 Review
Review

Affiliation:
1Specialist Haemostasis Unit, Department of Haematology, Cambridge University Hospitals NHS Foundation Trust, CB2 0QQ Cambridge, UK
2Department of Natural Sciences, Faculty of Science and Technology, Middlesex University, NW4 4BT London, UK
Email: gary.moore@addenbrookes.nhs.uk
ORCID: https://orcid.org/0000-0002-2987-281X
Explor Immunol. 2023;3:300–324 DOI: https://doi.org/10.37349/ei.2023.00104
Received: January 03, 2023 Accepted: March 29, 2023 Published: August 31, 2023
Academic Editor: Xiaofeng Yang, Temple University School of Medicine, United States
The article belongs to the special issue Autoantibodies Associated to Thrombosis and Hemostasis
Accurate lupus anticoagulant (LA) detection is crucial to antiphospholipid syndrome (APS) diagnosis. Detection is based on LA functional behavior in coagulation assays irrespective of epitope specificity. LA screening tests employ dilute phospholipids to accentuate in vitro inhibition by LAs, although they are not LA-specific and can be elevated by other coagulation abnormalities. Elevated screening tests are reflexed to mixing tests to distinguish between factor deficiency and inhibition. Confirmatory tests with high phospholipid concentration swamp LA to generate shorter clotting times than screening tests, whilst prolongation persists with non-phospholipid-dependent inhibitors. LA heterogeneity means that no single screening test detects every LA and the screen/mix/confirm medley must be applied to at least two assay types, usually dilute Russell’s viper venom time (dRVVT) and an LA-sensitive activated partial thromboplastin time (aPTT). Most laboratories restrict LA testing to these two assays, yet others, such as dilute prothrombin time (dPT), can perform with equal diagnostic efficacy, and additionally detect LA unreactive with dRVVT and aPTT. Converting clotting times to normalized ratios improves assay performance, and practitioners must choose between normal pooled plasma (NPP) clotting time denominators to reflect on-the-day assay performance, or reference interval (RI) mean clotting times to negate the effects of NPP variation. Cut-offs can be generated parametrically from normally distributed data, or different percentiles applied depending on the preferred balance between sensitivity and specificity. Sourcing sufficient donors for accurate cut-off estimations is problematic and transference exercises can be undertaken on low donor numbers. Analytical limitations of mixing tests have led to the adoption of alternative algorithms to the screen/mix/confirm test order, whilst some continue to rigidly apply the latter despite those limitations. Strategies to reduce or eliminate the effects of therapeutic anticoagulation have limitations, whilst the Taipan snake venom time (TSVT) screening test with an ecarin time (ET) confirmatory test is insensitive to vitamin K antagonist (VKA) and direct activated factor X anticoagulation.
Antiphospholipid syndrome (APS) is a systemic autoimmune disorder characterized by the non-specific clinical manifestations of arterial or venous thrombosis and/or pregnancy morbidity in patients with persistent antiphospholipid antibodies (aPL) [1]. The term aPL is something of a misnomer in that the antibodies are predominantly directed against phospholipid (PL)-binding proteins in complex with negatively-charged PLs rather than PL per se [2–4]. Although antibodies with numerous antigenic targets have been reported in patients with APS [5, 6], it is only the triumvirate of anticardiolipin antibodies (aCL), anti-β2 glycoprotein I antibodies (aβ2GPI), and lupus anticoagulants (LA) that are employed as criteria antibodies in diagnostic practice [1, 4, 7]. Persistent positivity for more than one criteria antibody has a stronger association with APS, and patients or carriers with the triple-positive profile, comprising concomitant positivity for aCL and aβ2GPI of the same isotype, plus LA, have a high risk of thrombosis recurrence or development of a first thrombosis respectively [7–10]. Of note is that persistent isolated LA is reported as a high-risk profile in some recent studies [11–13].
aCL and aβ2GPI are defined by their antigenic targets, whilst LAs are defined through functional properties irrespective of epitope specificities [4, 14]. LA functional activity is mainly attributed to autoantibodies against β2GPI and prothrombin, each separately contributing to the overall LA activity [14, 15]. Consequently, aCL and aβ2GPI detection is relatively straightforward since they can be detected and quantified with calibrated, isotype-specific, solid phase assays [1, 14], whilst LAs are detected by inference based on their behavior in a medley of coagulation assays [14, 16]. Reliance on coagulation assays to detect a specific abnormality is fraught with difficulties because interpretation is necessarily based on the assumption that no other causes of abnormal clotting times are present, which is not always the case. This is particularly problematic in a patient population where thrombotic manifestations are common as laboratory diagnostics may be initiated only after anticoagulation has commenced [17]. Furthermore, antibody heterogeneity and variability of the assays employed for the detection of LA activity create a situation where no single assay, or assay type, will detect every LA, warranting multiple-assay repertoires in diagnostic practice [7, 14, 16]. Two assays are typically employed in routine practice. One is the common pathway-based dilute Russell’s viper venom time (dRVVT), and the other is the intrinsic pathway-based activated partial thromboplastin time (aPTT) [14, 16], although other assays have been described and are commercially available for LA detection [18]. Guidelines from a number of expert bodies are available for LA testing, and although there is broad harmonization between them, there are areas of contention [16, 19–21]. This article considers different aspects of the analytical processes for identifying the presence of LA and discusses where opinion is divided on what constitutes optimal practice.
LA detection employs a sequence of modifications of each respective parent assay that are designed to exhibit different in vitro properties of LAs, in order that interpretation of the composite can differentiate LA from normality, and non-LA coagulation abnormalities [14]. The principles underlying each modification are depicted in Figure 1.
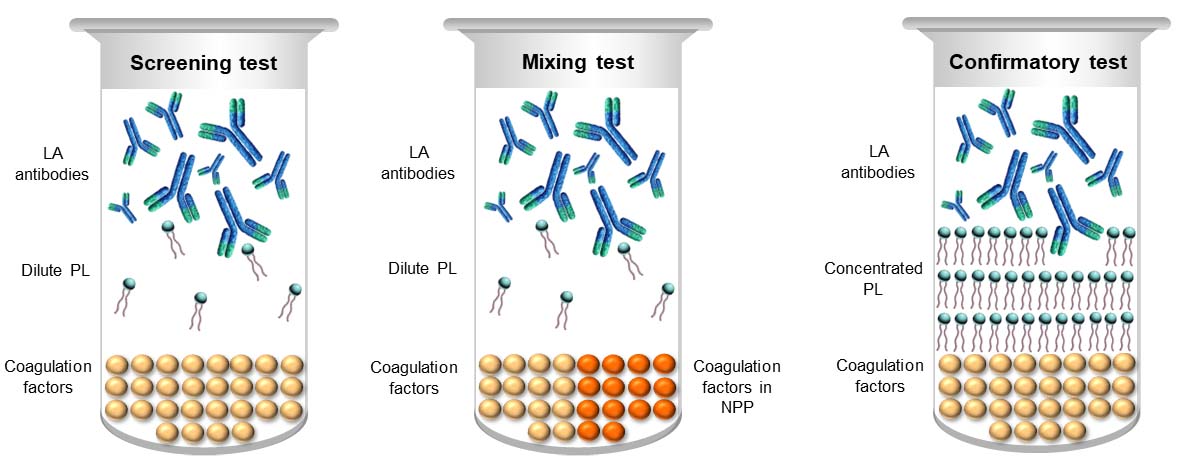
LA test medley. Dilute PL in the screening test accentuates the in vitro inhibitory effect of LA to maximise the chance of realizing an elevated clotting time. Performing the same screening test on an equal volume mixture of test plasma and normal pooled plasma (NPP) assists in distinguishing factor deficiency from inhibition. The NPP can be expected to correct factor deficiencies, thereby returning a normal result, whilst most inhibitors will persist in elevating the clotting time. PL-dependence of the inhibitor is ascertained by recapitulating the screening test but with an excess of PL to overwhelm an LA, thereby shortening the clotting time relative to that of the screening test
The search for an LA begins with the performance of screening tests to detect or refute the presence of an elevated clotting time. A key point here is that although reagents for assays such as dRVVT and certain aPTT formulations are described and marketed as LA assays, none are LA-specific. Thus, an elevated screening test in isolation evidences no more than the presence of one or more causes prolonged clotting times to which a given assay is responsive, and is not in itself diagnostic of the presence of an LA. An elevated screening test is a prompt to initiate subsequent assay modifications in the testing sequence.
