 Review
Review

Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-4925-2754
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0003-2175-0348
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
†These authors contributed equally to this work.
ORCID: https://orcid.org/0000-0001-9632-8614
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0000-0002-8878-0915
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0000-0002-9643-5396
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0000-0001-8315-255X
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0009-0007-2017-5771
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0009-0002-7398-8040
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
ORCID: https://orcid.org/0000-0003-1850-5907
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
Email: gauri@jcbose.ac.in
ORCID: https://orcid.org/0000-0002-5322-3481
Affiliation:
Division of Molecular Medicine, Bose Institute, P-1/12, Calcutta Improvement Trust Scheme VII M, Kolkata 700054, India
Email: tanya@jcbose.ac.in
ORCID: https://orcid.org/0000-0002-9583-0693
Explor Immunol. 2023;3:384–405 DOI: https://doi.org/10.37349/ei.2023.00108
Received: April 13, 2023 Accepted: June 12, 2023 Published: August 31, 2023
Academic Editor: Sunil K. Arora, Postgraduate Institute of Medical Education & Research, India
Cancer stem cells (CSCs) are a small subpopulation of cells that drive the formation and progression of tumors. However, during tumor initiation, how CSCs communicate with neighbouring immune cells to overcome the powerful immune surveillance barrier in order to form, spread, and maintain the tumor, remains poorly understood. It is, therefore, absolutely necessary to understand how a small number of tumor-initiating cells (TICs) survive immune attack during (a) the “elimination phase” of “tumor immune-editing”, (b) the establishment of regional or distant tumor after metastasis, and (c) recurrence after therapy. Mounting evidence suggests that CSCs suppress the immune system through a variety of distinct mechanisms that ensure the survival of not only CSCs but also non-stem cancer cells (NSCCs), which eventually form the tumor mass. In this review article, the mechanisms via which CSCs change the immune landscape of the tissue of origin, which contains macrophages, dendritic cells (DCs), myeloid-derived suppressor cells (MDSCs), natural killer (NK) cells, and tumor-infiltrating lymphocytes, in favour of tumorigenesis were discussed. The failure of cancer immunotherapy might also be explained by such interaction between CSCs and immune cells. This review will shed light on the critical role of CSCs in tumor immune evasion and emphasize the importance of CSC-targeted immunotherapy as a cutting-edge technique for battling cancer by restricting communication between immune cells and CSCs.
Despite the discovery of numerous diagnosis processes and modalities of cancer therapy, there is still scope for developing more effective and targeted therapeutic options of cancer. Revealing the origin of cancer, therefore, has become an extremely important issue in that it might shed light on successful and relapse-free treatment of this deadly disease. During the last 25 years, a plethora of evidences have suggested the candidature of a small subpopulation of cancer cells, named cancer stem cells (CSCs) with tumor-initiating capability, as the core origin of tumorigenesis [1, 2]. Like normal stem cells, CSCs take charge of a cancer hierarchy, harbouring stem cell-like properties involving self-renewal, to form identical daughter cells, and differentiation potential to create various types of progenies that finally generate non-stem cancer cells (NSCCs) to build up the total tumor mass [3]. Besides, CSCs have also been characterized as the drivers of metastasis [4, 5]. Enhanced invasiveness and metastatic capability of any tumor that ensure infiltration to the surrounding area and forming new tumor to the secondary organs, have been reported to be related to CSCs [4, 6]. In fact, quiescent CSCs that have travelled to distant places may cause metastases that arise many years after curative surgical treatment of a primary tumor. CSC hypothesis could, therefore, implies that targeting CSCs might be the most promising therapeutic strategy. However, despite frequent efforts to target CSCs, current anti-cancer therapies are still obstructed by highly drug-resistant CSCs. In fact, by application of therapeutic regimens such as chemo- or radiotherapy, NSCCs with a high proliferative capacity are targeted and killed causing a decrease in tumor size while rare CSCs survive these therapeutic regimens [7], explaining why local recurrence is usually always the result of effective treatment of solid tumors with radiation or chemotherapy [8]. Such high resistance thus provides CSCs not only a survival advantage over differentiated NSCCs [7], but also the chance to “recreate” the tumor after anti-cancer therapy [9]. At this point, it will not be out of context to mention that in a tumor mass, since majority of the tumor cells are NSCCs, they become more effective in immune evasion than CSCs. This information, together, signify the robust functionalities of this small subpopulation of tumor-initiating cells (TICs) as the major contributors in tumor immune evasion during tumor initiation, metastasis, and relapse.
It is acknowledged that during the early stages of tumor initiation, immune cells get activated and infiltrate to tumor microenvironment (TME) to mount an effector immune response, eliminating immunogenic tumor cells [10, 11]. Although above discussion portrayed CSCs as the TICs during primary initiation, metastasis, and relapse [2, 12], their survival during the initial ‘elimination phase’ of immune attack [13] still remains elusive. This tempted us to understand whether due to change in the immune system by CSCs ensures their own survival during elimination phase to finally build the whole tumor mass either during primary initiation, or during metastasis or recurrence.
This review discusses the recent studies investigating immuno-modulation of CSCs during tumor initiation, and the underlying molecular mechanisms as well as the impact of immune cells on CSCs. A deeper understanding of this bidirectional crosstalk that reshapes the immunological landscape and governs therapeutic responses, will expedite the improvement of cancer immunotherapy, in which CSCs might apply the same strategy of overpowering immune attack by suppressing the infiltrating active immune cells, thus generating immunosuppressive milieu.
