 Review
Review

Affiliation:
1Department of Life Sciences, University of Trieste, 34127 Trieste, Italy
Email: mariagiulia.spazzapan@phd.units.it
ORCID: https://orcid.org/0000-0002-4338-0942
Affiliation:
2Institute for Maternal and Child Health, IRCCS Burlo Garofolo, 34137 Trieste, Italy
ORCID: https://orcid.org/0000-0002-1819-7417
Affiliation:
2Institute for Maternal and Child Health, IRCCS Burlo Garofolo, 34137 Trieste, Italy
ORCID: https://orcid.org/0000-0002-8315-2258
Affiliation:
1Department of Life Sciences, University of Trieste, 34127 Trieste, Italy
ORCID: https://orcid.org/0000-0003-1737-5776
Explor Immunol. 2023;3:574–589 DOI: https://doi.org/10.37349/ei.2023.00122
Received: June 07, 2023 Accepted: November 25, 2023 Published: December 14, 2023
Academic Editor: Dominique J. Charron, Hospital Saint Louis, France
The article belongs to the special issue The Complement System in Health and Disease
The complement component C1q plays a role as a pro-angiogenic factor in different contexts, acting in a complement-independent way. For example, this molecule is able to foster the remodeling of the spiral arteries for a physiological pregnancy and to promote the wound healing process. It is also involved in angiogenesis after post-stroke ischemia. Furthermore, it has a role in supporting the tumor vessel growth. Given its role in promoting angiogenesis both under physiological and pathological situations, other studies are needed to understand its potential therapeutic implications.
C1q is mostly known for its role in activating the classical pathway of the complement system [1], but it is also responsible for the clearance of apoptotic cells and immunocell recruitment and is involved in the coagulation process [2]. Besides these actions, it exerts a broad range of non-canonical complement-independent functions. For example, in the central nervous system, it has a neuroprotective role interacting with microglial cells [3] and is also involved in synapsis pruning during fetal brain development [4]. In addition, it has a role in healthy pregnancy, being important for normal placentation [5]. Moreover, this molecule is linked to aging, since its serum concentration is increased in elderly people [6]. Furthermore, this protein can be considered an important pro-angiogenic factor.
The aim of this review is to examine the different contexts in which C1q could act as an angiogenesis-stimulating factor.
C1q has a distinctive structure, typically described as a bouquet of tulips; it is composed of six subunits constituted by a C-terminal heterotrimeric globular head C1q (gC1q) domain and an N-terminal triple-helical collagen-like C1q (cC1q) region, holding the subunits together as a bouquet. Each of these subunits is a heterotrimer formed by three chains, named C1qA, C1qB, and C1qC [7–9]. These three polypeptides are the product of three different genes located on chromosome 1p [10].
The major source of C1q is the monocyte/macrophage lineage [11], but evidence suggests that dendritic cells, fibroblasts, epithelial, mesenchymal [12], endothelial [13, 14], and lastly, trophoblast cells [15] are also able to produce it, at least under certain conditions.
The two domains of C1q permit its binding to a wide range of ligands. For example, the gC1q domain is able to bind to self-, non-self-, and modified self-ligands representing the key to C1q versatility [12, 16]. On the other hand, the cC1q domain can be recognized by several putative receptors and is involved in the C1q-mediated chemotaxis and in the clearance of apoptotic cells by macrophages [17].
C1q, together with two serine proteases C1r and C1s, is part of the C1 complex (C1qr2s2). The interaction of C1q with activators, such as antigen bound-immunoglobulin G (IgG) or IgM, necrotic or apoptotic cells, or proteins of the extracellular matrix (ECM), leads to conformational changes in C1q molecule that in turn activates C1r, which activates C1s, thereby initiating the classical complement cascade (Figure 1) [18].