LAs are reported to exert their in vitro anticoagulant effect by enhancing the binding of target proteins to negatively charged PL, thereby competing with activated coagulation factors for PL-binding sites [22, 23]. Because of this, the most effective screening tests for LA employ dilute PL to increase competition for limited binding sites, which accentuates the inhibitory effect of any LA present and increases the likelihood of achieving a prolonged clotting time. Sensitivity to LA is not dictated by PL concentration alone, but also the composition of the PL in a reagent in terms of the relative amounts of component PLs [24–26].
Given that dilution of PL enhances screening test sensitivity to LAs, a critical pre-analytical consideration is the avoidance of PL contamination of the plasma to be tested with PL from platelets in the primary sample [27]. Platelet counts in plasma being tested for LA must be < 10 × 109/L, otherwise the residual available PL may provide sufficient additional binding sites for coagulation factors to shorten the clotting time. This is particularly important when the plasma is frozen for inclusion in batched analysis at a later date, which is common practice [28], because the freeze/thaw process ruptures platelet membranes, releasing further PL into the plasma. The greater the degree of contamination, the greater the possibility of shortening what would have been an elevated clotting time into the reference range, because of additional PL and/or quenching of weaker LA, resulting in failure to detect the antibodies. Recommended practice to achieve the requisite platelet count reduction is double centrifugation, where the primary sample is centrifuged, the plasma is carefully removed while keeping the pipette tip some distance from the buffy layer, the supernatant plasma centrifuged in a secondary container, and the plasma carefully separated from any residual cellular material before testing or storage [19–21]. This is a resource-intensive procedure, and whilst ultracentrifugation might appear a tempting alternative, it is not recommended because the severity of centrifugation conditions can generate PL-containing microparticles [19, 29]. Similarly, plasma filtration through 0.22 µm filters will remove all platelets, but is not recommended as it can also generate microparticles, and additionally, may reduce or remove some coagulation factors [19, 20].
LAs are, by definition, in vitro inhibitors, so the traditional response to finding an elevated LA screening test is to perform the same screening test on a 1:1 mixture of test plasma and NPP [14]. The theory behind such mixing tests is that the NPP will replenish any deficient clotting factors such that the mixing test result corrects back to normality, whilst any inhibitor present, whether coagulation-factor specific or PL-dependent, will also inhibit the function of coagulation factors in the NPP and an elevated clotting time will persist, which may not be as marked as when tested undiluted [14, 30].
The combination of elevated screening and mixing tests narrows the interpretation to that of an inhibitor, and it is the confirmatory test that then secures the distinction between factor-specific inhibition and the factor-non-specific/PL-dependent inhibition of LAs [14]. This is undertaken by re-capitulating the screening test in a modification rendering it LA-insensitive, which is usually achieved by concentrating the PL to provide an antigen excess that will sufficiently swamp or overwhelm the LA to reduce or eliminate competition for PL-binding sites. By doing so, the clotting time is shortened relative to the screening test, often but not always returning into the reference range [14, 31, 32]. Non-PL dependent abnormalities, including factor-specific inhibitors, express their elevated clotting times irrespective of PL concentration and can normally be expected to elevate confirmatory tests to a similar or greater degree as that of the screening test. Interestingly, a recent study evidences that whilst prothrombin-directed LA competes with factor (F) Xa for PL-binding sites, inhibition by LA-β2GPI complexes arises from their direct binding to FV, which impedes FXa-dependent activation of FV [15]. Fortunately, the low and high PL screen and confirm assay design remains effective for these antibodies because their inhibition occurs only at limiting PL concentrations, and not at high PL concentrations.
The wealth of evidence that pairing dRVVT with a suitable aPTT has high detection rates validates recommendations in all guidelines, and common international practice, that they should be the first station on the diagnostic journey of LA detection [14, 18–21, 28, 33]. The dilemma stems from whether LA testing should start and end with dRVVT and aPTT, or if there is a place for other assays, at least in certain circumstances [14, 18–21, 34, 35]. The stance for using only dRVVT and aPTT stems mainly from the possibility of an increase in false-positive results as more tests are employed, which has a consequent effect of promoting common practices and reducing inter-laboratory variability [21]. Within this paradigm, it should be recognized that there is a between-reagent variation of dRVVT and aPTT preparations from different manufacturers, particularly aPTT, which can result in some LA being detected by the reagent for a particular assay type from one manufacturing source but not another [35–42]. Variations in reagent performance are mainly due to differences in PL composition [36], although differences can be apparent when using different analyser types [38], and venom heterogeneity can affect dRVVT [39]. The primary roles of aPTT reagents designed for routine coagulation screening are detection of factor deficiencies and heparin monitoring, and the PL component is not necessarily optimized for LA detection. The variable PL composition of routine aPTT reagents results in a continuum of LA-sensitivity [14], and few routine reagents contain diluted PL. LA testing is best done with an aPTT reagent specifically formulated for LA detection, even if it means having a separate reagent for routine coagulation screening.
The counterpoint to the false-positive argument is that LA testing does not stop at the screening test, so any false-positive screening test will be subjected to the rest of the three-step process, where a false-positive interpretation of the composite is less likely [14, 20, 35]. For instance, a factor deficiency or natural statistical outlier would generate an elevated screening test result, yet the mixing test would be normal and the confirmatory test similarly prolonged, so a false-positive composite interpretation would not ensue. Additionally, other assays have been shown to detect small LA sub-populations that do not manifest in dRVVT and aPTT, and can increase detection rates [43–50], although standardization issues exist for some of them. A summary of available LA assays is presented in Table 1.