A growing body of evidence suggests that malignant tumors are initiated and maintained by CSCs which have similar biological properties to normal adult stem cells [14]. The CSCs have specific surface signature-markers depending on their tissue of origin. The CSC hypothesis states that tumors, like adult tissues, arise from cells with the ability to self-renew and/or give rise to differentiated tissue cells [14]. According to the canonical hardwired stem cell/CSC hierarchy model, stem cells/CSCs are rare, relatively dormant, and defined primarily by intrinsic properties. When they divide asymmetrically, they produce one stem cell and one transient amplifying cell [15]. As the tumor evolves, these progenitor cells differentiate to NSCCs that proliferate quickly to form the tumor mass. Therefore, according to the CSC theory, at the early stages of tumor initiation very few CSCs are engaged to sculpt the tumor mass, which will eventually be built with the division of NSCCs generated by the asymmetric division of CSCs [15, 16].
To metastasize and initiate new tumor at the secondary site, cancer cells go through a “metastatic cascade”, consisting of extracellular matrix (ECM) disassembly by enzymes such as matrix metalloproteinases (MMPs), followed by remodelling of the ECM to drive growth in the metastatic niche [17, 18]. These obstacles make metastasis an inefficient process in which most tumor cells die in circulation or upon arrival at the distant tissue [11]. Only a subset of disseminated cells with CSC-like properties [19], overcome such hurdles and initiate tumor growth at distant tissues. These “metastatic stem cells” [20, 21] like primary tumor CSCs, have the ability to restart tumor growth at distant sites. According to our findings, non-migratory tumorigenic intrinsic CSCs generate C-X-C chemokine receptor type-4 (CXCR4)-positive migrating CSCs in the tumor’s periphery, possibly to help the migrating fraction withstand the rigours of the metastatic voyage, because depletion of this population resulted in the complete tumor’s metastatic activity being halted [22]. These reports support our hypothesis that CSCs adapt to new environments, initiate the formation of heterogeneous tumors, and explain the clinical course of metastasis and distant relapse [23, 24].
Tumor recurrence is a significant impediment to cancer treatment, even though its underlying causes are mostly unknown. The most commonly used antitumor therapies, such as chemotherapy and radiotherapy, have lethal effects on cancer cells that are highly proliferating. CSCs are slow-cycling by nature, in contrast to their transitory amplifying progeny, which actively proliferates prior to differentiation [25, 26]. CSCs have the ability to enter the quiescent cell cycle phase, which represents a reversible latent state in which cells are alive but not multiplying [25, 27]. As a result, non- or slow-proliferative latent CSCs escape chemotherapeutic cytotoxicity and persist after treatment. As the cytotoxic effects of the chemotherapeutics wear off, they re-appear and initiate tumors in the same or at distant new locations [13, 28, 29], eventually leading to tumor relapse. Moreover, this group has also reported the contribution of CSCs to acquired chemoresistance and higher residual risk of recurrence in cancer patients having acquired chemoresistance [13] since as a result of their inherent properties and adaptability to their respective TME, “drug-spared” CSCs play pivotal roles in fuelling tumor growth during relapse.
This discussion signifies the possibility of involvement of CSCs in initiating tumor during initiation phase, after metastasis, and during tumor relapse (Figure 1).
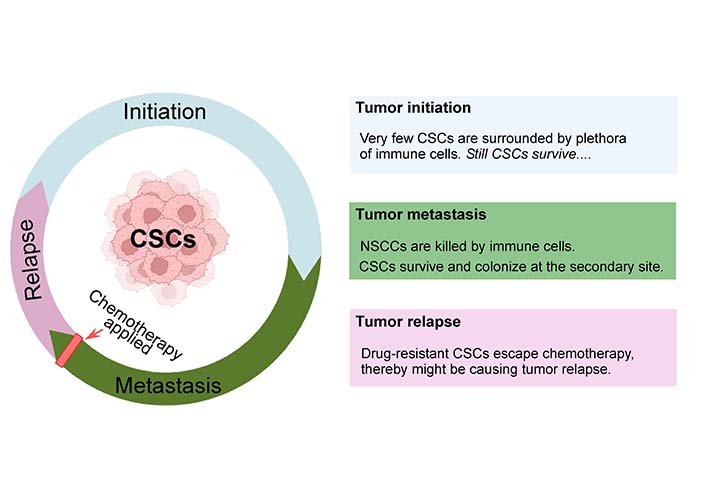
Intricate pattern of tumor initiation, progress, remission, and relapse. CSCs undergo substantial cellular proliferation while maintaining their stemness signature and produce differentiated progenies. They play instrumental roles in tumor initiation, metastasis, and relapse. CSCs evolve when the adult stem cells of our body acquire genetic/epigenetic alterations, and initiate tumor. During different stages of tumor progression, CSCs play a central role in facilitating tumor growth. This may also be accelerated by having dynamic interplay with the stem cell niche. During metastasis, although CSCs and NSCCs both are detected in the invasive front of primary tumors, only CSCs survive immune surveillance or conventional cancer therapy and cause distant metastasis. On the other hand, conventional therapeutic techniques kill NSCCs while CSCs escape the effect of chemotherapy and increase their repertoire to finally result in tumor relapse. Created with Biorender.com
A typical tumor shape is made up of the tumor core, as well as the surrounding stromal and lymphoid components [30, 31]. Immune infiltration can vary from patient to patient. Macrophages, dendritic cells (DCs), natural killer (NK) cells, mast cells, B lymphocytes, and T lymphocytes including T-helper-1 (Th1), T-helper-2 (Th2), T-regulatory (Treg) cells, and cytotoxic T lymphocytes (CTLs), are all common immune cell types found in TME [30, 32]. Within this immunological milieu, there are components that are advantageous to the patient as well as components that are harmful [32, 33] (Figure 2). Tumors create an array of neo-antigens, which allow the immune system to distinguish tumor cells from normal cells [34, 35]. Immune surveillance is the process by which malignant cells are identified, damaged and are then targeted for eradication by the immune system.