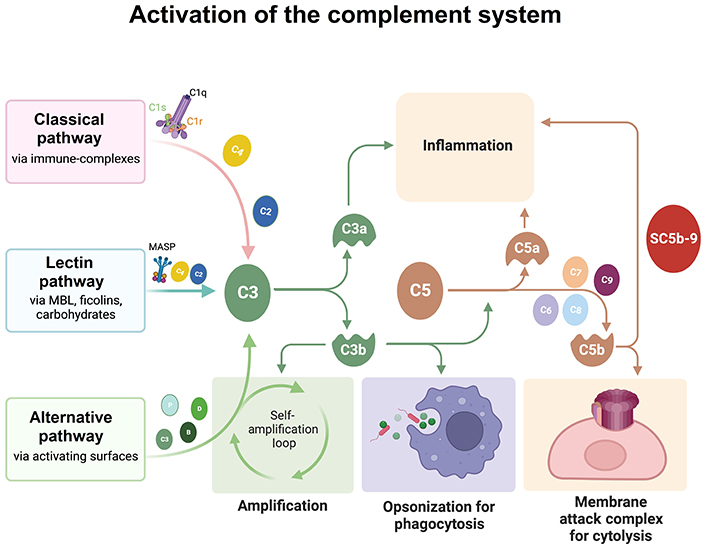
Schematic representation of the three activation pathways of the complement system. All of the three pathways converge on the cleavage of C3 in C3a and C3b. C3b attaches to C3, becoming C5 convertase, which in turn cleaves C5, originating C5b. C5b, together with C6, C7, C8, and C9 forms the membrane attack complex. The Figure was created with BioRender software (https://biorender.com/). MASP: mannose-associated serine protease; MBL: mannose-binding lectin
This process is tightly regulated by a C1 inhibitor (C1 INH) which acts in two ways: by binding to the precursor of C1r and C1s preventing autoactivation and by covalently interacting with the active sites of C1r and C1s. This latter mechanism inactivates the catalytic function of C1r and C1s, causing their dissociation from the complex and resulting in free-serum C1q [19–21].
Thanks to its different domains, C1q interacts with several cell types through distinct receptors, exerting multiple functions, associated with both innate and adaptive immunity, and involved in tissue differentiation and cancer [12, 22, 23].
The most known and well-characterized receptors for C1q are:
gC1q receptor (gC1qR). The receptor for gC1q, also known as hyaluronan binding protein 1 (HABP1), p32 or p33, or C1q binding protein (C1qBP). Interestingly, one of the transcripts of the gC1qR gene codifies for a truncated isoform of the receptor. This protein lacks the first 104 amino acids, constituting the C1q binding region. The result is a smaller protein with a molecular weight of 14–19 kDa, compared to 32–33 kDa of the main transcript. A bioinformatics approach predicted the membrane localization for this isoform, in contrast with the mitochondrial localization of the main transcript. Moreover, Kahraman et al. [24], by bioinformatic analyses, associated this isoform with a tumoral microenvironment.
cC1q receptor (cC1qR). It is the receptor for the collagen-like domain, known as the collectin receptor or calreticulin. Interestingly, both of them (gC1qR and cC1qR) are primarily localized at the level of intracellular organelles, such as the endoplasmic reticulum for the cC1qR and the mitochondria for the gC1qR. Nevertheless, they were also detected in the cell membrane of a variety of cells [7, 10, 25–27]. Biochemical studies had demonstrated that both receptors lack the transmembrane domain or lipid anchor, so their intracellular signaling cascades should be mediated by other molecules forming complexes, such as CD91, CD209, CD44, and β1 integrin [12, 23, 28–31].
Complement receptor 1 (CR1, CD35): named also the leukocyte C3b/C4b receptor, is present on erythrocytes and most peripheral blood leukocytes. It is a transmembrane receptor for the cC1q [32] and is primarily involved in the clearance of complement-opsonized components from the bloodstream, having a role in controlling the complement-mediate attack. CR1 gene is a member of the regulators of complement activation (RCA) family and is located in the “cluster RCA” region of chromosome 1. The extracellular domain of CR1 can be divided into 30 short consensus repeats (SCRs) also known as complement control protein (CCP) repeats or sushi domains. Besides C1q, CR1 also recognizes the activation products C3b and C4b and binds MBL and ficolins [33].
A broad range of complement-independent activation responses are stimulated by C1q; for example, it induces leukocyte oxidative response, phagocytosis, inhibition of B and T cell proliferation, fibroblast and endothelial cell (EC) adhesion, trophoblast migration, regulation of dendritic cells, chemotaxis of eosinophils, mast cells, neutrophils, and angiogenesis [12, 13, 15, 34–40].