LA assays
| Types of LA assay/principle | Assays | Non-LA causes of clotting time prolongation |
|---|---|---|
| Based on the intrinsic pathway | LA-sensitive routine aPTT Dilute activated partial thromboplastin time (dAPTT) Kaolin clotting time (KCT) Silica clotting time (SCT) | Isolated or combined deficiencies of coagulation factors II, V, VIII, IX, X, XI, XII, prekallikrein (PK), and high molecular weight kininogen (HMWK) Fibrinogen deficiencies Non-phospholipid-dependent inhibitors Therapeutic anticoagulation with vitamin K antagonist (VKA), unfractionated heparin (UFH), low molecular weight heparin (LMWH), direct thrombin inhibitor (DTI), and direct FXa inhibitor (DFXaI) |
| Based on the extrinsic pathway | Dilute prothrombin time (dPT) Activated Seven Lupus Anticoagulant (ASLA) assay | Isolated or combined deficiencies of coagulation factors II, V, X; also factor VII for dPT Fibrinogen deficiencies Non-phospholipid-dependent inhibitors Therapeutic anticoagulation with VKA, DTI, and DFXaI Therapeutic anticoagulation with UFH & LMWH if neutraliser capacity exceeded |
| Based on the common pathway (FX activation) | dRVVT Vipera lebetina venom time (VLVT) | Isolated or combined deficiencies of coagulation factors II, V, and X Fibrinogen deficiencies Non-phospholipid-dependent inhibitors Therapeutic anticoagulation with VKA, DTI, and DFXaI Therapeutic anticoagulation with UFH & LMWH if neutraliser capacity exceeded |
| Based on the common pathway (FII activation) | Taipan snake venom time (TSVT) Textarin time | Factor II deficiency; also factor V for textarin time Fibrinogen deficiencies Non-phospholipid-dependent inhibitors Therapeutic anticoagulation with UFH, LMWH, and DTI |
Every intrinsic pathway-based LA assay is no more than a modified aPTT, so they are not so many alternative assays as they are additional choices for the aPTT-based assay in a given repertoire. The KCT was once a mainstay since the design of kaolin contact-activation and reliance on residual plasma lipid and cell membrane fragments rather than a PL reagent make it exquisitely sensitive to LA [51, 52]. However, the minimal PL leads to long clotting times and hence poor reproducibility, and the inevitable between-patient variability in residual PL reduces standardization [18]. The turbid and rapid-settling kaolin is incompatible with analyzers employing photo-optical clot detection techniques, although low-opacity analyzer-friendly formulations are available. Crucially, there are no commercially available confirmatory tests for KCT, which compromises specificity, and the dilutions in NPP to evidence inhibition can reduce sensitivity to weaker antibodies [18]. Availability of better-defined aPTT reagents that can be used as screen and confirm pairs have led to a decline in the use of KCT. A solution to the incompatibility with some analyzers is the replacement of kaolin with colloidal silica in the SCT [53], which works well for the intended purpose but does not overcome the other assay limitations. A more analytically robust version of the SCT employs dilute exogenous PL in the screening test and concentrated PL in a paired confirmatory test [54, 55]. Provision of the SCT in that format converts it to a mainstream LA-sensitive aPTT, whereupon it is erroneous to classify it as a separate entity.
Alongside aPTT, the other PL-dependent routine coagulation screening test is the prothrombin time (PT), so perhaps inevitably, diluting the thromboplastin reagent has been employed to screen for LA, in the tissue thromboplastin inhibition test or dPT [56–58]. The use of a lower thromboplastin dilution, or undiluted reagent, operates as the confirmatory test. Initial versions of the assay employed tissue-derived thromboplastins, which exhibited good LA sensitivity, but concerns arose regarding poor reproducibility due to prolonged clotting times and between-reagent variation and compromised specificity arising from poor discrimination between LA and factor deficiencies and VKA therapy [18, 59, 60]. Marked improvements in reproducibility and reagent variation are achieved with recombinant thromboplastins, and specificity is increased with an integrated interpretive approach [43, 61]. Of clinical import is that some studies have evidenced that LAs detected with dPTs employing recombinant thromboplastin exhibit a higher association with thrombosis than those detected by dRVVT and aPTT [58, 62]. The only other extrinsic pathway LA assay that has been described is the ASLA assay, which employs activation of FX by recombinant human FVIIa independently of thromboplastin in screen and confirm reagents with low and high PL concentration respectively [45, 46]. It is sensitive and specific for LAs and detects LAs unreactive with dRVVT and aPTT, but it is not commercially available [45, 46, 63].
Similar to dRVVT, the VLVT employs a snake venom-derived FX activator, from the Levantine or Blunt nosed viper (now classified as Macrovipera lebetina), in screen and confirm reagents with low and high PL concentration respectively [64]. The assay is commercially available and has FDA clearance in the USA, although there are no published peer-reviewed studies to evidence diagnostic equivalence with dRVVT.
Two other common-pathway-based LA screening assays employing snake venom activators and dilute PL have been described, the textarin time [47] and the TSVT [48]. The textarin fraction from the venom of the Australian Eastern Brown Snake (Pseudonaja textilis), and the Oscutarin C fraction from the venom of the Coastal Taipan (Oxyuranus scutellatus scutellatus), directly activate FII in a PL- and Ca2+-dependent manner [65]. Textarin is also FV-dependent, whilst Oscutarin C is not. In place of a high PL concentration confirmatory test, the textarin time was instead paired with the ecarin time (ET) as a confirmatory test [47]. The ET employs the ecarin fraction of Indian saw-scaled viper (Echis carinatus carinatus) venom, a cofactor-independent FII activator [65]. The absence of PL in the assay renders LA incapable of interfering, making it an ideal confirmatory test for FII-activating screening tests. TSVT was first described with a high PL concentration, platelet material-derived confirmatory test, until improved performance with ET as a confirmatory test was evidenced [66], and is now the most commonly used pairing of FII-activating LA assays [14, 18]. ET can also be useful in instances where potent LAs partially or completely overcome the swamping effect of high PL concentration confirmatory reagents used for other assays [18, 67]. A diagnostically valuable commonality of these three venom fractions is that they are insensitive to the effects of VKA therapy because they can activate the resultant undercarboxylated FII, which is advantageous given that all other LA assays are affected by VKAs to varying degrees depending on the dose. Direct FII activation inevitably increases specificity and both textarin time and TSVT are sensitive LA screening tests, additionally capable of detecting LA unreactive with dRVVT and aPTT [38, 47–50, 68–70]. Standardized reagents formulated for LA testing are not commercially available for textarin time, but they are available for TSVT and ET in some countries.