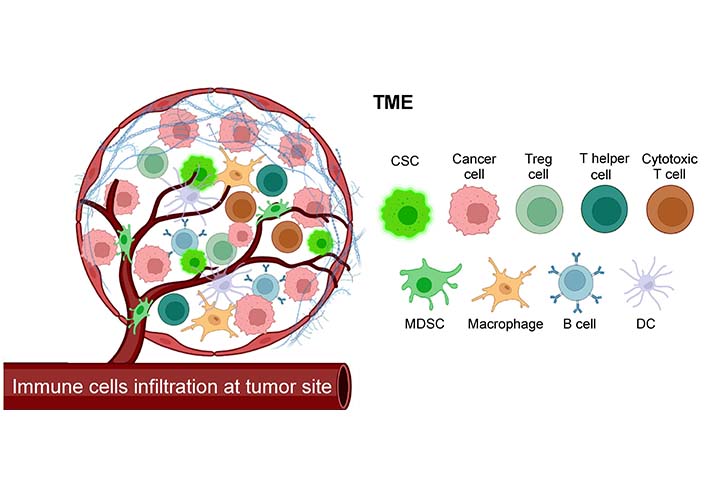
Components of the TME. TME is a complex ecology of NSCCs, CSCs, stromal cells, endothelial cells, tumor fibroblasts, ECM, and immune cells, living amid a network of dysregulated vasculature and collagen. TME contains immune cells from both the myeloid and lymphoid lineages that infiltrate the tumor. Created with Biorender.com. MDSC: myeloid-derived suppressor cells
Growing evidences suggest that immune cells can both prevent the formation of precancerous cells and facilitate the regression of established tumor burden [34]. Substantial evidences claim that certain innate and adaptive immune cell types, effector chemical molecules, and immunogenic pathways can sometimes operate as extrinsic tumor-suppressor mechanisms in concert. Antitumor effector immune cells such as macrophages, DCs, and other myeloid-derived polynuclear cells not only phagocytose and present neoplastic altered cells (and/or foreign pathogens) to Th cells, but also mount antitumor immunity by leaking tumoricidal cytokines [36, 37]. Continuous approaches and technologies have precipitated several important discoveries related to the adaptive handle of tumor immune response. These reports demonstrating T cell mobility within different lymphoid and non-lymphoid compartments, effector functions after antigen presentation, priming other immune cells for eliminating foreign pathogen and/or neoplastic cells via secreting plethora of cytokines, advanced our understanding of effective immunosurveillance [38, 39].
This paradigm has recently been expanded to include the concept of immunoediting, in which altered self-cells give rise to tumor formation despite having selection pressure from immune system [40, 41]. However, some tumors escape the immune clearance. This incident highlights the seminal role of tumor in reshaping the immune cells into immune-suppressive pro-tumorigenic cells to ensure their own survival [42].
Macrophages are highly plastic cells that perform a wide-range of biological functions, including tissue development and homeostasis, clearing cellular debris, eradication of pathogens, and modulation of inflammatory response [43]. M1 macrophages are generated by cytokines such as interferon gamma (IFNγ), tumor necrosis factor alpha (TNFα), granulocyte-macrophage colony-stimulating factor (GM-CSF), as well as the bacterial toxin lipopolysaccharide (LPS). Pathogen-associated molecular patterns (PAMPs) that activate toll-like receptor (TLR) signaling also promote M1 macrophage polarization [44, 45]. M1 macrophages show antitumor effects, which can distinguish tumor cells from normal cells. M1 macrophages secrete molecules such as reactive oxygen species (ROS) and nitric oxide (NO), which have tumoricidal properties. M1 macrophages are also known to promote a pro-inflammatory Th1 response [43, 46].
It is known that tumors recruit macrophages from the periphery by chemotaxis and differentiate into tumor-associated macrophages (TAMs) in local environment [47–49]. Current studies have shown that TAM population oscillates between M1 (classically-activated macrophage) and M2 (alternatively-activated macrophage) types [48, 50]. The nature and concentration of distinct signals in tumor milieu dictate the proportion of each form. TAMs express a variety of cytokines which in turn stimulate proliferation and survival of tumor cells [51]. TAMs are also known to promote metastasis directly via producing soluble factors [52]. These cells, generally found in hypoxic areas of tumor, promote neo-angiogenesis [53]. The cytokines produced by tumor cells also promote the differentiation process of TAM from M1 macrophages, thus forming a positive feedback loop between TAMs and epithelial-mesenchymal transition (EMT) [54, 55]. Besides, TAM can regulate the function of other immune cells and recruit Tregs through chemokine ligand 22 (CCL22) which exhibits further suppression of T-cell mediated antitumor immune responses [56].
CSCs recruit TAMs to their advantage from blood (when they are monocytes), with the help of the factors such as CCL2, CCL5, CSF1, chemokine (C-X-C motif) ligand-12 (CXCL12) and CCL8 [48, 57, 58]. Interestingly, CSCs also skew such recruited TAMs into pro-tumor state with the help of multiple immunosuppressive cytokines [48, 57]. As already discussed, the alternatively activated M2-macrophages help in promoting cancer as CSCs polarize them by releasing interleukin 4 (IL4), IL13 and transforming growth factor-β (TGFβ) [59–61]. CSCs shift TAMs towards M2 by releasing IL6 and IL10 [57, 62, 63]. CSCs employ multiple mechanisms, one of them being low expression of major histocompatibility complex (MHC) molecules and high expression of programmed death-ligand 1 (PDL1) checkpoint molecule during tumor immune evasion [57, 64]. Another notable way to avoid immune destruction is the high-expression of cluster of differentiation 47 (CD47) (don’t eat me marker), which upon binding to the signal regulatory protein-1α (SIRP1α) on these phagocytic cells, curbs the process of phagocytosis [65]. Therefore, not only do CSCs bring the TAMs to the tumor-site, they guide them towards to pro-tumor subtype and moreover, exploit tools to escape them. Reciprocally, TAMs help in maintenance of CSCs via secretion of cytokines, chemokines, soluble factors, and direct interaction. TAMs support CSCs by releasing IL6 and TGF. Both IL6 and TGF have been reportedly known to aid in EMT and acquisition of stemness in various cancers [66, 67]. Vascular endothelial growth factor (VEGF) and epidermal growth factor (EGF) are also secreted by M2 macrophages which promote angiogenesis, proliferation and stem like phenotype [66, 68]. Chemokines CCL2 from TAMs are also implicated in augmenting stemness factors and related genes in breast cancer [69]. Other distinctive factors such as pleiotrophin and milk-fat globule-EGF-VIII (MFG-E8) also demonstrated tumor growth via CSC increase [67]. MGF-E8 from TAMs activates signal transducer and activator of transcription-3 (STAT3) and Sonic hedgehog (SHH) pathway in CSCs and contributes to drug resistance [67]. The physical interaction between TAMs and CSCs is thereby essential too in maintenance of stemness. CD11b on macrophages, when in contact with CD90 bridged with ephrin type-A receptor-4 (EphA4) on cancer cells led to the production of IL6, IL8 and GM-CSF which supports augmenting the CSC phenotype [57, 66]. Direct contact between these two cell types also elicits an activation of Notch, SHH, STAT3 and nuclear factor κ-B (NFκB)-signaling cascades which are reported to increase the CSC population [57].
