 Review
Review

Affiliation:
SH/Scientific-Hemostasis, Scientific Director and Consultant in Hemostasis and Thrombosis Diagnostics, 95130 Franconville, France
Email: jean.amiral@scientific-hemostasis.com
ORCID: https://orcid.org/0000-0002-0680-9172
Explor Immunol. 2024;4:34–58 DOI: https://doi.org/10.37349/ei.2024.00127
Received: August 14, 2023 Accepted: December 18, 2023 Published: February 05, 2024
Academic Editor: Dominique J. Charron, Hospital Saint Louis, France
The article belongs to the special issue Autoantibodies Associated to Thrombosis and Hemostasis
Many acquired bleeding and thrombotic complications are provoked by autoantibodies to blood coagulation factors, or to hemostasis inhibitors and regulatory proteins. If occurrence of those antibodies remains rare or ultra-rare, affected patients are not always well-identified and associated pathologies are not always understood. Today, autoantigens tend to be better characterized. New available methods allow investigating structural changes of body components, responsible for auto-immunization. This renders it possible to develop laboratory assays for detecting autoantibodies and estimating their blood concentration. This review analyzes the major autoantibodies reported to be associated with hemorrhagic or thrombotic pathologies and their possible inducing causes when known. Pathogenicity is strongly patient- and context-dependent and is related to autoantibodies’ concentration, avidity, and capacity to bind to autoantigen structures in-vivo, misdirecting the immune system to the own body’s cells or organs. Identification of autoantigens allows for developing laboratory methods for testing autoantibodies and following their evolution kinetics. In-vitro investigations concern functional assays, to evaluate autoantibody’s capacity to inhibit physiological activities, or autoantigen-capture-based assays to detect autoantibodies, like with enzyme-linked immuno-sorbent assay (ELISA) methods. Exploring patients with autoimmune complications remains difficult as few specific assays are available. They mainly concern diseases with the highest incidence, like anti-phospholipid antibodies, lupus anticoagulants, or heparin-dependent antibodies. The present understanding suggests that antibodies to ubiquitous components, like phospholipids or polysaccharides, are actually targeted to proteins with a strong affinity binding to those components: Autoantibodies are not directed to phospholipids, but to phospholipid-binding proteins, and heparin-dependent antibodies are not directed to anticoagulant polysaccharides, but to platelet factor 4. Most pathogenic autoantibodies are of immunoglobulin G (IgG) isotype, but in some cases, IgM or IgA isotypes can be involved. Identification and characterization of autoantibodies associated to hemorrhagic or thrombotic pathologies remains complex at the laboratory level, although they are of high relevance for the right management of concerned patients.
In some cases, the occurrence of bleeding or thrombotic disorders can be induced by the development of autoantibodies; though frequently transient, they can become chronic in some exposed patients [1–4]. The pathophysiological contexts which can generate such autoantibodies in affected patients are not always well-understood. However, the mechanism has been identified in many cases, and the disease development kinetics has been described [5, 6]. Most often, the development of autoimmune pathologies is associated with various conditions: infectious diseases, some at-risk drugs, replacement therapies for patients with a specific protein deficiency, or metabolic disorders including malignancy and tissue alteration, and ageing [7–9]. These conditions lead to exposure of cryptic or unusual epitopes on proteins and are at risk for developing autoimmune responses. All these circumstances are associated with the unusual presentation of a self-component, which deceives the host immune system and is then recognized as non-self, and the apoptotic mechanism, which controls lymphocytes targeted to self-components, fails [10–12]. Briefly, what can deviate the immune response from the specific recognition of non-self-structures is the intimate association of self-components with foreign factors or the contact with an external substance sharing some epitopes with a self-component. However, not all autoantibodies cause health burdens and associated autoimmune pathology is only detected when it becomes symptomatic and causes disease.
When diseases present with unexpected bleeding or thromboembolic complications, or necrosis at skin or limb extremities, autoantibodies, which interfere in the hemostasis system, must be suspected. The causative factors and the subsequent pathogenic consequences depend on the targeted autoantigen. In autoimmune pathologies, when thrombotic or bleeding complications develop, autoantibody targets are often exposed to platelets (Plts) or other blood or endothelial cells. This binding leads to the occurrence of thrombocytopenia, Plt or blood cell activation and destruction. In other cases, targets are hemostasis proteins involved in the activation and regulation of the coagulation cascade: factors, cofactors, or inhibitors. Pathogenic consequences depend on the physiological function of the autoantibody targeted factor. When it concerns coagulation factors or cofactors, autoantibodies tend to induce bleeding, although frequently they can paradoxically provoke thrombosis [1, 13]. When targeted to coagulation inhibitors or regulators, they generate thrombotic complications [14, 15]. However, more complex mechanisms can be involved, and targeted proteins behave like cargos for carrying autoantibodies to blood cells, endothelium, or tissues, where the autoimmune response is deviated, with its harmful effects focused on these sites [16–18].
This review article summarizes the major circumstances favoring the generation of autoantibodies, how they become pathogenic, which laboratory tools are available for their identification, and how autoimmune pathology can be managed to limit disease consequences. This report is entirely focused on autoimmune diseases affecting the hemostasis system and associated with bleeding or thrombotic complications. This only concerns a small part of autoimmune pathology. Fortunately, not all circumstances that generate autoantibodies lead to disease development in affected individuals, suggesting the contribution of individual or genetic risk factors. Therefore, only very few patients exposed to potential autoantigens develop an autoimmune response, and yet in fewer ones, it produces symptomatic pathologies, and the incidence depending on the trigger itself and individual factors is not fully understood.