Antibodies undetectable in dRVVT and aPTT can be clinically significant [43–45, 62, 63, 66, 71, 72], yet it is arguable whether every diagnostic facility should implement a repertoire beyond the two mainstay assays. Indeed, some contend that LA testing should remain restricted to dRVVT and aPTT, for the aforementioned reasons of reducing variability and false positivity, despite acceptance of comparable diagnostic strength of other assays [21]. This should be a clinical and scientific concern because, in the absence of reference plasmas and gold standard assays that are truly LA-specific, it is incongruous to acknowledge marked antibody heterogeneity whilst at the same time restricting the tools of diagnostic enquiry. Pragmatically, a middle ground is required in that dRVVT and aPTT will suffice to detect or exclude LA in a majority of patients, whilst assays such as dPT can be added to first-line testing to achieve a full suite of intrinsic/extrinsic/common pathway assays [44, 62], or be employed as second-line assays for patients where clinical suspicion is high but dRVVT and aPTT are negative. Standardized dPT screen and confirm reagents are commercially available [44, 71]. TSVT can be added to second-line testing [49], and be additionally available for testing VKA anticoagulated patients [73]. The obvious venues for extended repertoires are reference laboratories and those with high LA workloads, and local decisions can be made in other facilities whether to embrace wider repertoires or refer appropriate samples to reference centers.
Assays for detecting functional LA activity are coagulation-based, so the analytical end-point is a fibrin clot, expressed as the time in seconds from the addition of the final reagent to clot formation. Expressing routine PT and aPTT results as clotting times is common and familiar practice, which some practitioners prefer to do with LA assays too [36, 40]. This is problematic in LA testing because the crux of LA detection is the discordance between the screen and confirm results that reveal an abnormality to be PL-dependent, whilst they are ostensibly concordant in normality and with non-PL-dependent abnormalities. However, differences in PL concentration between screen and confirm reagents commonly result in different clotting times for a given normal plasma, the dilute PL in screen reagents often generating longer clotting times than for the associated or paired confirm reagent. Consequently, there is a risk of interpreting a screen that is more prolonged than its’ confirm as an LA when it occurs purely as a function of reagent properties and design. The solution, which is a recommended practice in all guidelines, is the conversion of screen, mix, and confirm clotting times to normalized ratios [19–21]. This not only renders screen and confirm results directly comparable, thereby facilitating accurate assessment of concordance/discordance, but it also reduces intra-assay, inter-(same type) assay, and between-laboratory variation [14, 35].
The classical approach to normalization is to employ the clotting time of the NPP analyzed concurrently with test samples as the denominator in the ratio calculation [14, 74]. Normalizing in this way reduces assay performance variations by minimizing the effects of operator variability, analyzer variance, accounting for reagent quality and stability issues, and between-batch variation in NPP clotting time [14, 35, 74, 75]. This approach uses the NPP as a marker of on-the-day analytical system performance, from which the patient result is derived via the ratio calculation. An under-appreciated disadvantage of normalizing against NPP values is that different NPP preparations, or even different batches of the same preparation, do not necessarily generate the same clotting times with a given reagent, or with different reagents of the same test type [20, 76–78]. The greater the distance of the clotting time of a given NPP from the assay and analyser-specific reference interval (RI) mean clotting time, the greater the likelihood of systematic bias towards false-positive or false-negative ratios [20, 74, 77, 78]. In that respect, in the same way that a locally derived, fixed geometric mean normal PT is used in the calculation of the international normalized ratio for VKA monitoring [79], a local reagent and analyser-specific RI mean clotting time is a viable alternative for the denominator in LA assay ratios [14, 20, 28, 70, 75, 77, 80–82]. Whilst this approach circumvents the significant issue of NPP variability, detractors rightly indicate that it does not compensate for day-to-day system variation [21], yet this is easily overcome by analyzing the NPP as an LA-negative control to confirm that assays are operating within acceptable limits, as is done with normal controls for INR and countless other haemostasis assays [14]. Normalization against different denominators will often produce different RIs yet maintain similar diagnostic efficacies [83], but RI viability can be compromised with NPP batch or manufacturer changes if NPP denominators are used, or with reagent batch or manufacturer changes if RI mean clotting time denominators are used [77, 78]. Additionally, mixing tests should employ the clotting time of the undiluted NPP in which the test plasma is mixed as the denominator in order that the mixing test ratio expresses the test result as a function of the plasma in which it is mixed [84].
Well-defined cut-offs for each step in the sequential procedures of LA testing are crucial in facilitating the effective interpretation of the composite. Reagent variation, and differences in performance on different analyzers, mean that common RIs cannot be applied and they should be locally derived, the upper limits being employed as cut-offs [38, 70, 85–87]. As is common for many haemostasis assays, LA assay RIs have historically been derived parametrically from ± 2.0 standard deviations (SDs) from the mean [19, 88]. Parametric evaluation assumes a Gaussian distribution, and whilst LA assay RIs are often reported as such [14, 20, 70, 75, 82, 85, 87], this is not always the case [86]. Population distribution data should be checked for Gaussian distribution by histogram inspection and/or statistical normality calculations, or for whether it can be transformed, before parametric limits are applied [20, 88]. Recent guidelines from the International Society on Thrombosis and Haemostasis (ISTH) recommend blanket use of non-parametric 99th percentile cut-offs in an attempt to reduce false-positives, (mean +2SD equates to 97.5th percentile when data are normally distributed), with the recognition that the resultant increase in specificity is accompanied by a statistically inevitable decrease in sensitivity [21], which has been demonstrated in clinical studies [86, 89, 90]. Of note is that the 99th percentile of normally distributed data equates to mean +2.3SD, so the differences are minimal unless a distribution is markedly skewed. Other expert guidelines, such as from the British Society for Haematology (BSH) and the Clinical and Laboratory Standards Institute (CLSI) are open to parametrically derived RIs [19, 20], so practitioners are free to adopt the approach that matches their preferred balance of sensitivity and specificity. Population distributions must always be checked for outliers by statistical methods such as Reed-Dixon or Tukey before analytical limits are set [21, 87, 88, 91].
Although a potentially viable parametric estimate of an RI can be generated from as few as 39 normal donor plasmas, generating RIs with small sample sizes risks inaccuracies and a minimum of 120 normal donor plasmas are required to generate statistically valid RIs/cut-offs [88]. Sourcing such large donor numbers is difficult for most diagnostic facilities and a more practical alternative is available in the form of a transference validation study of the cut-off or RI from the reagent manufacturer, providing the manufacturer data are appropriately generated [19–21, 88]. Validating the transfer requires as few as 20 donors, or up to 40, who reasonably represent the laboratory’s healthy population and satisfy any exclusion criteria. Once analyzed, the data are checked for outliers within the donor population tested, using statistical methods, and any outliers are replaced with new specimens until a homogenous group without outliers is obtained. If no more than 10% of the results of the outlier-free population are outside the 95% reference limits, the manufacturer’s RI and cut-off can be adopted locally.