The sprouting of new blood vessels can either refer to vasculogenesis or to angiogenesis and those are two distinct events [41]. The term vasculogenesis indicates the formation of the vascular network during embryo development; in this period, endothelial progenitor cells (EPCs) migrate, differentiate, and assemble into new vessels. In particular, mesoderm-derived hemangioblasts move to the yolk sac; here, they organize themselves into blood islands originating from endothelial and primitive blood cells. These islands fuse and form the extra-embryonic blood network. Within the embryo, precursor cells proliferate and connect in a net that then differentiates into the primary vascular plexus, which will originate the rest of the vasculature [42]. In some cases, it is also possible that vasculogenesis occurs after birth, for example, in ischemic tissues and tumors. In fact, EPCs might be recruited from the bone marrow (BM) and incorporated into new vessels. Additionally, these progenitors may also release pro-angiogenic factors stimulating vessel growth. The mobilization and the recruitment of BM-derived progenitors to the site of vasculogenesis implicates molecular mechanisms that induce the expression of growth factors and specific receptors, adhesion molecules, cytokines, and matrix metalloproteinases (MMPs) [43, 44].
Unlike vasculogenesis, the angiogenic process consists of the sprouting of new capillaries and vessels from pre-existing vasculature [45]. Besides ECs, it involves the stabilization of new vessels by pericytes or vascular smooth muscle cells. This process plays a fundamental role in pathological and physiological events, such as development and reproduction. In fact, the regulation of placental angiogenesis is fundamental for physiological course of pregnancy; furthermore, new vessel formation is essential in the ovarian context to transform the ovulated follicles into the corpus luteum, and in the uterus to renovate the endometrium in order to ready for embryo implantation [46]. In addition, during the wound healing process, both angiogenesis and vasculogenesis occur to restore damaged vessels [47].
New vessel growth happens as sprouting angiogenesis (SA) or intussusceptive angiogenesis (IA) [48]. This latter mechanism is characterized by the splitting of existing blood vessels [49]. During this process, ECs of opposing inner vessel walls connect forming a transluminal bridge and intracellular junctions reorganize themselves to perforate the endothelial bilayer creating an interstitial pillar core. Lastly, myofibroblasts and pericytes start to cover the interstitial wall of the pillar and this structure fuses with adjacent pillars to split one capillary into two parallel capillaries (Figure 2A) [50]. IA is the fastest mechanism to expand a plexus and, being almost independent of EC migration and proliferation, it is supposed to be less energetically demanding [51]. On the other hand, SA is the most efficient mechanism. The development of new capillaries is led by specialized ECs, named tip cells, located on the top of the growing vessel. This type of cell senses a gradient of angiogenic factors [e.g., vascular endothelial growth factor (VEGF); see below] released by oxygen- and nutrient-deprived microenvironment and becomes activated [48, 52]. These cells start to secrete MMPs with the aim of degrading the basal membrane and invading surrounding hypoxic tissues [53]. They have a low proliferative ability but are characterized by lamellipodia and filopodia, which allow them to actively migrate in the ECM. Tip cells are followed by stalk cells, which are in contrast high proliferating cells, mediating lumen formation, and establishing adherent and tight junctions to provide stability to the newly formed vessel [46, 54, 55]. Once two tip cells meet, they anastomose to form a perfused new vessel and pericyte recruitment stabilize the vessel. Furthermore, the deposition of the basal membrane starts (Figure 2B) [52]. When vascularization occurs, the levels of pro-angiogenic factors diminish, and ECs reestablish their quiescent phenotype [46].
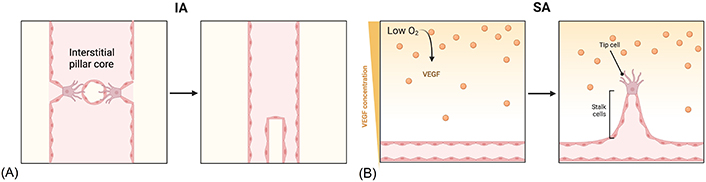
Alternative mechanisms of angiogenesis. (A) IA starts with protrusion from opposite walls into the lumen. The endothelium becomes centrally perforated and a transluminal pillar is formed and it increases in girth after being covered by pericytes; (B) SA begins when ECs sense a hypoxic microenvironment and a VEGF gradient; after the basal membrane degradation, tip cells start to migrate towards the angiogenic gradient, followed by stalk cells; when two tip cells meet, they anastomose, a new vessel is formed, and ECs return to a quiescent phenotype. The Figure was created with BioRender software (https://biorender.com/)
At a molecular level, the control of tip and stalk cell fate occurs via the Notch pathway. In tip cells, the activation of VEGF receptors (VEGFRs) stimulates the expression of Notch ligand delta-like protein 4 (DLL4), which triggers the Notch signaling cascade in adjacent stalk cells, which prevents those cells from acquiring tip cell behavior by the suppression of the VEGFR and the concomitant upregulation of a lateral inhibitory mechanism [46, 52, 56].