The immune system is a major defense mechanism, which protects the body’s integrity, especially from viral and microorganism infections, like bacteria, viruses, or fungi, but also from any introduction of non-self-component, organ, or cell, as illustrated by the grafts’ rejection [19]. Its action mode is a progressive time-dependent response, which first involves the innate immune system, followed a few days later by the adaptive immunological response, whose intensity develops with time of exposure to the non-self-component: stimulation of T and B lymphocytes, generation of IgM antibodies with moderate specificity and weak affinity, and progressively IgG antibodies with high specificity and increasing avidity. In some cases, the immune response can be anamnestic, with a direct or rapid generation of IgG isotypes. When this defense mechanism deviates and the immune system is misdirected to target self-components, autoimmune diseases can develop [20, 21]. Many causes have been suspected or reported as possible triggers for the generation of autoantibodies. The following paragraphs specifically present and discuss the autoimmune complications which provoke bleeding or thrombotic burdens. The most frequently reported clinical contexts associated to this pathology are summarized in Table 1, which however is non-restrictive.
Some pathophysiological contexts favoring the development of autoantibodies
| Inducing cause | Pro-thrombotic autoantibody generated |
|---|---|
Drug-induced Heparin Protamine sulfate Quinine Substitutive therapy | Anti-HPF4, anti-PF4 antibodies Anti-protamine sulfate antibodies (Plt activation) Anti-GP1b-IX (Plt activation, thrombocytopenia) Anti-FVIII, anti-FIX |
Infectious diseases Varicella Adenovirus Pharyngeal infection Herpes, megalovirus, EBV infections, etc. | Anti-PS, LA Anti-PT, LA Anti-PF4 Anti-GP-IIb-IIIa, GPVI, GP1b-IX-V, etc. |
Degenerative disease Malignancy Others | LA Anti-β2GP1 Anti-PT Anti-annexin V Anti-FXIII Anti-FVIII, anti-vWF, etc. Others |
Various pathological conditions Inflammation, NETs | ANCAs Anti-cytokines Anti-interferon Others |
Chemicals Environmental pollution Chemicals | LA, anti-β2GP1 Anti-FXIII Others |
Physiological conditions Ageing Genetic predisposition | Miscellaneous |
ANCAs: anti-neutrophil cytoplasmic antibodies; anti-GP1b-IX: anti-glycoprotein 1b-IX; anti-PT: anti-prothrombin; anti-FVIII: anti-factor VIII; anti-HPF4: anti-heparin-Plt factor 4 (PF4) complexes; EBV: Epstein Barr virus; LA: lupus anticoagulant; NETs: neutrophil extravascular traps; PS: protein S; vWF: von Willebrand factor
Briefly, primary autoimmune causes occur when infectious or foreign components are introduced into the body and are able to bind strongly with self-proteins in solution or on the cell surface [22, 23]. A complex is formed and a continuum between non-self and self-protein structures is generated [5]. Although the host immune system can usually distinguish between non-self and self-epitopes, in a subset of patients this selection does not operate efficiently, and the immunological response is extended to the host proteins or components present in these complexes (Figure 1). This mechanism is of special relevance in infectious diseases, especially viral ones. Viruses need to bind onto cell receptors to enter host cells and deviate the cell machinery for their growth, replication, and proliferation throughout the body, or in the infected organ. These receptors are cell-surface proteins, with a strong affinity for some viral proteins. A recent example is the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), whose spike protein binds with high affinity to angiotensin converting enzyme 2 (ACE2) through its receptor binding domain (RBD). ACE2 is expressed in lung and nasopharyngeal epithelial cells and is also present in many organs, like kidney, heart, brain, and on cells like endothelial cells. The viral infection is then ubiquitous if not controlled rapidly by the body’s innate and adaptive immune responses. ACE2 is therefore a potential target for autoantibodies, and they have been confirmed in a subset of patients with coronavirus disease 2019 (COVID-19) [24–27]. Viral diseases provoke cell destruction and systemic inflammation at infected sites, with recruitment and expansion of leukocytes, mainly neutrophils, and lymphocytes [natural killers (NKs) and helpers], and later production of specific T and B lymphocytes. High concentrations of inflammatory cytokines are produced to stimulate and regulate the function of immunological cells. Cell debris is released into blood circulation, and can contribute to autoimmune stimulation. Furthermore, autoantibodies to cytokines can be generated to regulate their excess, as it has been reported for interferons and for interleukine-8 (IL-8) for example [28–31].