A screening test ratio above the locally-derived or validated cut-off indicates an elevated clotting time, warranting initiation of subsequent tests in the medley. Although non-PL-dependent abnormalities are normally expected to realize confirmatory test ratios concordant with that of the screening test, any discordance requires mathematical assessment to evidence that the degree of reduction of the confirmatory test ratio relative to the screening test ratio is due to a pathology and not merely between-assay variance [14, 35]. Confirmation or exclusion of PL-dependence is achieved by calculating the percentage correction of the elevated screening test ratio by the confirmatory test ratio, as follows:
Cut-offs for percentage correction should also be locally derived, and are typically between 10–20% [38, 70, 85–87, 92]. Reagents for dRVVT are commonly available as directly paired screen and confirm assays such that the reagent components are identical other than the PL concentration. Such paired reagents can alternatively be interpreted by dividing the screen ratio by the confirm ratio to generate a third ratio, the normalized LA ratio, which indicates PL-dependence if elevated [21]. Other assays, such as SCT and dPT, are available as directly paired reagents and can be interpreted in this way. TSVT with ET as the confirmatory test can also be interpreted with either percent correction or the normalized LA ratio [70]. Discordance in interpretations for PL-dependence between percentage correction and normalized LA ratio are rare if cut-offs are derived locally [70, 81, 87]. One version of aPTT merely assesses the reduction in clotting time in seconds with the confirmatory reagent with respect to the screening test clotting time [16, 20].
Numerous mathematical models have been used for interpreting routine PT and aPTT mixing tests to distinguish between factor deficiencies and inhibitors, but none are 100% effective [30, 93]. The index of circulating anticoagulant (ICA) calculation, or Rosner index, has been widely used for interpreting LA assay mixing tests [90, 94–96], but recent studies have demonstrated that a mixing test-specific cut-off, ideally expressed as a normalized ratio and not the raw clotting time, is more sensitive to LA-induced inhibition than ICA [97–99]. Mixing test-specific cut-offs for normalized ratios are lower than those for undiluted plasma because mixing normal donor plasmas with NPP compensates for samples with clotting times at the extremes of normality [100]. The most recently published expert guideline, from the ISTH, recommends mixing test-specific cut-off over ICA because it produces less false-negative mixing test results [21].
There is a clear logic to the classical LA algorithm in that the screening test reveals an abnormality through an elevated clotting time, the mixing test then pinpoints the inhibitory nature of the abnormality, and the confirmatory test demonstrates its PL-dependence. This places the mixing test at a crucial diagnostic juncture because a normal result precludes completion of the assay medley, and in the presence of an elevated screening test, potentially initiates a separate search down a diagnostic blind alley. As is known for mixing tests with routine PT and aPTT testing, LA assay mixing tests are not 100% effective in detecting inhibition, because like any haemostasis assay, they have analytical limitations [101–103]. Principal amongst them is the unavoidable dilution effect that can lead to false-negatives, but other factors also contribute to variable reliability of mixing tests, such as reagent sensitivity and specificity, variability in NPP quality and suitability, (i.e. an NPP with a short clotting time requires more potent LA to prolong the result beyond the cut-off), and interpretive inconsistencies [20, 76, 96, 100–103]. Numerous studies have evidenced unacceptably high levels of false-negative mixing studies in LA testing, some concluding that a change in attitude and focus is indicated [14, 19, 20, 35, 76, 81, 96, 100, 103–106], and external quality assurance agencies report false-negative mixing tests as a common cause of misclassification of weaker LA [36, 104, 107]. The BSH, CLSI, and ISTH all acknowledge the potential for false-negative mixing tests, to the point where the BSH and CLSI recommend deviation from the classical algorithm, albeit only in specific circumstances. The BSH guideline states that “Mixing tests are a criterion for LA and improve the specificity. However, they introduce a dilution factor and may make weak LA samples appear negative. In the absence of any other causes of prolonged clotting times, such samples should be considered LA positive if the screen and confirmatory tests on undiluted plasma give positive results.” The CLSI guideline goes one step further and re-prioritizes test order to screen/confirm/mix, the mix only being performed if it can further contribute to unravelling the cause(s) of an elevated screening test, providing a more iterative approach. Criteria for deducing the presence of an LA without a mixing test are as follows:
(i) Elevated LA screening test.
(ii) No evidence for other causes of prolonged clotting times.
(iii) Confirmatory test fulfills mathematical criteria for PL-dependence and is not itself elevated.
A normal routine coagulation screen, which employs an LA-insensitive aPTT, and confirmation that the patient is not anticoagulated, suffice to exclude non-LA causes of elevated LA screening tests. The normal confirmatory test result is essential in the decision to omit the mixing test because it excludes assay-specific -interferences or -causes of an elevated clotting time. An elevated confirmatory test in this scenario can be due to a co-existing abnormality, such as a mild factor deficiency missed in routine coagulation screening, undisclosed anticoagulation, or an LA sufficiently potent to partially or completely resist the overwhelming effect of a high PL concentration confirm reagent [20, 31, 103, 108, 109]. In such situations, a mixing test can be invaluable as it will correct factor deficiencies and the effects of some anticoagulants, and can make PL-dependence more obvious by diluting the antibody titer [103, 109]. In acknowledgment of the occurrence of false-negative mixing tests, the ISTH guideline recommends performance of both mixing test and confirmatory test in response to the finding of an elevated screening test in place of strict adherence to sequential testing [21]. However, advice on the interpretation of an elevated screen corrected by the confirm but negative mix is ambiguous as it merely states to comment on the LA result (providing factor assays are normal), repeat LA testing, and interpret LA results in conjunction with aCL and aβ2GPI results, but without indicating whether the LA results should be reported as positive or negative.
Recognition that LA can be detected without mixing tests has seen a rise in the use of integrated testing, where the paired screen and confirm assays are performed in parallel and directly assessed for concordance or discordance, usually with the normalized LA ratio [80, 81, 109–113]. For most patients among whom other causes of elevated clotting times have been excluded, demonstrating PL-dependence of an elevated screening test is sufficient to conclude the presence of an LA because the mixing test is not required to distinguish between the inhibitor and factor deficiency [20, 103, 104, 110, 111]. Integrated testing has additional value in that it detects weaker antibodies where clotting time elevation above a patient’s baseline is insufficient to exceed the cut-off but screen and confirm discordance reveals the LA [113–115]. Reducing LA detection to a single parameter, the normalized LA ratio, has led to some practitioners jettisoning mixing tests altogether [36, 104, 116, 117], some contending that the screen and confirm discordance accurately detects LA even if the confirmatory test is elevated [80, 118]. Whilst this is certainly true for some patients, integrated testing oversimplifies LA detection in others, particularly where there are co-existing abnormalities, emphasizing that there is not a one-size-fits-all algorithmic solution to LA testing [14, 103]. Performing a mixing test increases specificity, especially in situations where other or co-existent abnormalities are suspected, but they also increase diagnostic certainty and confidence in otherwise straightforward cases if desired. Assays with innately higher specificity tend to require mixing tests less often to conclude the presence of an LA [80, 111]. In more complex cases, performing both screen and confirm assays on 1:1 mixes with NPP further improves specificity because it aids in distinguishing between potent LA, LA + concomitant abnormality, and non-LA abnormality [14, 19, 35, 73, 109, 110]. Some LAs are so potent that testing on undiluted plasma gives concordant screen and confirm results, whereupon dilution of antibody potency in the mixing test permits PL-dependence to manifest, and integrated testing returns a false-negative result [109]. The relatively rare phenomenon of the LA cofactor effect, the paradoxical finding of greater prolongation of the screening test clotting time in the mixing test than when tested undiluted, is inevitably undetectable in integrated testing. It was originally thought to be due to a deficiency of a cofactor necessary for LA to achieve their in vitro anticoagulant effect that is corrected on mixing with normal plasma, the inevitable suspects being prothrombin [119] or β2GPI [120], or alternatively, a prozone effect [121].