Multiple VEGFs (VEGF-A, VEGF-B, VEGF-C, and VEGF-D) interact with VEGFRs, such as VEGFR1 [fms-like tyrosine kinase-1 (Flt-1)], VEGFR2 [Flt-2 or kinase insert domain receptor (KDR)], and VEGFR3 (Flt-4) [57]. This protein family also includes placental growth factor (PlGF), originally discovered in the placental tissue and not highly expressed in normal embryonic or adult tissues [58, 59].
Little is known about VEGF-B, aside from its role in coronary artery development. VEGF-C and -D are crucial for lymphatic system development and they can also induce angiogenesis and increase vascular permeability [60, 61]. However, VEGF-A is the major angiogenic factor that controls vessel formation. Its expression is strictly regulated by different factors, including hypoxia, through the transcriptional factor hypoxia-inducible factor 1 (HIF1), other growth factors, cytokines [e.g., transforming growth factor-β (TGF-β)], hormones (e.g., estrogen and progesterone), and expression of oncogenes and tumor-suppressor genes [62]. The alternative splicing of the VEGF-A gene results in several transcripts; the most studied one is the VEGF-A165. Nevertheless, the different isoforms exert many of the same functions: They increase vascular permeability, stimulate EC proliferation, migration, and survival, and provide anti-senescence signals [63–65]. These effects are mediated through the binding to specific tyrosine kinase receptors. VEGFR-1 (Flt-1) binds VEGF-A, VEGF-B, and PlGF; VEGFR-2 (Flt-2 or KDR) recognizes and binds VEGF-A and processed forms of VEGF-C and -D; VEGFR-3 (Flt-4) binds unprocessed forms of VEGF-C and -D [61]. In addition, a non-kinase receptor, neuropilin-1 (NRP-1) binds PlGF and VEGF-A165 [66]. VEGFR-1 and -2 are extensively expressed in vascular endothelium under physiological situations but became upregulated in pathological angiogenesis induced by VEGF-A [67]. VEGF-A165 has a role in mobilizing ECs and hematopoietic precursors from BM precursors which also express VEGFR-1 and -2 [68]. A soluble form of VEGFR-1 [soluble Flt (sFlt)] is present in serum and maintains VEGF-A and PlGF-binding activity. It is associated with a pathological role in pre-eclampsia, a complication of pregnancy characterized by vascular abnormalities in the kidney and other organs [69]. VEGFR-1 is also present in uterine smooth muscle cells [70], and in monocytes, cells that may be responsible for pathological angiogenesis by expressing VEGF-A and other cytokines and growth factors [71]. Evidence suggests that some VEGF-A receptors, particularly VEGFR-1 and NRP-1, are expressed by some tumor cells, suggesting an autocrine loop that may stimulate tumor cell growth and migration [72]. Usually, the binding of VEGF-A triggers a dimerization of VEGFR-2 and activates crucial signaling pathways, including the phospholipase Cγ (PLCγ)-extracellular signal-regulated kinase 1/2 (ERK1/2) pathway with a role in vascular development and adult arteriogenesis; phosphatidyl-inositol 3-kinase (PI3K)-protein kinase B (PKB/AKT)-mechanistic target of rapamycin (mTOR) pathway involved in cell survival, regulation of vasomotion and of barrier function; and SRC kinases (SRC proto-oncogene, non-receptor tyrosine kinase) and small G-proteins (guanine nucleotide-binding proteins) that control cell migration, polarization, and endothelial junctions [56].
PlGF is a member of the VEGF family able to bind to VEGFR1 or its soluble form but fails to bind to VEGFR2 [73]. PlGF increases the proliferation, migration, and survival of ECs [58], but can also stimulate the proliferation of mesenchymal fibroblasts [74] and recruits myeloid progenitors and macrophages to growing vessels [75, 76]. PlGF is also able to up-regulate the expression of angiogenic factors such as VEGF, basic fibroblast growth factor 2 (FGF2), platelet-derived growth factor β (PDGFβ), and MMPs [77]. However, it seems that PlGF plays a minor role in physiological angiogenesis and is more associated with pathological conditions, like pre-eclampsia or the tumoral context [78].