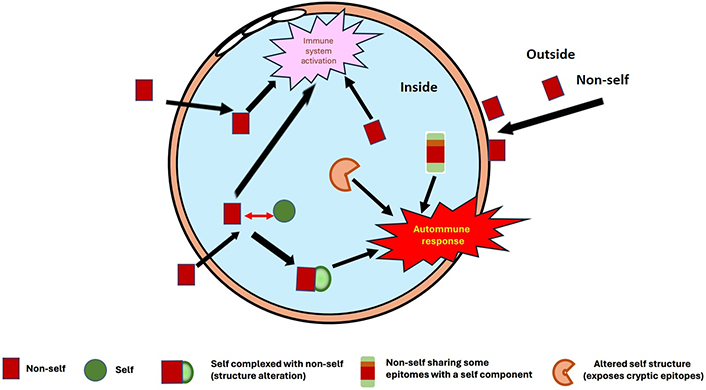
Different possible presentations of self-components/proteins, which interact with non-self-components (infectious agents, drugs, chemicals, etc.) or pathological sites (inflammation, malignancy, etc.), and provoke structure alterations: This can misdirect the immune response, inducing the generation of autoantibodies
This deviation of the immune system fulfills then a regulatory role to control the excess cytokines and their harmful effect on body. These antibodies should not be pathogenic, even if they can induce complications in some special circumstances. For example, this was reported for autoantibodies to IL-8, which can become pathogenic during heparin therapy. Presence of heparin favors their binding to blood cells, like Plts and endothelial cells, targeting then the immune response to those cells [28–31]. Furthermore, autoantibodies can be generated in pathologies inducing cell destruction: Cell components, which are released into blood circulation, can be exposed in an unusual way and exhibit cryptic epitopes. A complementary cause of autoimmune responses concerns drug treatments, like as heparin or quinidine. When in blood circulation, drugs can form complexes with proteins, and slightly modify their presentation, rendering them antigenic [31–34]. For example, when heparin meets in blood circulation, or in a micro-environment with endothelial lesion or injury, a stoichiometric PF4 concentration, HPF4 complexes are formed and can stimulate an immune response [35]. When antibodies are present, they can bind onto stoichiometric HPF4 complexes, expressed at the Plt or endothelial cell surface, where they focus the immune response, provoking heparin-induced thrombocytopenia (HIT) sometimes associated with heparin-induced thrombosis (HITT). Quinine can bind to Plt GP1b-IX and GP-IIb-IIIa, and stimulate an immune response, which is then targeted to Plts exposing these GP motives complexed with quinine, then inducing thrombocytopenia, and Plt activation [36].
Autoantibodies develop in presence of an autoimmune trigger, and they expand as long as this trigger remains present in the body. First, they use IgM isotypes, evolving progressively to IgG isotypes, but anamnestic immune responses can induce the rapid generation of IgGs (like in HIT). Simultaneously, presence of IgA isotypes can be frequently observed, as it has been reported in many different situations [26, 33, 35, 37]. For some autoimmune diseases, IgA isotypes remain rare, but in other cases, they are frequently observed [26, 33]. Specific clinical complications can be associated with these IgA isotypes, as is the case for renal complications (thrombotic microangiopathy/nephropathy) in patients with phospholipid-dependent antibodies or autoantibodies of other specificities. When the autoimmune trigger disappears, autoantibodies remain for several weeks or months, and then their concentration progressively decreases [38, 39]. This kinetics results from the half-life of the specific B lymphocytes generated, and from that of antibodies in blood circulation. When there is no more autoimmune stimulation, autoantibodies remain as long as they are synthesized by B lymphocytes, which progressively disappear from blood circulation. Autoantibodies are no more produced and their clearance kinetics depend on their in-vivo half-life. In many cases, antibodies become undetectable 3 to 6 months after the onset of the autoimmune response, but they can last for a prolonged period if the immunostimulant inducer remains [40–43]. When autoantibodies are symptomatic and cause clinical burden, pathogenic complications develop later and disappear sooner than the autoantibodies. In a few cases, the autoimmune response can remain at a pathogenic level over time, and the autoimmune disease then becomes chronic [44, 45].
Acquired coagulation factor deficiencies are detected clinically when a sudden hemorrhagic episode occurs in the absence of any obvious inducing cause in patients who previously were not characterized to be at hemorrhagic risk [46]. Development of autoantibodies to a coagulation factor or cofactor is the most frequent cause, and this induces a decreased or absent activity. Causes can be diverse and are often concomitant with some pathologies or therapies: infectious diseases, use of some drugs, tumoral diseases, metabolic disorders, among others. These contexts favor the development of autoantibodies [47]. Here after, we discuss the most frequently reported immune coagulation inhibitors, which expose affected patients to severe and recurrent hemorrhages, as summarized in Table 2 and discussed in the following paragraphs.