Practically speaking, where automated analyzers can automatically perform reflex testing, initiating both the confirmatory test and mixing test(s) in response to an elevated screening test provides maximum information on each patient [14, 19, 21]. If such equipment is unavailable, the screen/confirm/mix sequence is best applied, to avoid the performance of unnecessary mixing tests, providing that criteria for excluding non-LA causes of elevated clotting times can be met. A summary of the classical, iterative, and integrated algorithms is depicted in Figure 2.
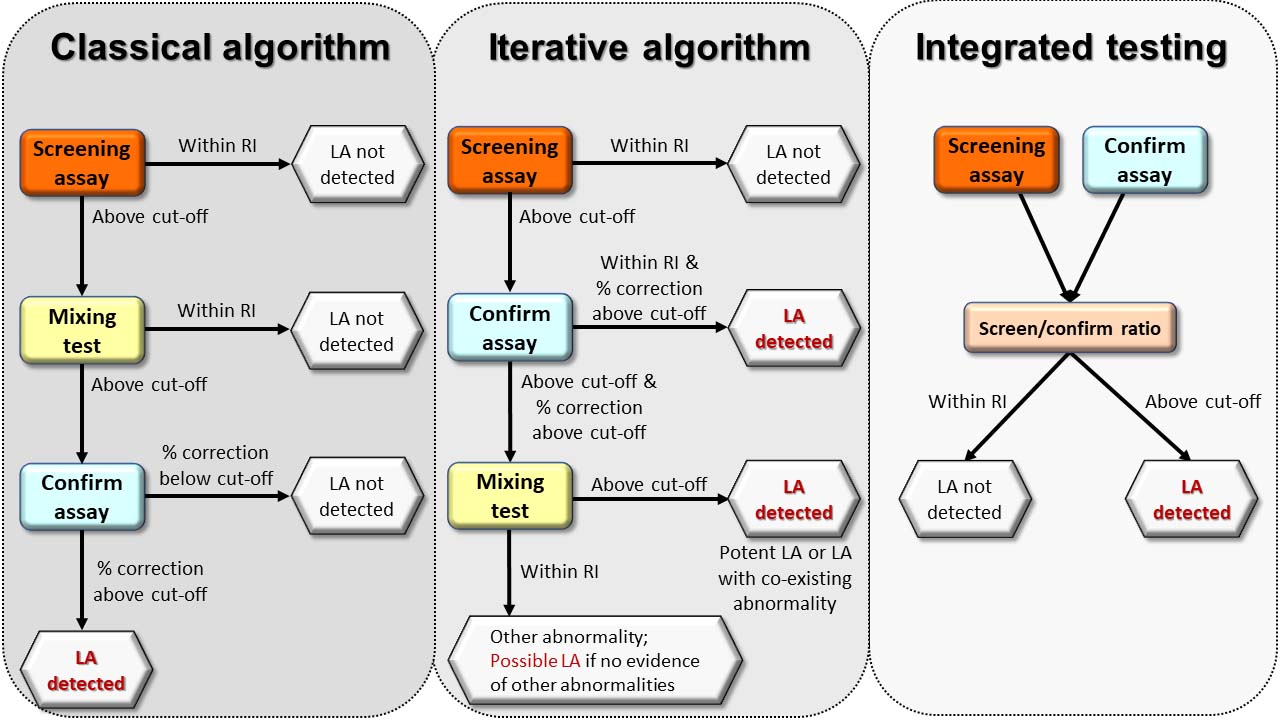
Algorithms for LA detection. The classical and iterative algorithms employ the screening test alone as the starting point, a normal result indicating that no LA is detected with that screening assay. Upon finding an elevated screening test, the classical algorithm proceeds to a mixing test to distinguish between factor deficiency and inhibitor, whilst the iterative algorithm proceeds to the confirm assay to immediately assess for phospholipid dependence. An elevated mixing test with the classical algorithm prompts the performance of the confirm assay to confirm phospholipid-dependent inhibition, whilst a normal mixing test suggests an LA is not present. Given the analytical inconsistencies of mixing tests, the iterative algorithm permits the conclusion of the presence of an LA if the confirm ratio significantly corrects the elevated screen ratio and the confirm ratio is normal, the latter excluding other causes of elevated clotting times in conjunction with normal coagulation screening and confirmation that the patient is not anticoagulated. Mixing tests are valuable in the iterative algorithm when other causes of elevated clotting times are suspected or known to be present as the NPP will commonly correct factor deficiencies and permit any LA to manifest if mixing test limitations are overcome. Integrated testing immediately assesses for phospholipid dependence irrespective of whether the screening test is elevated or not, which improves the detection of weaker antibodies, but oversimplifies more complex cases unless a mixing step is included
Note. Adapted from “Testing for lupus anticoagulants,” by Moore GW. Semin Thromb Hemost. 2022;48:643-60 (https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0042-1744363). © 2022 Thieme.
Acute venous and arterial thrombosis of numerous etiologies are common causes of morbidity and mortality in developed countries [122]. Venous thrombosis is treated with anticoagulants that target coagulation factors, resulting in many patients being investigated for APS after anticoagulation has been initiated. All anticoagulants interfere with at least some coagulation assays used for LA detection. Whether and to what degree a given anticoagulant affects specific assay types depends on the therapeutic target(s) of the drug, the dose of the drug, and the assay principle and design [14, 16–21]. This complicates LA testing and increases the possibility of false positive and negative interpretations, and testing for LA in anticoagulated patients is largely discouraged [17, 19–21]. LA testing is ideally performed prior to anticoagulation, or deferred until it is discontinued [17, 21]. Brief interruption of anticoagulation, or bridging with an anticoagulant less likely to interfere (i.e. LMWH with the sample taken at the time of trough level), can be considered, but is not without clinical risk [21]. However, attempting to detect LA in anticoagulated patients can be valuable for decisions regarding therapy duration and choice of type of anticoagulant in view of the aPL profile [17, 123, 124]. Consequently, diagnostic laboratories require a suitable assay repertoire to maximise the ability to detect LA in these circumstances, whilst maintaining the requisite expertise to recognize when the results are reliable, and when they are not.
VKA anticoagulation affects all LA assays to some degree, with the exceptions of textarin time, TSVT and ET [18, 47, 70]. The reliability of screen and confirm results from undiluted plasma of VKA anticoagulated patients, particularly with dRVVT, is disputed [80, 118, 125–129]. Applying integrated testing with a higher normalized LA ratio cut-off than for non-anticoagulated patients has been shown to improve specificity, but with a reduction in sensitivity [130, 131].