Besides the VEGF/VEGFR pathway, angiopoietins (ANGs) trigger angiogenesis through the interaction with tyrosine kinase with Ig-like and epidermal growth factor (EGF)-like domains (TIE) receptors, TIE1 and TIE2 [or tyrosine endothelial kinase (TEK)]. The ANG family includes well-characterized isoforms such as ANG1 and ANG2, and less studied isoforms such as ANG4, as well as ANG3. TIE1 and TIE2 are co-expressed on both vascular and lymphatic ECs. TIE2 is constitutively expressed on ECs, whereas the transcription of TIE1 appears to be more regulated. TIE receptors are expressed by circulating hematopoietic cells and by hematopoietic stem cells resident in the BM niche. These receptors are also expressed by several tumor cell populations [79]. TIE2 is the main binding receptor in the ANG/TIE signaling pathway for all ANGs. On the other hand, TIE1 does not bind any ANGs [80]. ANG1 is mainly produced by peri-ECs (pericytes and smooth muscle cells) and is a potent TIE2 activator that supports vascular stability and endothelial barrier function [81]. At a molecular level, in quiescent vasculature, when ANG1 binds to TIE2, the receptor becomes phosphorylated and triggers the activation of the PI3K-AKT-mTOR pathway resulting in the inhibition of the forkhead transcription factor 1 (FOXO1). The consequence is the expression of genes involved in vascular stability and maturation, and the suppression of transcription of factors involved in vascular destabilization, such as ANG2 [81–83]. In contrast, ANG2 acts both as an agonist and an antagonist of TIE2 in a context-dependent manner; it is expressed only by ECs and usually stored within the Weibel-Palade bodies. Under healthy conditions, ANG2 overexpression stimulates vascular remodeling without leakage or destabilization, suggesting ANG2 agonism of TIE2 signaling [84]. Its production is triggered by hypoxia, VEGF, shear stress, and increased levels of inflammatory cytokines [85]. During inflammation, ANG2 competitively inhibits ANG1 activation of TIE2 signaling, causing vessel destabilization and leakiness [86]. The mechanism involves the suppression of the PI3K/AKT signaling, leading to the activation of FOXO1 and ANG2 expression [87].
The interaction between the complement system and ECs triggers physiologic changes in cell shape and behavior [88, 89]. Those modifications could be similar to the decidua remodeling occurring during pregnancy. There is accumulating evidence suggesting a pro-angiogenic role of C1q, at least in some specific microenvironment. For example, in the female reproductive system, Bulla et al. [14] found the positivity for C1q on the endothelium of the spiral arteries in healthy placental tissue. Moreover, it was demonstrated that, during pregnancy, decidual ECs (DECs) start to produce and bind C1q onto their surface. In addition, the inability to detect C4 leads to the understanding that the presence of C1q on DECs was independent of complement activation. C1q present on DEC membrane could be recognized by gC1qR on trophoblasts migrating into the spiral arteries, acting as a molecular bridge between decidual endothelium and endovascular trophoblasts [14]. Masat et al. [90] suggested that C1q expressed by DECs may also contribute to maintaining the anti-inflammatory behavior of this type of ECs, characterized by strong constitutive activation of the non-canonical nuclear factor kappa B (NF-κB) pathway and low responsiveness of the canonical pathway to lipopolysaccharides (LPS).
To summarize, the results of these studies indicated that C1q was produced by invading trophoblasts and by ECs of the spiral arteries (Table 1). At a molecular level, C1q was able to interact with both gC1qR and α4β1 receptors that are present on trophoblasts. Its binding induces the phosphorylation and the subsequent activation of the ERK1/mitogen-activated PK (MAPK) signaling, a pathway involved in cell adhesion and migration [15].
Pro-angiogenic role of C1q in various contexts
| Angiogenic role of C1q in: | Key finding(s) | Model(s) | Reference |
|---|---|---|---|
| Pregnancy | C1q acts as a molecular bridge between DECs and invading trophoblast, triggering the activation of ERK1/MAPK signaling cascade | In vitro assays (human): DECs, trophoblasts | [14] |
| Wound healing | C1q is expressed at the wound site and has a pro-angiogenic effect similar to VEGF; involvement of gC1qR | In vitro assays (human): HUVECs Ex vivo assays (rat): aortic ring assay | [13] |
| Post-stroke | C1q stimulates the formation of new capillaries, having a pro-angiogenic effect; involvement of LIAR1 | In vivo assays (rat): pMCAO rat model In vitro assays (human and rat): RBMECs and HBMECs | [91] |
| Cancer | C1q stimulates angiogenesis in some types of tumors | In vivo assays (mouse): C1qa–/– mouse | [92] |
HUVECs: human umbilical vein ECs; pMCAO: permanent middle cerebral artery occlusion; RBMECs: rat brain microvascular ECs; HBMECs: human brain microvascular ECs; LIAR1: leukocyte-associated Ig-like receptor 1
The wound healing process is characterized by four well-orchestrated phases: hemostasis, inflammation, proliferation, and remodeling. After the acute inflammatory phase, cells of the immune system start to produce growth factors that stimulate the proliferation of ECs and fibroblasts. During this phase, the synthesis of collagen also occurs to restore the integrity of ECM. The next stage involves the beginning of angiogenesis and the formation of granulation tissue. Then, keratinocytes migrate in the granulation tissue and start the re-epithelization of the wound. Chronic wounds are defined as barrier discontinuity after damage that lasts longer than 42 days. This condition is characterized by an imbalance in the production of pro- and anti-inflammatory cytokines and by the failure to progress beyond the inflammatory phase, precluding the proliferation of ECs. Given the fundamental role of angiogenesis in wound healing, its stimulation could represent a potential therapeutic approach for treating chronic wounds [93, 94].