Major reported autoantibodies, inducing bleeding complications, and their laboratory interference
| Targeted autoantigen | Clinical context | Biological effect | Interference in laboratory assays |
|---|---|---|---|
| PT | Bleeding, bruise, peripheral thrombosis | Accelerated FII clearance, inhibition of coagulation cascade | LA, apparent FII, V, VII, IX, X, XI, XII deficiencies |
| FV | Severe bleeding (ultra-rare; association with antibiotics) | Decreased FV activity, coagulation cascade inhibition | LA, apparent FV, VII, IX, X, XI, XII deficiencies |
| FVII | Asymptomatic to severe bleeding (ultra-rare) | FVII inhibition | Prolonged PT, FVII deficiency |
| FVIII | Severe recurrent hemorrhages (frequent in hemophilia A) | FVIII inhibition | Prolonged APTT and mixing tests, FVIII deficiency |
| FIX | Severe bleeding (can occur in hemophilia B) | FIX inhibition | Prolonged APTT and mixing tests, FIX deficiency |
| FX | Ultra-rare, bruise | FX inhibition | Prolonged APTT, FX deficiency |
| FXIII | Delayed bleeding (can be fatal) | Delayed bleeding, intracranial hemorrhages | Decreased FXIII activity, variable FXIII-A antigen |
| AT | Enhanced AT activity, bleeding tendency | “Heparin-like” behavior enhances AT activity | Prolonged thrombin time |
| vWF | Severe bleeding | Reduced Plt adhesiveness | Decreased vWF activity (collagen binding) |
| GP-IIb-IIIa | Thrombocytopenia, bruise, bleeding | ITP | MAIPA assay, Plt count, flow cytometry |
| GP1b-IX | Thrombocytopenia, bruise, bleeding | ITP | MAIPA assay, Plt count, flow cytometry |
| GPVI | Bruise, bleeding | ITP | MAIPA assay, Plt count, flow cytometry |
APTT: activated partial thromboplastin time; AT: antithrombin; ITP: immune thrombocytopenic purpura; MAIPA: monoclonal antibody-specific immobilization of Plt antigens
Substitutive therapies are frequently used for treating hemophiliacs during bleeding episodes, or as prophylaxis to supply the missing coagulation factor, either blood extracted or recombinant. Patients can detect it as a foreign substance, especially when they totally lack FVIII or FIX synthesis. Autoantibodies develop then and render the substitutive therapy ineffective [48]. When this complication occurs, it is no longer possible to correct hemorrhages by infusing the deficient coagulation factor, and alternative therapeutic strategies must be used [49, 50]. The highest incidence for these autoantibodies concerns patients with severe hemophilia A, unable to synthesize FVIII, even in the inactive form (hemophiliacs A): They can develop high to very high titers of FVIII neutralizing antibodies. It is not rare to observe patients with antibodies able to neutralize various thousand-folds of the normal body’s FVIII concentration. Yet, FVIII concentration is very low, of only about 0.100 μg/mL, and antibodies are able to neutralize several hundreds of μg/mL of FVIII. This situation is especially illustrated by the introduction of prophylaxis with substitutive therapies, which cannot be used for hemophiliacs with inhibitors. In about 20% to 40% of severe hemophiliacs A, a different therapeutic management is required, because of the presence of inhibitors, sometimes transient, but often persistent [50]. Furthermore, many cases of acquired FVIII inhibitors have been reported, and they also require an alternative treatment [51]. When dramatic hemorrhagic events occur, they must be treated using an FVIII bypassing activity (FEIBA) strategy, like that with activated coagulation factors [FEIBA or activated PT complex concentrate (PCC)] or more recently with recombinant activated FVII (rFVIIa), Novoseven® [51]. Recently, a new engineered drug has been introduced for curative or prophylactic treatment of patients with FVIII inhibitors (neutralizing FVIII antibodies). It is a biphasic antibody, emicizumab (Hemlibra®), targeted to both FX and FIX/activated FIX (FIXa), and mimics FVIII activity by closing FX and FIX on the phospholipid surface, therefore bypassing FVIII [52]. This drug has a much longer half-life than blood-extracted, recombinant, or even engineered long-lasting FVIII, and it benefits from an extended half-life in blood circulation. Furthermore, it is injected subcutaneously only every 2 to 4 weeks to prevent bleeding, whilst FVIII requires up to 2 injections per week. This drug then offers an improved quality of life to patients. The long-term use of this drug is being followed closely now and it needs a retrospective evaluation for tolerance and safety, as patients with FVIII coagulation inhibitors require a life-long treatment. Autoantibodies to FIX have also been described in severe hemophiliac B, but this complication remains much rarer than that associated with FVIII inhibitors.
Other autoantibodies to coagulation factors, associated with bleeding diathesis, have been described in the literature. They concern most of the coagulation factors, including fibrinogen [53], PT [54], FX [55], FVII [56], FV [57, 58], vWF [59, 60], and FXIII [61, 62]. When induced by infectious episodes, they are transitory and can provoke various clinical disorders associated with hypocoagulability, like bruises, gum bleeding, intracranial hemorrhages, or more severe bleeding complications. In some cases, they can become life-threatening or fatal. The induced clinical complications result from a double action: i) the direct effect through the inhibition of the targeted coagulation factor, which induces a factor deficiency; ii) the indirect effect resulting from the immune complex binding to body’s cells or organs, focusing the immune response onto them: The deleterious effects include tissue injury, inflammation, thrombo-inflammation, paradoxically disseminated intravascular coagulation (DIC), and thrombosis for some autoantibodies (like those to thrombin), although targeted to clotting factors [63, 64]. Autoantibodies can react with the protein active site and then inhibit directly the coagulation factor or cofactor function, or they react with non-functional epitopes and therefore accelerate the protein clearance, which induces deficiency. They can interfere with the coagulation cascade through their binding to phospholipids, which produces a steric hindrance, preventing the clotting factors from interacting with phospholipids.
Autoantibodies can develop to very specific or particular epitopes, for yet non-identified reasons. This is the case for a reported autoantibody to AT, which mimics the AT binding to heparin and can induce hemorrhages [65]. Other autoantibodies can be targeted to thrombin exosites and are associated with bleeding tendencies by impairing the thrombin clotting activity [66]. These antibodies totally differ from those generated by thrombin itself, like those produced when fibrin glue prepared with bovine topical thrombin was used for healing injuries or during surgery and paradoxically induced thrombosis [63, 64]. They are presented in Table 2, and are more extensively discussed in “Autoimmune thrombotic complications”.
