatic gain control
ECs: emerging contaminants
ESI: electrospray ionization
FA: formic acid
LOD: limits of detection
LOQ: limits of quantification
NCE: normalized collision energy
PE: process efficiency
PhACs: pharmaceutically active compounds
PPCPs: personal care products
PRP: polar-modified reversed phase
RSDr: repeatability relative standard deviation
RSDR: reproducibility relative standard deviation
SPE: solid phase extraction
TPs: transformation products
ΜE: matrix effects
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/101061_sup_1.pdf.
This research was co-financed by Greece and the European Union (European Social Fund-ESF) through the Operational Programme «Human Resources Development. Education and Lifelong Learning» in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” (MIS-5000432). Implemented by the State Scholarships Foundation (ΙΚΥ). The authors would like to thank the Unit of Environmental. Organic and Biochemical High-Resolution Analysis-Orbitrap-LC-MS of the University of Ioannina for providing access to the facilities.
AK: Validation, Formal analysis, Investigation, Writing—original draft, Writing—review & editing, Visualization. CK: Methodology, Formal analysis, Visualization, Writing—original draft, Writing—review & editing. TA: Conceptualization, Methodology, Visualization, Supervision, Resources, Funding acquisition.
The authors declare no competing interests.
Not applicable.
Not applicable.
Not applicable.
All relevant data is contained within the manuscript.
This research is co-financed by Greece and the European Union (European Social Fund-ESF) through the Operational Programme “Human Resources Development. Education and Lifelong Learning” in the context of the project “Strengthening Human Resources Research Potential via Doctorate Research” [MIS-5000432]. Implemented by the State Scholarships Foundation (ΙΚΥ). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Authors 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Stella A. Ordoudi, Maria Z. Tsimidou
Nikolaos Nenadis, Maria Z. Tsimidou
David Heath ... Nives Ogrinc
Alexandros Nakas ... Andreana N. Assimopoulou
Gabriel Mustatea, Elena L. Ungureanu
Florinda Artuso ... Fabio Pollastrone
Sandra Gueifão ... Inês Coelho
Despoina Langari, Fani Th. Mantzouridou
Eleni Zymvrakaki ... Urania Menkissoglu-Spiroudi
Arianna Latini ... Patrizia Galeffi
Pierpaolo Di Bitonto ... Sabina Tangaro
Maria Z. Tsimidou ... Claudia Zoani