In general, the interaction between C1q and the endothelium induces an inflammatory response, but, surprisingly, it can also stimulate angiogenesis via its globular heads. Its binding to cC1qR and gC1qR which are present on the ECs, stimulates the spreading of these cells and the production of adhesion molecules [35], as well as the synthesis of chemotactic factors such as interleukin-8 (IL-8) [95]. Furthermore, C1q is able to recognize apoptotic ECs [96].
Since previous studies reported that C1q is associated with vessel development during the placentation acting as a molecular bridge between the invading trophoblast and the spiral arteries, and given the fact that this process involves deep changes in the local tissues, it was hypothesized that this molecule could be involved in microenvironment associated with tissue remodeling [10, 14, 15, 88].
Wound healing is an ideal process for studying angiogenesis. Bossi et al. [13] demonstrated that C1q was locally expressed in the vessels of granulation tissue, but not in normal skin, and the complement system downstream molecules (e.g., C4 and C3) were absent in the wound, supporting the hypothesis that this molecule was not derived from the activation of the classical pathway, but had a role independent of complement system activation. Its presence was associated with vascular remodeling and a pro-angiogenic environment as confirmed by real time-quantitative polymerase chain reaction (RT-qPCR) and in situ hybridization. Results of these assays demonstrated the C1q expression in the wound tissue, but not in intact skin. The angiogenic effect was investigated by permeability, proliferation, and migration assays conducted with HUVECs. C1q stimulation induced vascular leakage by loosening the cell junction and activated cell proliferation, evaluated by single-cell analyses of the proliferation marker Ki-67. The migration ability of C1q-stimulated ECs was evaluated by scratch assay and transwell model system and results confirmed a pro-angiogenic role of C1q. Tube formation assay demonstrated the ability of this complement component to induce the formation of capillary-like structures by ECs seeded onto Matrigel. Ex vivo aortic ring assays showed that C1q stimulated not only the microvessel formation but also the placement of pericytes around the new vessels. Finally, C1q-deficient mice showed limited vessel formation in wound skin samples compared to the skin of the wild type (WT) mice. Remarkably, the angiogenesis was restored when C1q was added locally. To identify which was the receptor of C1q involved in its pro-angiogenic role, anti-gC1qR or anti-cC1q antibodies were used in the migration assay. The greatest blocking effect was obtained by using antibodies against gC1qR, whereas the use of anti-cC1qR antibodies did not exert an inhibitory result. Based on previous findings by Agostinis et al. [15], it was hypothesized that the main pathway mediating angiogenesis activation could be ERK1/MAPK pathway. The investigation of phosphorylated status of ERK1, assessed by immunoblotting, confirmed this idea. In conclusion, C1q stimulated angiogenesis, in a VEGF comparable level [13] and C1q-mediated angiogenesis seems to be fundamental for proper wound healing (Table 1).
Further evidence suggested that C1q is also involved in post-stoke-associated angiogenesis. Fan et al. [91] used pMCAO rat model to demonstrate that after 5 days of brain ischemia, the C1q expression increased compared to control rats without pMCAO. Furthermore, when the expression of C1q was silenced through silencing RNA (siRNA), the number of capillaries in the ischemic brain zone decreased compared to the control group. Regarding the in vitro studies, viability and angiogenic behavior of RBMECs and HBMECs were evaluated by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), transwell system migration, and tube formation assays. Results indicated that C1q-treated cells increased their viability, their migration, and the ability to form capillary-like structures. The pro-angiogenic behavior was inhibited when anti-VEGF antibodies were used, suggesting its involvement in the C1q stimulation. Fan et al. [91] hypothesized the involvement of LIAR1, a transmembrane protein belonging to the C1q receptor group. The study by Fan et al. [91] showed that LIAR1 silencing in C1q-treated RBMECs diminished the C1q-enhanced VEGF expression, migration, and tube formation. At a molecular level, the expression of HIF1α, the transcriptional factor responsible for the VEGF expression under hypoxic conditions, was also reduced, suggesting its involvement in this pathway (Table 1) [91].