VKAs achieve their anticoagulant effect by inducing an acquired deficiency of the vitamin K-dependent coagulation factors, such that mixing tests can be employed to correct the deficiencies whilst the LA abnormality remains [14, 19–21, 102, 103]. This is an example of how adding a confirm mixing test to the medley can be invaluable, because the confirm ratio on undiluted plasma will often be elevated, more so for dRVVT and dPT than aPTT-based assays. The LA will elevate the screen mixing test providing it overcomes mixing test limitations, whilst a normal ratio in the confirm mixing test evidences both NPP correction of the factor deficiencies, and reagent correction of the LA [14, 19, 20, 73]. In VKA anticoagulated patients, an elevated screen mixing test accompanied by significant discordance with a normal confirm mixing test is diagnostic for the presence of an LA, but crucially, negative mixing tests do not exclude an LA due to the dilution effect [14, 19, 20, 73]. Similarly, the dilution effect may reduce screen elevation such that discordance with the confirm is insufficient to exceed the percent correction or normalized LA ratio cut-off, whereupon an LA cannot be confirmed [100].
The obvious solution is to test with VKA-insensitive assays in the first instance. Although commercial versions of textarin time are not available, they are for TSVT and ET. The TSVT and ET pairing has been recently validated for use in non-anticoagulated patients and those anticoagulated with VKAs and DFXaIs (see later section on direct oral anticoagulants (DOACs)) in a large, international, multi-center study published under the auspices of the ISTH Scientific and Standardization Committee subcommittee on LA/aPL [70]. It must be recognized that TSVT as an isolated screening test is affected by LA heterogeneity, so whilst positive TSVT/ET testing is diagnostic for LA, a normal TSVT ratio alone does not exclude LA, and mixing tests for other assays can then be undertaken.
All screen and confirm assays are affected by UFH, with the sole exception being ET, because steric hindrance prevents the heparin-antithrombin complex from inhibiting meizothrombin, the product of ecarin-activated prothrombin [70, 132, 133]. In response, commercial dPT and VLVT reagents, most dRVVT reagents, and some aPTT reagents specifically formulated for LA detection, contain heparin-neutralizing agents capable of quenching UFH and LMWH to within or just above therapeutic ranges, commonly up to 0.8–1.0 IU/mL [14, 16, 17]. A practical problem arises at the point of interpretation because a heparin level measurement is rarely undertaken during routine LA screening yet the interpreter needs to know if reagent quenching capacity has been exceeded, potentially rendering the results unreliable. Before initiating resource-expensive anti-Xa assays to quantify the heparin level, assuming they are locally available, scrutiny of the confirmatory test result can be informative in paired reagents as they contain the same neutraliser concentration. If the screen is elevated but the confirm is normal, and discordance significant, the normal confirm result evidences quenching of the heparin by the neutraliser, and of the LA by the confirm reagent [14, 16, 20]. If there is discordance in undiluted plasma but the confirm is elevated, mixing tests can serve to dilute the heparin to a quenchable level, permitting an LA to manifest if mixing test limitations are overcome. If heparin is the only cause of an elevated clotting time, screen and confirm results are normally concordant and the mixing test may or may not be elevated depending on dose and reagent heparin sensitivity, and interpretation of the composite would not suggest the presence of an LA [134]. Confirmatory reagents employing platelet-derived PL can shorten clotting times due to heparin neutralization by platelet factor 4, leading to a false impression of PL-dependence [14]. Care is required when using LA-sensitive and LA-insensitive routine aPTT reagents as screen and confirm reagents because they do not contain heparin neutralisers and will have different heparin sensitivities, potentially leading to false-positive and -negative interpretations for LA. Low phosphatidylserine or phosphatidylinositol content correlates with increasing heparin sensitivity [135], whilst increasing phosphatidylserine content correlates with decreasing LA sensitivity [24].
LMWHs, danaparoid, and fondaparinux tend to exert less interference on LA assays than UFH unless levels are supra-therapeutic [116, 134], although false-positives have been reported in dRVVT, aPTT, and dPT [128, 134, 136–138], and for TSVT with LMWHs and danaparoid [70]. The likelihood of interference is reduced if samples are taken at a distance from LMWH administration, such as 12 h or more [116].
DTIs (dabigatran, argatroban, bivalirudin) interfere with every LA assay in a dose-dependent manner [70, 128, 138–140]. DFXaI (rivaroxaban, apixaban, edoxaban) interfere with all LA assays except textarin time, TSVT, and ET, because direct FII activation bypasses inhibition of FXa [70, 128, 137, 141, 142]. The inhibitory action of DTI and DFXaI commonly results in elevated mixing test results. dRVVT and dPT tend to be more responsive to DFXaI than aPTT [116, 143].
False-positive interpretations due to greater elevation of screen results compared to confirm results have been reported for dRVVT and aPTT-based assays with DTI and DFXaI [116, 137, 138, 141, 142], and for TSVT/ET with DTI [70]. The degree to which this phenomenon manifests is reagent, drug, and dose-dependent, it being a particular problem with some but not all dRVVT reagents in the presence of rivaroxaban [116, 137, 143–146], where false-positives can occur even at trough levels [144, 147]. False negative dRVVT and TSVT/ET have been reported with apixaban and argatroban respectively, due to greater interference with confirm assays than screen assays [70, 148].
The DOACs (dabigatran and the three DFXaIs) and parenteral DTI are unaffected by heparin neutralizers. Instead, several products employing a pre-analytical procedure involving adsorption of the drugs by activated charcoal are available to facilitate LA testing (and other haemostasis assays) unencumbered by anticoagulant interference [149–153]. They are reported to be effective in facilitating reliable dRVVT and aPTT analysis [149–153], but less beneficial for dPT [154]. Some studies report incomplete DOAC adsorption in some samples, sometimes at drug concentrations that would normally result in full removal, and altered clotting times in some samples from non-anticoagulated patients, and VKA or heparin anticoagulated patients [150, 151, 155–158]. Consequently, the recent recommendations state that LA testing on patients receiving DOAC should not be done unless an adsorption procedure has been performed, and that the adsorption should only be done on samples from patients known to be DOAC anticoagulated [17, 155]. It is additionally advisable to measure post-adsorption drug levels to ensure adequate removal, yet importantly, there is no guarantee that the effects on clotting times sometimes encountered in non-DOAC samples are not occurring in DOAC samples [14, 155]. Altered formulation dRVVT reagents have recently become commercially available that are less affected by VKA and DOAC anticoagulation because they return similarly elevated screen and confirm results in the absence of LA [131, 159], although there may be a degree of reduced LA sensitivity [159, 160].
In view of the limitations of strategies to reduce or eliminate anticoagulant interference in LA assays, a testing hierarchy for each type of anticoagulant is suggested in Figure 3.