DCs have long been considered as one of the critical factors in antitumor immunity due to their effective antigen-presenting capability [70, 71]. However, despite promoting antitumor responses, tumor-associated DCs are largely defective in their functional activity thereby contributing to immune-suppression in TME [70]. In TME, DCs acquire, process and present tumor-associated antigens (TAAs) on MHC molecules (signal-1) [72, 73] and provide co-stimulation (signal-2) [71, 74] as well as soluble factors (signal-3) to modulate T cell responses [71, 72].
In recent years, the presence of immune-suppressive regulatory DCs in the TME has been documented [75]. Tumor-infiltrating DCs (TIDCs) inhibit both innate and adaptive immune effectors through a variety of methods [76, 77]. The programmed cell death protein-1 (PD1) ligand, PDL1, is constitutively expressed on activated DCs and is further increased by the TME [63, 66, 78]. Since TIDCs co-express both PD1 and PDL1, suppression of T cell activation might be mediated by any of these molecules present on T cells [78–80]. Furthermore, blocking of PDL1 selectively on murine TIDCs improved TIDCs’ ability to induce T cell activation [79].
Although myeloid cells serve primarily to defend the host against infections, cancer progression or chronic inflammation, few cells transform into myeloid-derived suppressor cells (MDSCs), which play a significant role in suppressing antitumor immunity [81].
Myeloid progenitor cells found in the bone marrow give rise to MDSCs. MDSCs develop when the myeloid progenitor cells’ ability to differentiate is compromised [81]. The TME greatly upregulates the MDSCs’ immunosuppressive activities [81–83]. Numerous inflammatory chemicals support MDSCs. The key tumorigenic properties of cancer cells like invasion, proliferation, survival, and adherence are mediated by several chemokines, cytokines, and enzymes that are released by MDSCs [84–86]. Stemming theories state that CSCs and MDSCs have reciprocal impacts on the growth of cancer [87, 88]. The enhanced tumor-initiating and tumor-repopulating capability of CSCs is known, in part due to their notable capacity to avoid immune detection. Liu et. al. [87] have shown that triple-negative breast CSCs bypass immunosurveillance upon recruitment of MDSCs, according to a newly discovered aldehyde dehydrogenase (ALDH)-dependent mechanism.
Several malignancies, particularly breast cancer, show high expression of the CSC marker ALDH1A1. Phosphorylation of TGFβ-activated kinase-1 (TAK1), activation of NFκB-signaling, and an increase in GM-CSF production were the mechanisms by which ALDH1A1 lowered the intracellular pH in breast cancer cells, causing MDSC growth and immunosuppression [87]. Apart from that, patient-derived CSCs selectively promoted MDSC-mediated immune-suppression in co-culture trials. Using a cytokine screen, it was shown that CSCs expressed numerous elements that encouraged this activity, including the highly abundant macrophage migration inhibitory factor (MIF). In MDSCs, MIF addition enhanced arginase-1 production in a CXCR2-dependent manner, but MIF blockade decreased arginase-1 production. Targeting tumor-derived MIF gave tumor-bearing animals a survival benefit and boosted the CTL response inside the tumor [88] (Figure 3).
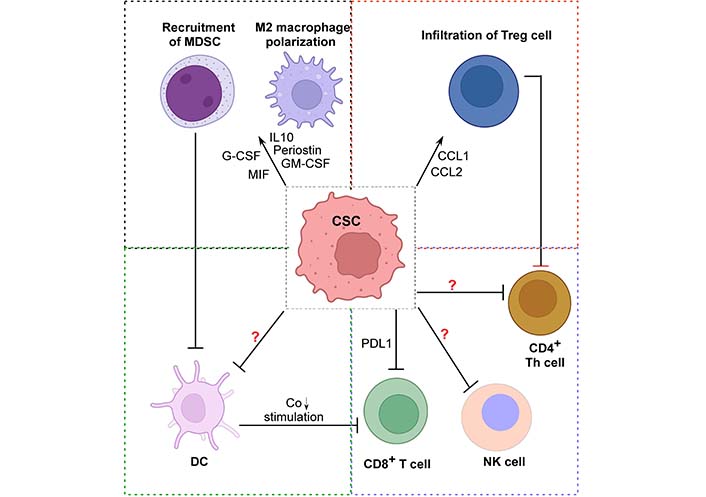
Mechanisms of immune-suppression in TME by CSCs. The schematic diagram depicts CSC-induced immune-suppression. CSCs might be re-educating immune cells like Treg cells, CTLs, MDSCs, promoting M2 macrophage polarization during tumor initiation, thus escaping elimination phase and orchestrating a pro-tumorigenic environment with the help of the suppressive immune cells. However, there is lack of information on how these CSCs actually modulate or inhibit the antitumoral actions of NK cells, CD4+ Th cells, and DCs. So, more research is needed to explore this new area of CSC-driven immune-suppression. Created with Biorender.com. →: promote; ┤: inhibit. G-CSF: granulocyte colony-stimulating factor
Th cells are attributed for their crucial function of generating an immune response either through antigen-specific effector cells or by activating the innate immune cells [89]. Although Th1 cells exhibited stronger cytotoxicity than Th2 cells, both cell types can eradicate tumors [90]. Th1 cells eradicate the tumor mass by releasing TNFα and IFNγ which directly induce cancer cell death. Additionally, cytokines released by Th cells induce a pro-inflammatory response in other immune cells as well, amounting to a concerted antitumor effect [91]. Th cells are also known to promote CD8+ CTL activation and effector functions along with providing co-stimulatory activation to augment the expression of MHC class molecules and eliciting humoral response [91]. Th2 subset is mainly recognized for its role in creating a pro-tumor environment; however, few reports suggest Th2 also carries out recruitment of macrophages and eosinophils as well as, augmenting humoral immune responses to the tumor-site [92].