Neoangiogenesis is a crucial process that supports tumor growth. Since the tumor microenvironment is highly hypoxic, tumor cells secrete pro-angiogenic factors to improve oxygen supply, including VEGF; the resulting activation of ECs leads to the secretion of MMPs, which digest basal membrane and allow cells to migrate and form new vessels. Newly formed tumor blood vessels are convoluted, disorganized, and do not have a well-developed EC lining. Thus, the tumor vessels are leaky and ineffective. Furthermore, the tumor engulfs surrounding normal vasculature, incorporating it into tumor vessels [42, 52, 97, 98]. Bulla et al. [92] investigated 30 invasive malignant tumor samples (including melanoma, colon, lung, breast, and pancreatic cancers) by immunohistochemistry analysis for the presence of C1q, C1s, C3, and C4. The staining was clearly positive for C1q, whereas the other complement components were faintly present, indicating that C1q deposition was independent of complement activation. In particular, C1q staining was detected in stroma, ECs, and infiltrating leukocytes. The expression of C1q was associated with newly formed vessels, as vasculature was positive both for that complement component and CD34. The C1q pro-tumoral effect was also evaluated with in vivo mice model of melanoma and lung cancer. C1qa–/– animals showed a prolonged survival compared to WT animals and compared to C3- and C5-knock out (KO) animals. Immunohistochemical staining on tumor specimens showed a marked C1q deposition on vascular endothelium and stroma, in the absence of C4 staining. The vascular density of WT animals was significantly higher than C1qa–/– mice, in particular in the peri-tumoral area. It is well-known that BM-derived cells are the main source of circulating C1q, although other cell types can synthesize this molecule. In order to understand the contribution of stromal-derived C1q to tumor development, a reciprocal radiation BM-chimeras (between C1qa−/− and WT mice) was generated. BM cells from WT mice transplanted in C1qa–/– animals, despite their ability to restore serum C1q levels, had only a minor effect on tumor growth suggesting that non-BM-derived cells were the main source of tumor microenvironment C1q (Table 1) [92].
The dependence of tumor growth on angiogenesis has prompted researchers to further study anti-angiogenic therapies directed specifically against the anti-VEGF pathway. One of the VEGF function-blocking antibodies, known as bevacizumab, a monoclonal antibody (mAb), was the first one to show efficacy in colorectal cancer patients in combination with chemotherapy [99]. Although the initial enthusiasm, clinicians observed that anti-angiogenic therapies were not active across all tumor types. Drugs indeed induced a hypoxic state, which in turn activated a series of escape mechanisms [46]. New anti-angiogenic treatments would require improving efficacy, preventing resistance, and reducing toxicity. Nevertheless, the discovery of new antigens with complementary functions, other than growth factors, could really improve treatments. Orlandini et al. [100] identified a novel antibody directed towards CD93, a transmembrane protein mainly expressed by ECs fundamental for their proliferation, migration, and differentiation. Since cycling ECs express a different antigen profile compared to the quiescent state, this novel target has been discovered through the immunization of mice with proliferating HUVECs. By flow cytometry, antibodies that were able to recognize surface protein on ECs were selected and purified. Those antibodies were then evaluated in angiogenic assays, such as proliferation, migration, differentiation, and tube formation assays, for their ability to inhibit those processes. mAb 4E1 was the only antibody that inhibited all those angiogenic functions. Orlandini and colleagues [100] also confirmed the data in vivo, with a Matrigel plug assay in mice. To identify the target of this mAb, cell extracts of proliferating HUVECs were co-immunoprecipitated with 4E1, and electrophoretic bands were excised and analyzed by mass spectrometry. The bands corresponded to CD93, also known as phagocytic C1qR (C1qRp), which is abundantly expressed onto ECs independently of the cell-cycle state. Flow cytometric analyses with propidium iodide demonstrated that mAb 4E1 was not cytotoxic for ECs, but only cytostatic. To understand the involvement of this molecule in the angiogenic process, the effect of the CD93-silencing was then evaluated in HUVECs. CD93-silenced HUVECs showed a decreased thymidine uptake in response to growth factors stimulation, meaning a reduced ability to synthesize DNA. Cell adhesion and VEGF-stimulated migration also resulted impaired in silenced ECs [100, 101]. Despite the binding between CD93 and C1q is still not completely clarified, this study could represent a further piece of the puzzle of the angiogenic role of C1q in the tumor microenvironment and the discovery of this molecule could represent a novel anti-angiogenic target for anti-cancer treatment [102].