Some contact phase proteins’ deficiencies are induced by autoantibodies to FXII, or Hageman factor, or more frequently to FXI, or Rosenthal factor. Bleeding disorders are mainly observed in some FXI deficiencies, as the result of an autoimmune response [67–69].
Lastly, autoantibodies can be targeted to Plts, especially onto their surface proteins and receptors, and are then associated with thrombocytopenia, which can induce bleeding episodes in severe cases. Some viral infections have been identified as possible causes for the generation of these anti-Plt GPs’ autoantibodies, like those with Herpes, EBV, or cytomegalovirus. Various autoantibody-targeted GPs have been identified, mainly GP1b-GPIX, GP-IIb-IIIa, GPV, and GPVI [70, 71]. These autoantibodies induce thrombocytopenia and are of major concern during pregnancy. They can cross the placenta, and newborns require special caution and follow-up, as they can last some weeks.
Autoantibodies can be the cause of thrombotic episodes occurring in unusual contexts, in the absence of any currently identified trigger or pathology. Development of these thrombogenic autoantibodies can occur after an infectious episode, frequently viral, or as an iatrogenic complication to drug therapies. They can also be generated in other pathologic conditions, like metabolic disorders or malignant diseases. The common factor within all these contexts is a stimulation of the inflammatory system and/or the development of an immune response, innate and adaptive, to control pathology [72]. The immunological conflict develops at pathological sites or in a diffuse manner throughout the body. Modifications of self-components occur through complex formation with non-self-products generated by infection, or drug interaction, or result from the exposure of cryptic or normally masked epitopes during malignant or metabolic pathologies. The normal immune response is then misdirected to self-components, which are not correctly discriminated from foreign substances as illustrated in Figure 2. As described in the here above paragraph “Autoantibodies inducing bleeding events”, other autoantibodies can conversely be associated to thrombotic complications. This pathology can result from 2 distinct processes: i) the inhibition of the anticoagulant or antithrombotic protein function; ii) the binding of immune complexes onto blood cells, endothelium, or organs, where the immune response is targeted and can induce cell activation and/or destruction [73, 74]. This latter mechanism is of special relevance when immune complexes bind to Plts or endothelial cells. The most frequently reported autoantibodies associated with thrombosis are presented in Table 3.
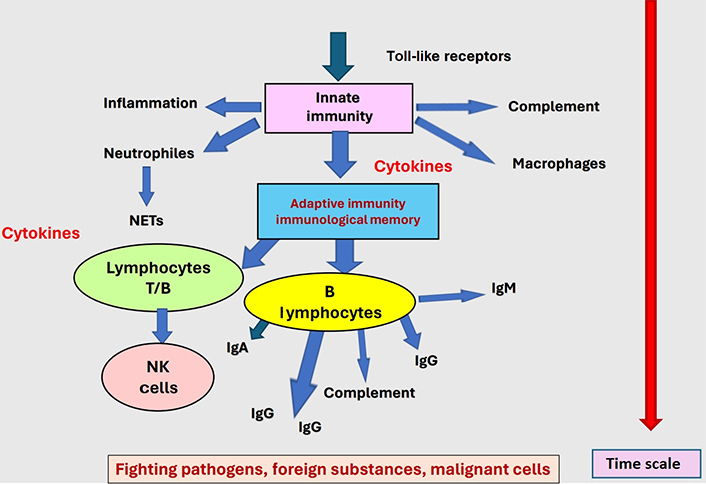
The immune response develops as a key body’s defense against pathogens, foreign substances, and malignant cells. When deviated and misdirected as shown in Figure 1, generation of autoantibodies may occur and induce pathology
Target antigens of autoantibodies, which can induce thrombotic diseases, and their interference in laboratory testing
| Autoantigen target | Clinical context | Possible cause and biological effect | Laboratory impact |
|---|---|---|---|
| β2GP1 | Thrombosis, fetal loss | Interferes in coagulation cascade, Plt activation, targets immune system to blood cells | LA, positive ACA or APA assays |
| PT | Thrombosis, fetal loss | Viral infection, interferes in coagulation cascade, targets immune system to blood cells | LA, decreased FII, apparent deficiencies in FVII, V, VIII, IX, X, XI, XII; positive anti-PT ELISA |
| PS | Thrombosis, purpura fulminans | Varicella, inhibits PS anticoagulant activity | LA, decreased free PS, positive anti-PS ELISA |
| PC | Severe thrombosis | Very rare, activated PC resistance | LA, decreased PC activity, positive anti-PC ELISA |
Annexin V, PZ | Thrombosis, abortions | Rheumatoid arthritis, systemic sclerosis, fetal loss, microangiopathy | LA, positive anti-A5 ELISA, flow cytometry |
| TM | Thrombosis | Maligancy, inhibition of PC activation | Inhibits PC activation, positive anti-TM ELISA |
| PF4 | Severe thrombocytopenia, thrombosis | Adenovirus vector vaccine, targets immune system to Plts and endothelial cells | Positive PAT and PF4 ELISA |
| HPF4 complexes | Thrombocytopenia, thrombosis | Targets immune system to Plts and endothelial cells | Positive PAT and HPF4 ELISA |
| ADAMTS-13 | TTP | Inhibits ADAMTS-13, high MW vWF multimers | Plt hyper-aggregability |
| IL-8 | Peripheral thrombosis, DIC | Targets immune system to Plts and blood cells | Variable positive PAT and positive IL-8 ELISA |
| Thrombin | Thrombosis, DIC | Inhibition of PC pathway | Prolonged clotting tests, positive AT ELISA |
| GP1b-IX quinine | Immune thrombocytopenic purpura, Plt activation | Thrombocytopenia, thrombosis | Plt count, Plt activation, flow cytometry |
| Alloantibodies to PLA | Thrombocytopenia, Plt activation, neonatal complications | Transfusion, allo-immunization | Plt activation, flow cytometry |
A5: annexin 5; ACA: anti-cardiolipin-antibodies; ADAMTS-13: A disintegrin and metalloprotease with thrombospondin type I repeats-13; APA: anti-phospholipid antibody; MW: molecular weight; PAT: Plt aggregometry test; PLA: Plt alloantigen; TM: thrombomodulin; TTP: thrombotic thrombocytopenic purpura