Another significant subtype is Th17, which is activated by IL6 and TGFβ operates context-dependently in cancer. Its antitumor activity includes the release of IFNγ and the recruitment of DCs, CD8+ T cells, and NK cells [93]. In response to a multitude of cytokines in TME, Th cells can differentiate into a number of subsets [91].
Treg, a subtype of CD4+ T cells designated as CD4+CD25hiCD127loForkhead box P3+(FOXP3+), are well recognized for their immune-suppressive function, prevent autoimmunity, and inhibit antitumor immune response [93–95]. The presence of CSCs and Tregs in cancers have been found to be positively correlated, indicating potential cross-talk between these cell types in maintaining an immunosuppressive environment [96]. In general, tumor-produced chemokines like CCL1, CCL2, and CCL5 take active part in recruiting Treg cells in TME [97–99]. Interestingly, studies have found that SRY-box 2 (SOX2) positive breast CSCs and glioblastoma CSCs can secrete chemokines like CCL1, CCL2 in tumor environment [99, 100]. These chemokines are also crucial for migration of myeloid cells into the tumor milieu, that could promote infiltration of Treg cells, and this might be suggesting a combination of direct and indirect mechanisms of CSC-driven Treg cell accumulation [98]. FOXP3, the lineage-specific transcription factor of Treg cells, plays an important role as a transcription of C-C chemokine receptor type-4 (CCR4), the regulator of Treg infiltration in tumor-site [94]. Moreover, tumor cells derived indoleamine 2,3-dioxygenase 1 (IDO1) as well as TGFβ also have a role in Treg cell infiltration and immune-suppression. Studies have shown that tumor-initiating CSCs express elevated levels of IDO and TGFβ signaling-related genes which play critical roles in immune evasion by helping recruitment of Treg cells [63, 66, 101]. It has also been observed that co-stimulatory molecule PDL1, soluble galectin-3 in CSCs promote Treg cell infiltration [99, 102]. In return, Tregs secrete TGFβ and IL17 to induce self-renewal ability and EMT formation toward tumor progression and invasion [103, 104]. Tregs in hypoxic TME secrete VEGF, as a mediator of angiogenesis that supports CSC survival, stemness, and self-renewal [105]. Infiltrating Tregs, on the other hand, exhibit metabolic changes in the TME, which enables their selective survival over others [106]. Besides classical Treg cells, reports from the laboratory work indicates to the acquisition of immunosuppressive properties by Th17 cells in TME by expressing Treg lineage-specific transcription factor FOXP3 and ectonucleotidase CD73 [107]. CSCs have been suggested to modulate the Th17/Treg balance by modifying the production of cytokines such as IL6 and IL8 and chemokine CCL5 in the tumor milieu [93]. It has been found that IL17-secreting Tregs promote the development of stemness markers, such as CD44, CD133, CD166, epithelial cell adhesion molecule (EpCAM), and ALDH, and drive cells to develop into CSCs demonstrating the capacity of Tregs to support CSC development [108]. It has also been observed that prostaglandin-E2 released by Tregs induces CSC expansion and metastasis [109]. Therefore, CSCs promote Treg infiltration by expressing co-stimulatory molecules and secreting chemokines in the TME, whereas Tregs alternatively promote CSC proliferation and expansion directly or indirectly through angiogenesis and EMT. Various CD8+ T cell subsets with regulatory properties have been found in multiple experimental systems and dubbed as “CD8+ Tregs”. CD8+FOXP3+ Tregs were found to be augmented in breast tumor milieu [110]. Multiple studies have explained apparently distinct mechanisms of immune regulation by a variety of CD8+ Treg subsets, such as the direct killing of the target cells, secretion of multiple chemokines and cytokines such as IL10, TGFβ, IL16, and CCL4 [111]. At an early stage of tumor onset, this tumor-induced IFNγ-producing FOXP3–CD8+CD25+CD127– T-cell subset is shown to enhance immune surveillance by targeting human leukocyte antigen-E (HLA-E)-restricted CD4+ Treg cells while keeping the effector T-cell population untouched [111].
CD8+ CTLs patrol practically every region of our body for foreign antigens [112]. Crosstalk between peripheral immune system and the TME is crucial for effector CD8+ T cell mediated antitumor immune responses [32]. CD8+ T cells use granzyme B, perforin, and IFNγ for its cytotoxicity, ultimately results in killing of cancer cells [112]. Additionally, CD4+ Th cells can boost antitumor CTL responses by promoting clonal expansion at the tumor-site, avoiding activation-induced cell death, and acting as antigen-presenting cells (APCs) for CTLs to selectively produce immunological memory cells [113]. Lactic acidosis by Warburg effect is common to most solid tumors and has been found to affect infiltrating immune cells [114]. Lactic acid in TME inhibits CTL activity involving complete blockage of cytokine production and partial impairment of lytic granule exocytosis [115].