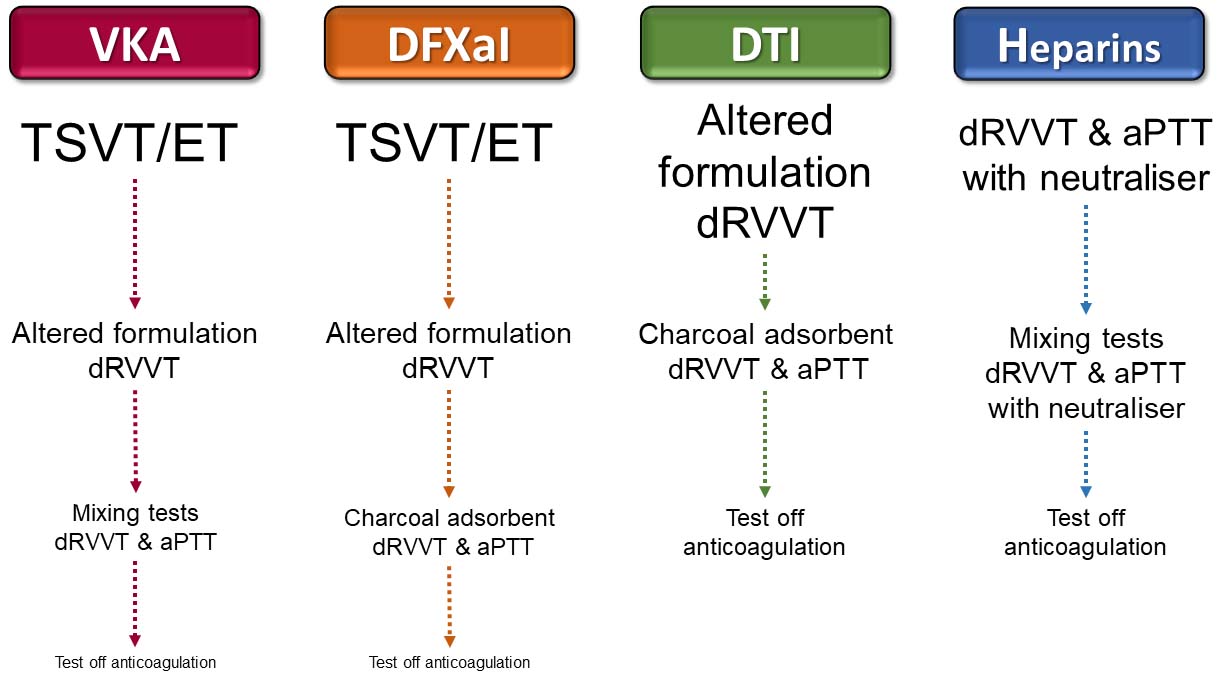
Suggested testing hierarchies for anticoagulated patients. For VKA and DFXaI anticoagulated patients, TSVT/ET are suggested as the first assays as they are unaffected, where a positive result negates the need to look further with potentially compromised assays. Altered formulation dRVVT reagents are suggested next as they will detect some TSVT-unreactive antibodies, but if negative (or the assays are unavailable), mixing tests that can detect LA in some VKA anticoagulated patients, or post-adsorption dRVVT and aPTT for patients on DFXaIs, are suggested to detect LA unreactive with TSVT and altered formulation dRVVT. Altered formulation dRVVT is suggested as first-line testing for DTI anticoagulated patients, but if negative (or unavailable), post-adsorption dRVVT and aPTT are indicated. Patients on heparins can be directly tested with routinely used dRVVT and aPTT reagents if they contain neutralisers, with close attention paid to whether neutraliser quenching capacity has been exceeded. If so, mixing tests can reduce the heparin level to within capacity, permitting detection of LA that overcome mixing test limitations. It should be noted that a negative result with each step in the assay hierarchies may not fully exclude LA in some patients and testing off anticoagulation could be the only viable option. In some patients, bridging with LMWH and testing at the time of trough with reagents containing heparin neutralisers may also be an option
Aside from avoiding LA testing in anticoagulated patients wherever possible, it should also be a consideration at the point of a thrombotic event and its immediate physiological aftermath, and in other clinical scenarios involving an acute phase response, such as infection, inflammation, or trauma [19, 21, 161]. The increased C-reactive protein can interfere with aPTT-based assays, giving rise to false-positive results [162], whilst the elevated FVIII and fibrinogen [20, 21, 163, 164] can shorten aPTT, giving rise to the potential for false-negatives. Elevated FVIII can also be encountered in pregnancy [165] and malignancy [166], and LA results can fluctuate during pregnancy [167, 168]. Transient LAs in response to infections are well described [169–171], whilst infections are reported as a common precipitating factor of catastrophic APS in patients with existing APS [172].
Accurate LA detection is crucial to effective diagnosis and management of APS but it remains hampered by the lack of specific assays. Functional detection of heterogeneous antibodies necessitates the use of a battery of clotting-based assays that are inevitably compromised by other causes of altered clotting times. Antibody heterogeneity and reagent variability affect all assays, including the favored dRVVT and aPTT pairing, yet overreliance on two familiar and well-performing assays solves some problems but ignores others. Applying composite testing to each assay type improves specificity but interpretation can be challenging when trying to unravel the possible presence of an LA where there are other abnormalities that may mask, mimic, or co-exist. There are variations on how to apply the composite testing model, particularly regarding the place of mixing tests, which can be invaluable where other abnormalities are present, but a hindrance when design limitations of the mixing test principle give misleading information. It is inconsistent that we accept and respond to assay limitations in other areas of hemostasis testing, such as variants of non-severe Hemophilia A manifesting only in one-stage or two-stage assays but not both [173], or the inability of antigenic assays to detect qualitative deficiencies of hemostasis participants such as protein C, protein S, and FXIII [174–176], yet some practitioners rigidly permit a normal mixing test result to conclude LA-negativity when other evidence (and theory) suggests otherwise. Strategies to detect LA in anticoagulated patients are improving. Interpretation of the presence of an LA can be relatively straightforward in uncomplicated cases, very complex and challenging in others, and at times, insurmountable. It is crucial that practitioners involved in performing and reporting LA testing maintain sufficient expertise to robustly set up their assays to maximise analytical performance, and to interpret results in light of the clinical picture, results from related assays, and in view of LA assay principles and limitations.
aCL: anticardiolipin antibodies
aPL: antiphospholipid antibodies
APS: antiphospholipid syndrome
aPTT: activated partial thromboplastin time
aβ2GPI: anti-β2 glycoprotein I antibodies
BSH: British Society for Haematology
CLSI: Clinical and Laboratory Standards Institute
DFXaI: direct FXa inhibitor
dPT: dilute prothrombin time
dRVVT: dilute Russell’s viper venom time
DTI: direct thrombin inhibitor
ET: ecarin time
FII: factor II
FV: factor V
FXa: activated factor X
ISTH: International Society on Thrombosis and Haemostasis
KCT: kaolin clotting time
LA: lupus anticoagulant
LMWH: low molecular weight heparin
NPP: normal pooled plasma
PL: phospholipid
RI: reference interval
SCT: silica clotting time
SDs: standard deviations
TSVT: Taipan snake venom time
UFH: unfractionated heparin
VKA: vitamin K antagonist
VLVT: Vipera lebetina venom time
GM: Conceptualization, Visualization, Writing—Original Draft, Writing—Review & Editing
G.W. Moore reports consultancy fees from Technoclone.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Masahiro Ieko ... Akitada Ichinose
Osamu Kumano ... Jean Amiral
Fariza A. Cheldieva ... Tatiana M. Reshetnyak
Tiffany Pascreau ... Marc Vasse