The most frequent autoimmune-induced thrombotic complications concern the development of APAs, especially those targeted to β2GP1, or to PT, usually reported as APAs, which are responsible for the anti-phospholipid syndrome (APS). These phospholipid-dependent antibodies induce the prolongation of clotting times in coagulation testing and are reported as LA. Although these antibodies are anticoagulant in laboratory tests, they paradoxically provoke a thrombotic tendency in vivo and can be highly thrombogenic [75, 76]. In extreme cases, they can lead to catastrophic APS (CAPS), with disseminated blood activation and coagulation and multi-organ failure, a high-risk clinical complication, with often a fatal issue if not rapidly and appropriately controlled [77]. If β2GP1 and PT are the preeminent targets in APS or CAPS, some other phospholipid-binding proteins, like coagulation PS or A5 have been reported in this disease. In addition, other vitamin K-dependent proteins have been reported to be seldomly associated with APAs, like coagulation PC or PZ, although their incidence remains rare [78, 79]. If APAs generate LA activity, the latter can result from other autoantibodies and is not always associated with thrombotic complications. LA activity can be produced by autoantibodies which induce bleeding complications, like autoantibodies to FV or to PT [57, 80]. Moreover, autoantibodies to PT can induce opposite clinical manifestations, and be hypo-or hyper-coagulant. The mechanisms are yet poorly understood, but they depend on the generating trigger. These anti-PT autoantibodies may be targeted to different protein epitopes or domains, resulting in different consequences [81, 82]. Transitory anti-PT autoantibodies usually produce mild bleeding and are developed following viral infections. In a few cases, they become chronic, and tend to produce thrombotic complications, disseminated or at various body sites, and are then very difficult to manage and control [83, 84]. The inducing primary cause is not always clearly identified. This can result from an abnormal exposure to PT due to a genetic predisposition (HLA system), and the immune system lacks the efficiency to select self-components from the non-self-components, or to correctly recognize altered self-components [85, 86].
Another major group of autoantibodies associated with thrombotic complications concerns anti-PF4-dependent immune reactions, responsible for HIT. In a significant number of cases, they are associated with thrombosis and were recently described as the causative factor of the ultra-rare vaccine-induced thrombotic thrombocytopenia (VITT) [87, 88]. PF4 is a chemokine present in Plt α-granules, which is early released upon Plt activation as a high MW complex formed by 8 PF4 tetramers bound to 2 proteoglycans [89]. PF4 half-life in blood circulation is short (a few minutes). When released from Plts, it is displaced from Plt proteoglycans and binds to endothelial cell glycosaminoglycan (GAGs), for which the affinity is higher. When patients are anticoagulated with heparin, that drug displaces PF4 from the endothelial cell storage pool and forms high-affinity HPF4, whose presentation depends on the respective concentrations of heparin and PF4. When stoichiometric concentrations are reached [about 27 international units (IU)/150 μg of heparin per mg of PF4], heparin, a molecule with a high density of electronegative sulfate groups, wraps tightly around the ring of electropositive charges of the PF4 tetramers. This binding alters the PF4 structure and exposes new epitopes, which can become antigenic and generate heparin-dependent antibodies, targeted to complexes of heparin and PF4. These heparin-dependent antibodies provoke deleterious effects when targeted to Plts, blood, or endothelial cells exposing HPF4 complexes on the cell surface [90, 91]. Therefore, HIT pathogenicity results from a “double misfortune”: i) release of PF4 to a concentration which forms stoichiometric complexes with heparin, and which stimulates the immune response; ii) permanence of HPF4 stoichiometric complexes in blood circulation, which bind onto Plt, blood, or endothelial cells and react with the heparin-dependent antibodies, thus provoking the adverse complications. Stoichiometric HPF4 complexes are formed for HPF4 respective concentrations of 0.027 IU/mL of heparin per μg/mL of PF4. In practice, PF4 in blood circulation is present at very low concentrations (< 10 ng/mL), and stoichiometric complexes can only be formed for very low heparin concentrations (0.000027 IU/mL for 10 ng/mL PF4). They are then present at a too-low level for becoming antigenic [87, 90, 91]. However, at the beginning of heparin therapy, PF4 is released from the endothelial cell storage pool, and its concentrations can then be much higher in blood circulation, of various hundreds of ng/mL. PF4 concentration is positively correlated to the hypercoagulability status of heparin-treated patients, and this drug favors PF4 released from Plt α-granules [92, 93]. When high levels of PF4 are in blood circulation (up to 1.0 μg/mL), stoichiometric complexes can be formed with heparin concentrations from 0.0027 IU/mL to 0.027 IU/mL, which still remains very low. But PF4 can accumulate in