In contrast to their instrumental roles in mounting antitumor immunity, CTLs are also known for their pro-tumorigenic functions as imposed by the TME. Communication between CSCs and CD8+ T cells can be broadly divided into two categories: evasion of T cell-mediated death and suppression of antitumorigenic capabilities of T cells. Effective execution of the first area can occur by reducing CSCs’ ability to offer neoantigens to T lymphocytes through downregulation of MHC-I expression in CSCs [116, 117]. Mechanistic studies demonstrated that these CSC avoided cytotoxic T cell death by upregulating CD80 in a TGFβ-dependent manner [63]. On the other hand, how CSCs control T cell activities, is a more sophisticated and well-defined mechanism that encompasses many avenues via both direct and indirect contact. CSCs impede cytotoxic T cell function by selectively enriching inhibitory checkpoint ligands in comparison to non-CSC tumor cells [63, 118]. It is reported that, higher PDL1 expression on breast CSCs helps in antitumor immune evasion [119]. While the whole spectrum of co-inhibitory receptors expressed by CSCs has not been studied, multiple investigations have demonstrated that breast [120], colon [121], head and neck squamous cell carcinoma [122], glioblastoma [123, 124] are characterized by their expressing higher amounts of inhibitory checkpoint receptors, thereby augmenting immuno-suppressive environment.
Amongst other immune cells, NK cells and NK T cells are endowed with a unique property of rapidly killing multiple adjacent cells with varying oncogenic cell surface markers [125]. Along with this cytotoxic property, their ability to increase antibodies and T cell responses suggest their role as effective anticancer agents [126]. Current studies involving pre-clinical animals and clinical trials indicate that both NK and NK T cells have potent antitumor activity, which makes them potential targets for cancer immunotherapy [125, 127, 128].
Growing evidences suggest that CSCs can over-shadow NK cells via augmenting their rate of apoptosis, increased degranulation, reducing their cytotoxic ability, and diminishing the expression of activating receptors on the NK cells [63, 66]. Thus, CSCs develop several mechanisms to evade NK cell-mediated immune surveillance by modulating cell surface molecules that help recognize them and also by releasing immunosuppressive soluble factors, e.g., TGF, human leukocyte antigen-g (HLAg), prostaglandins, and adenosine, in TME [129, 130].
B cells (bursal-derived lymphocytes) are key participants in humoral immunity due to their ability to produce immunoglobulin (Ig) or antibodies [131]. In the majority of cancer types, B cell infiltration is associated with a favourable outcome [132]. To begin with, tumor-specific antigens are recognized by the B cell receptor (BCR), which is made up of membrane-anchored antibodies and co-receptor molecules. When exposed to an antigen for the first time, naive mature B cells become activated, capable of proliferating and differentiating into plasma cells, which generate and release antibodies [133]. Additionally, B cells contribute to immunity in ways other than antibody generation, most notably through the secretion of cytokines that activate T cells, DCs, lymphoid tissue remodelling, and neogenesis. B cells show their potential antitumor activities via serving as an APC or by directly killing cancer cells [134]. B cells also interact with T cells and cluster in tertiary lymphoid structures (TLS) within tumors, where naive T cells are assumed to be activated, implying that B cells may play a role in antitumor immunity by collaborating with T cells [135]. Beside antitumoral activity of B cells, an emerging body of evidence has recently demonstrated a pro-tumoral B cell-subset are designated as B-regulatory (Breg) cells [136]. It was also found that Breg cells deregulates class switching recombination to suppress antibody response in cancer patients [137].
Early detection of neoantigen-expressing cells engaging several immune cell types, is a complex process [35]. Stemming examples demonstrate the innate immune system’s instrumental and decisive functions in eliciting antitumor immunity within the tumor onset window period [34, 36, 38, 39]. APC subtypes coordinate and produce an immune response against neoantigen-expressing cells even in the absence of PAMPs which are generally necessary to licence APCs to present foreign or self-antigens as immunogens [33]. When mutated gene products are broken down and displayed as neoepitopes on the patient’s MHC molecules, they can behave as tumor neoantigens. This immunological complex allows surveying monocytes to present neoantigens to CD4+ T cells as immunogens. CD4+ Th cells use CD40L to licence cross-presenting DCs to induce a CTL response against neoantigen-expressing cells [138]. Th cells direct the development of antigen-primed B cells in secondary lymphoid tissues which secrete natural IgM required for the detection of neoantigen-expressing cells and the beginning of adaptive immunity [138]. Thus, adaptive immune responses take some time to get primed, expand, and secrete antibodies against an antigen. But the number of cancerous cells increase during this time course since they have endless proliferating capacity. Without making any delay, our body triggers innate immune responses to engulf the changed self-cells or eliminate the premalignant cells via perforin-granzyme mediated cytotoxicity [34, 36].
So far, this review has discussed that CSCs are often responsible for tumor initiation and progression. However, the question is always raised that despite having strong immune surveillance mechanisms, how could CSCs get undetected by the immune system? Generally, CSCs play comprehensive roles in modifying the body’s immune response in favor of its own survival by creating immunosuppressive conditions via multiple mechanisms (Figure 3). By producing GM-CSF, periostin, and cyclooxygenase-2, CSCs recruit TAMs and polarize them into the pro-tumor M2 phenotype [139–141]. Studies reported that CSCs produce various substances that aid in infiltration as well as formation of MDSCs to generate an immune-suppressive milieu [87, 88, 142]. Tumor-initiating CSCs also express various chemokines, like CCL1 and recruit immunosuppressive Treg cells via NFκB-CCL1 signaling axis [100]. Such findings shed light on the functional relationship between Tregs and CSCs, thereby pointing towards a promising future cancer therapeutic method. It has been already mentioned that cancer cells attract immunosuppressive cells via generating numerous factors such as IDO [143], TGF [63, 144], IL10, and PDL1, thereby avoiding immune responses [63, 66, 96]. Remarkably, current studies show that CSCs, express the same set of markers as IL10 [145], IL4 [61], IDO [101], and PDL1 [118, 119]. Therefore, the above-mentioned immunosuppressive environment might also be generated by the CSCs during initiation.