In conclusion, accumulating evidence has shown the involvement of C1q in several physiological and pathological processes connected to angiogenesis. Given the fact that, in animal models, C1q deficiency is correlated with pre-eclamptic-like symptoms, a gestational syndrome characterized by angiogenic dysfunction, this protein seems to be crucial during the development of the placental tissue for a healthy pregnancy. Moreover, Bossi et al. [13] demonstrated that C1q also showed a pro-angiogenic role in the wound healing process, being localized in granulation tissue independently of complement activation and by stimulating permeability, proliferation, and migration of ECs. C1q is also involved in the angiogenic process occurring during the post-stroke ischemia recovery. In these contexts, C1q could be used as a pro-angiogenic factor, opening new scenarios to innovative therapeutic perspectives. On the other hand, as many studies demonstrated that it could also drive tumor angiogenesis, anti-C1q immunotherapies could be useful when combined with standard anti-cancer treatments (Figure 3) [92, 103].
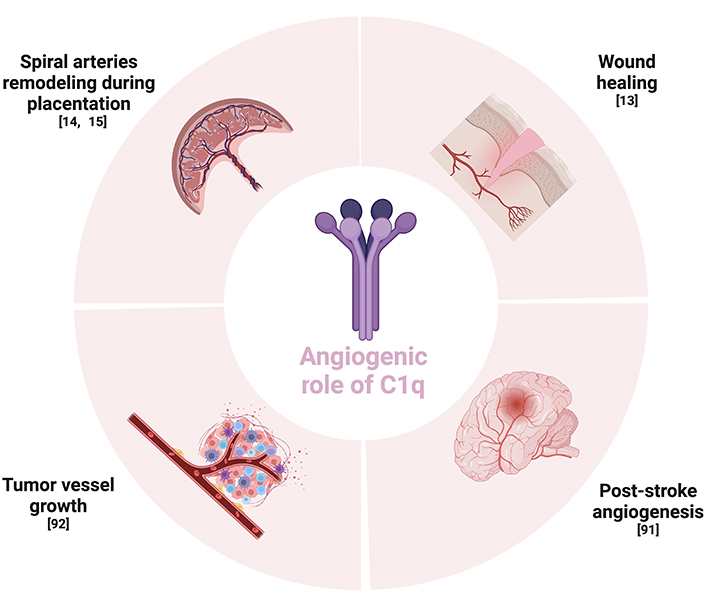
Schematic summary of the context regarding the angiogenic role of C1q. The Figure was created with BioRender software (https://biorender.com/)
Further studies are needed to identify the precise molecular mechanisms that are activated by the stimulation of C1q in each of these contexts.
AKT: protein kinase B
ANGs: angiopoietins
BM: bone marrow
cC1q: collagen-like C1q
cC1qR: collagen-like C1q receptor
CR1: complement receptor 1
DECs: decidual endothelial cells
EC: endothelial cell
ECM: extracellular matrix
ERK1/2: extracellular signal-regulated kinase 1/2
Flt-1: fms-like tyrosine kinase-1
gC1q: globular head C1q
gC1qR: globular head C1q receptor
HUVECs: human umbilical vein endothelial cells
IA: intussusceptive angiogenesis
IgG: immunoglobulin G
LIAR1: leukocyte-associated immunoglobulin-like receptor 1
mAb: monoclonal antibody
MAPK: mitogen-activated protein kinase
MMPs: matrix metalloproteinases
PI3K: phosphatidyl-inositol 3-kinase
PlGF: placental growth factor
pMCAO: permanent middle cerebral artery occlusion
RBMECs: rat brain microvascular endothelial cells
SA: sprouting angiogenesis
TIE: tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains
VEGF: vascular endothelial growth factor
VEGFRs: vascular endothelial growth factor receptors
WT: wild type
MS: Writing—original draft, Visualization. SP and CA: Writing—review & editing. RB: Conceptualization, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2023.
Copyright: © The Author(s) 2023. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Martin Kolev ... Pascal Deschatelets
Célia Dos Santos ... Analía Sánchez-Luceros
Jeyaparthasarathy Narayanaperumal, Ganesh Gopal