pathological sites with inflammation or blood activation, where it can reach very high concentrations, of several orders of magnitude higher than in blood circulation. Chances to generate stoichiometric complexes are then much higher, and compliant with heparin therapeutic levels, especially since heparin concentrations vary over time following intra-dermal injections. Immunogenicity can then develop more easily. Heparin blood concentration remains stable in treated patients only when the drug is infused intravenously. This mechanism explains the low patients’ propensity to develop HIT during heparin therapy and the association of incidence with clinical contexts, although any individual can develop an immune reaction when exposed to stoichiometric HPF4. Interestingly, many individuals develop asymptomatic antibodies just after the onset of heparin treatment, whilst only a few of them evolve to HIT, possibly complicated by thrombosis [94]. Conversely, pathological states with high grades of Plt and blood activation, like traumatic surgery or cardiovascular diseases, are associated with a higher frequency of HIT, with an incidence of up to 5% of heparin-treated patients. Thrombotic complications occur at pre-existing pathological sites, especially those associated with inflammation or blood activation, and with chemo-attraction of leukocytes and Plts, like infectious or malignant diseases, metabolic disorders, impaired vessel patency, and atherosclerotic deposits [95]. Disease then develops at sites of former existing disease. In addition, HIT incidence and severity increase with the HPF4 antibody concentration and affinity. These site-targeted and concentration-dependent complications observed in HIT also occur in other autoimmune-associated thrombotic diseases, like those induced by LA and APAs. However, in HIT and also probably in other autoimmune diseases, all autoantibodies are not pathogenic, and many remain asymptomatic. Following the purification of heparin-dependent antibodies in patients with HIT, we demonstrated that only a few of them are able to bind and activate Plts, whilst the vast majority remain asymptomatic [96].
Recently, PF4-dependent autoantibodies have been demonstrated to be responsible for VITT. The pathogenic mechanism and the targeted epitopes on PF4 are totally different from those involved in HIT. Whilst in HIT the immunogen is the HPF4, which expose new PF4 epitopes, in VITT the reactive PF4 epitopes are located on the protein domain which reacts with heparin, i.e., the ring of positive charges [87, 97, 98]. Heparin, even at a very low concentration, is able to displace these antibodies through its strong binding to the involved PF4 epitopes. The causes which induce an autoimmune response to PF4 following the adenovirus vector SARS-CoV-2 vaccine, are unknown and concern only very rare individuals, which suggests that an unusual event has occurred. This is not a general response to the vaccine as in the case of heparin therapy. No genetic predisposing factor has been identified until now. Noteworthily, the vaccine contains an adenovirus vector, but without any replication possibility. The usual route for the vaccine is intradermic, with no contact with blood circulation. A possible cause could be in very rare cases; the vaccine entrance into blood vessels, and adenovirus can then react with Plts and PF4, through the electronegative adenoviral cell-surface hexon moieties. This could then induce a potent immune response to PF4 complexed with adenovirus through an epitope-spreading mechanism [87]. In VITT, the anti-PF4 concentrations are very high, and antibodies are much more reactive to PF4 than to HPF4 complexes. Conversely, in HIT, antibodies are first generated to HPF4 complexes and can be extended to PF4 alone, especially with time of exposure, but the preeminent reactivity to complexes remains. This is a convenient way to differentiate PF4 autoantibodies developed in HIT or VITT by testing their respective reactivity to PF4 or to HPF4 complexes. The autoantibody reactivity optical density (OD) ratio [OD (HPF4):OD (PF4)] is then far greater than 1.00 for HIT and far smaller than 1.00 for VITT. Using a chimeric human-mouse recombinant monoclonal antibody, K070 provided by HYPHEN BioMed (Neuville sur Oise, France), or using affinity-purified antibodies to PF4, we were able to estimate the plasma weight concentrations of those antibodies in HIT and VITT. They are about from 10 μg/mL to > 100 μg/mL in VITT, and from < 5 μg/mL to > 10 μg/mL in pathogenic HIT.
Lastly, anti-PF4 autoantibodies have been described in other pathological complications, favoring the development of thrombotic complications, often intra-cranial, and are paradoxically worsened by heparin therapy, which induces thrombocytopenia and thrombosis. This was observed in some pharyngeal infections, as reported previously in a young child, and confirmed later as “spontaneous HIT” [98, 99]. Antibodies persist for more than 6 months but progressively decrease with time and become asymptomatic. Recently, similar antibodies have been reported in patients with severe COVID-19 evolution or other infectious diseases, and their presence and concentration have been described to be associated with illness severity [17, 100].