Recent report showed that EMT-β-catenin-STT3 (oligosaccharyltransferase complex)-PDL1 signaling-axis enriches PDL1 in CSCs [119]. This axis is also used by the general cancer cell population, although it has a far greater impact on CSCs than NSCCs to promote immune evasion [119]. CSCs have been shown to downregulate the expression of transporter associated with antigen processing (TAP), a vital player in antigen processing and presentation machinery in order to mask the recognition by adaptive immune system [66, 146]. CSCs also avoid the cytolytic effects of NK cells by downregulating NK group-2, member D ligands (NKG2DL) expression [147]. Such immune evasion is essential for the replication of TICs which further gives rise to differentiated tumor cells, resulting in formation of a heterogeneous tumor bulk [5]. Similar incidents take place when a tumor relapses after chemotherapy. Besides the immune-regulatory effects of CSCs, as mentioned above, the stressed immune system following chemotherapeutic shock, allows drug-resistant CSCs to resurface from lymph nodules, and initiate tumor [9]. As the arena of research involving CSC-immune cross talk is a recently emerged field, the information regarding how CSCs re-educate the antitumor immune system to generate pro-tumor one, is not yet complete. More intense research is needed to know in depth the full mechanisms underlying CSC-mediated immune-suppression during tumor development and progression.
Strong evidences suggest that CSCs give unique inputs to the TME in order to positively govern their survival and behaviour, dictating their ability to remain stem-like, invasive, and resistant to drug-induced apoptotic signals. Because CSCs have different treatment resistance modalities by nature, eliminating CSC pools is a valuable strategy for achieving a relapse-free cancer cure [63, 148] (Figure 4). Exploiting a variety of factors that make immune effectors more immunogenic towards CSCs and other resting cells in the tumor niche could aid in the development of novel therapeutic approaches. Several treatments have been developed with the goal of eliminating CSCs, including (i) specifically targeting CSCs, (ii) modifying their stemness, and (iii) influencing the microenvironment. In this case, monoclonal antibody-based therapies are best suited to target the markers commonly used to isolate and detect CSCs [149].
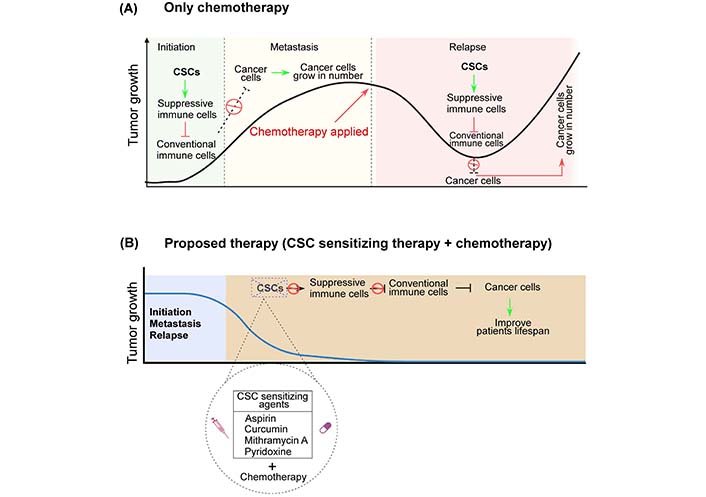
Hypothesis. Schematic diagram showing: (A) Recent CSC theory portrays CSCs as the driver of tumor initiation and progression. Having the potential to kill cancer cells only, conventional chemotherapies not only render the CSC pool unaffected but also increase their numbers. After escaping chemotherapeutics, these drug-resistant CSCs might cause tumor remission via differentiation or self-renew, aggravating tumor condition; (B) these tumor-initiating CSCs must thus be targeted or sensitized for better therapeutic outcome. First sensitizing CSCs using various small molecule inhibitors like curcumin, mithramycin A, pyridoxine etc. followed by their killing utilizing conventional chemotherapy, for tumor regression from the root. Created with Biorender.com
CSCs can also be selectively suppressed by compounds that inhibit drug-efflux pumps and detoxifying enzymes [1]. By targeting CSC-specific pathways including Hedgehog, Wnt, Notch, and focal adhesion kinase by CSC-specific cytotoxic drugs, positive results have been obtained [150]. All-trans retinoic acid [151, 152], histone deacetylase inhibitor [153], bone morphogenetic proteins [154, 155], and cyclopamine [156] are a few of the compounds that induce cell differentiation while suppressing CSCs’ specialized stemness. Furthermore, various small molecule inhibitors like mithramycin A, curcumin, andrographolide [157–160] or conventional drugs have been repurposed to sensitize CSCs prior to chemotherapy [161]. In this regard, repurposing aspirin for successfully sensitizing breast CSCs towards chemotherapy has been reported recently from this group [13, 29, 162, 163]. Targeting the interaction between tumor-stroma and nearby cancerous cells by inhibiting paracrine signaling molecules such as VEGF, hypoxia-inducible factor, CD44v, and CXCR4, also holds alluring prospects for the successful eradication of CSCs [164]. Furthermore, the chemotherapeutic drug gemcitabine and the ALDH1A1 inhibitor disulfiram have been reported to suppress breast tumor development by increasing T-cell immunity and eliminating ALDH+ TICs [87]. All these findings reveal novel therapeutic approaches for targeting cancer from its “root”.
Cancer immunotherapy is an emerging treatment strategy that re-educates host’s immune system to fight against cancer [165]. Although several immunotherapeutic strategies are employed in cancer treatment, low patient response rates remain a significant obstacle. In fact, plethora of basic science discoveries and subsequent clinical translation finally could undeniably demonstrate the supremacy of immune system modulation in tumor regression. Further research involving regulation of T cells and other immune cells, e.g., APCs and NK cells, might allow the scientists to improve the efficacy of this approach [166–168]. The effects observed in clinical trials of checkpoint blockade agents [169, 170], adoptive T cell transfer therapies [170], and cancer vaccines [171, 172] in “difficult to treat” tumors were far greater than the most effective chemotherapeutic agents. Even though immune-related side effects are common, these novel immuno-targeting therapies are more well-tolerated than traditional chemotherapeutic agents but definitely is cost-effective [173].
The field of NK cell-based cancer therapy has advanced significantly and is now a critical area of immunotherapy innovation. The development of NK cell-directed therapeutics should prioritize two objectives: increasing the source of therapeutic NK cells for adoptive transfer and increasing their in vivo cytotoxicity and durability while avoiding CSC-mediated suppression [174–176]. Immunotherapy that combines the benefits of cellular and antibody-based therapies employs chimeric antigen receptor-expressing T (CAR-T) cells [177, 178]. CAR-T therapy involves the administration of autologous CD4+ and CD8+ lymphocytes that have been modified to express CAR on their cellular surfaces. This allows the treated cells to recognize and respond to a specific antigen without requiring an earlier antigen-presenting procedure. There is also evidence that CAR-Ts that specifically target markers such as CD44v6, CD133, EpCAM, and CD54 can effectively eradicate CSCs [179–182]. But CSCs can limit the activity of CAR-T cells regardless of whether they are targeted or sensitized.
This review highlights the role of tumor-initiating CSCs in modulating the immune-landscape of the cancer patients, raise the important issue that any effort to boost host via present-day available cancer immunotherapy might finally activate the pro-tumor immunity and not the antitumor immune response of the patient through CSC-induced re-education of the same, thereby leading to failure of the high-cost therapy. Disrupting the deadly liaison between CSCs and immune cells should therefore be the cutting-edge research target to finally achieve fully successful immune-therapy of cancer in near future.
This review establishes the instrumental role of CSCs during tumor initiation, metastasis to a secondary organ, and tumor relapse. CSC-regulated re-education of immune cells for ensuring the survival of these TICs during tumor initiating phases bolsters the paramount significance that CSCs uphold in tumor formation, propagation, and immune-modulation. This review article has discussed how the anti-cancer immune-subsets infiltrate the tumor site and eradicate cancer cells via multitude of mechanisms. However, tumor-initiating CSCs manipulate the TME in its favor by promoting infiltration of pro-tumor immune-subsets such as Treg and TAMs, as well as, by re-educating the phenotype from pro-inflammatory to an immune-suppressive one, thereby, establishing a deadly liaison between CSCs and pro-tumor immune cells. As a result, the tolerogenic immune milieu is formed, further supporting CSCs, thereby ensuring tumor initiation and growth. Hence, it becomes imperative to develop therapies targeting CSCs and their interaction with immune cells rather than already established conventional therapeutic regimens for getting successful and relapse-free survival of cancer patients. Consequently, it highlights few treatment avenues which either specifically sensitize CSCs or attack the CSC-immune cross-talks. However, future studies are essential to be conducted for effective targeting of CSCs to successfully eliminate tumor. To that end, appropriate model systems employing authenticated cell surface markers and/or reporter systems, properly validated for CSC activities, are required to investigate the interaction of these cells with the immune system. Without acquiring the modalities for targeting CSCs and understanding CSC-mediated complexities of the TME, it will be difficult to develop effective immune-modulatory treatment procedures of cancer including the flourishing field of cancer immunotherapy. In fact, the prospective success stories of cancer immunotherapy, emphasize the requirement of coherence between basic CSC research and clinical practice. This review demonstrates how a bench-to-bedside approach, based on CSC research, will pave the way for a successful fight against one of human’s most feared diseases.
ALDH: aldehyde dehydrogenase
APCs: antigen-presenting cells
CAR-T: chimeric antigen receptor-expressing T
CCL: chemokine ligand
CD: cluster of differentiation
CSCs: cancer stem cells
CTLs: cytotoxic T lymphocytes
CXCR4: C-X-C chemokine receptor type-4
DCs: dendritic cells
ECM: extracellular matrix
EGF: epidermal growth factor
EMT: epithelial-mesenchymal transition
FOXP3: forkhead box P3
GM-CSF: granulocyte-macrophage colony-stimulating factor
IDO1: indoleamine 2,3-dioxygenase 1
IFNγ: interferons gamma
MDSCs: myeloid-derived suppressor cells
MHC: major histocompatibility complex
MIF: migration inhibitory factor
NFκB: nuclear factor kappa-B
NSCCs: non-stem cancer cells
PDL1: programmed death-ligand 1
TAMs: tumor-associated macrophages
TGFβ: transforming growth factor-β
Th: T-helper
TICs: tumor-initiating cells
TIDCs: tumor-infiltrating dendritic cells
TME: tumor microenvironment
Treg: T-regulatory
VEGF: vascular endothelial growth factor
The authors gratefully acknowledge Indian Council of Medical Research (ICMR), and Council of Scientific & Industrial Research (CSIR) for financial supports.
TD: Conceptualization, Supervision, Writing—review & editing. GS: Conceptualization, Supervision, Project administration, Writing—review & editing. SC: Writing—original draft, Investigation. SM: Writing—original draft, Data curation. UB: Writing—original draft, Validation. SP: Visualization, Resources. Apratim D: Methodology, Resources. Saikat D: Investigation. SD: Methodology. TS: Validation. AG: Formal analysis.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was funded by Indian Council of Medical Research (ICMR) [74/1/2020-Pers] and Council of Scientific & Industrial Research (CSIR) [27(0355)/19/EMR-II]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