Among complications linked with heparin therapy, cases of autoantibodies to IL-8 have been described [101]. Autoantibodies to IL-8 can develop in presence of inflammation, especially chronic, and their presence can contribute to regulating the excess of that inflammatory chemokine. This mechanism was reported for the regulation of other inflammatory cytokines, and it is not expected to complicate the pathological state. However, if any condition targets the cytokine-antibody complex in cells, this can focus the autoimmune response onto these cells, and then autoantibodies become deleterious. For IL-8, this was described in a patient with malignancy and a tendency to develop thrombosis. Hypercoagulability was not reversed by heparin therapy, but by contrast, it worsened and disseminated thrombi were formed [102]. Anti-IL-8 antibodies pre-exist the heparin therapy, but heparin treatment then favors the binding of IL-8 to various blood cells and Plts, which therefore react with autoantibodies: This focuses the autoimmune response onto these cells, provoking thrombocytopenia, although milder than in typical HIT, cell and coagulation activation, and finally thrombosis.
Another possible at-risk context concerns the use of protamine sulfate to reverse heparin therapy, especially for extra-corporeal circulation (ECC) or extra-corporeal membrane oxygenation (ECMO). For these indications, heparin is used at high concentrations, up to 5 IU/mL in plasma, and is then neutralized by protamine sulfate at the end of the process. In a few observed cases of HIT-like syndrome, but with negative anti-HPF4 antibodies, investigations lead to identifying antibodies to protamine sulfate. They are not autoimmune as protamine sulfate is not a self-component but is usually extracted from salmon sperm [103]. Pathogenicity of some anti-heparin sulfate antibodies results from the binding of heparin protamine sulfate complexes to many blood cells, especially when heparin is slightly in excess. When anti-protamine sulfate antibodies are present, they are then targeted onto these cells, where they focus on the immune response and can generate thrombocytopenia and thrombosis. Nevertheless, many patients exposed to protamine sulfate (to neutralize heparin, or as a component in some insulin preparations used for diabetes treatment) can develop antibodies, but only a very small proportion of them develop pathogenic complications [104, 105]. This tends to occur when patients are exposed various times to protamine sulfate, especially for ECC or ECMO. Yet, antibody concentration is an important factor in the development of pathogenic complications.
Autoimmune thrombotic complications can be observed in some rare cases of autoantibodies developed to anticoagulant proteins, like PS, PC, or TM, or to thrombin, which impairs the PC activation pathway as discussed in the chapter “Autoimmune thrombotic complications”. Many were already discussed along with the phospholipid-dependent antibodies. Autoimmune thrombotic complications can result from various factors, often combined between i) inhibition of the antithrombotic protein activity by the autoantibody; ii) focus of the autoimmune response onto blood cells, which induces cell activation and destruction, and sometimes thrombosis, especially when cell targets are Plts or endothelial cells; iii) inflammation and blood activation through chemoattraction of neutrophils, and release of NETs, which contribute to development of thrombotic complications. The 3 isotypes, IgG, IgA, or IgM isotypes can be present.
When autoantibodies reduce the activity of a coagulation factor, the major clinical risk is first bleeding, with the occurrence of severe hemorrhages, which can become life-threatening. This is especially the case for anti-FVIII autoantibodies generated in hemophilia A patients, or more rarely for anti-FIX autoantibodies in hemophilia B patients [50, 51]. These autoantibodies, which can also be acquired in non-hemophilic patients, especially with malignant diseases, render the usual therapy with FVIII or FIX substitutive drugs ineffective, and another treatment strategy must be used [49]. In practice, treatments which allow FEIBA are used, and they allow the coagulation process to be operational without FVIII. Various drugs have been successfully proposed under strict clinical control to avoid side effects: FEIBA, activated PCC, or rFVIIa/Novoseven®. Recently, a bifunctional antibody, with an antibody site targeted to FIX/FIXa and the other to FX (emicizumab, from Chugai/Roche, Japan & Germany) has been introduced with very encouraging results [49, 50]. Emicizumab is injected subcutaneously and offers a much-extended half-life than FVIII, rendering this treatment also possible for hemophilia A prophylaxis, even in the absence of autoantibodies, although the possible consequences and side effects during life-long treatments must be assessed.
Presence of autoantibodies associated with thrombosis is usually suspected or detected following the characteristic symptoms in patients with thrombosis, often in the absence of the usual thrombotic triggers. They can be preceded by thrombocytopenia, like in the case of HIT or VITT. However, thrombocytopenia by itself is not a criterion of autoimmune thrombotic risk, and it can be associated with bleeding, depending on the autoantibody target on the Plt surface receptor or protein. Clinical signs of impaired blood patency can lead to further investigations to identify a developing thrombotic process or the presence of peripheral thrombosis and skin necrosis due to circulation blockade in micro-vessels. The immediate clinical behavior, when possible, is first to suppress the autoimmune trigger, or to control the autoantibody targets, then to symptomatically treat the clinical complication, for example with an anticoagulant compliant with the patient status. In presence of HIT suspicion, heparin must be immediately withdrawn and replaced by another anticoagulant, especially a direct oral anticoagulant (DOAC), or sodium danaparoid, or eventually argatroban, to immediately stop the immune harmful effects and protect affected patients from the occurrence or progression of thrombosis. It has been successfully used in many different pathologies, like LA, APAs, anti-PS, anti-PT, or anti-A5 autoantibodies. This treatment can be completed by corticoids or infusion with immunoglobulin concentrates to lower the immune response, and therefore the autoantibody concentration. Other options concern anti-Plt drugs, or humanized monoclonal antibodies, like in the case of autoimmune TTP [106–108].
For autoimmune bleeding complications, supplementing patients with the deficient factor infusion is usually not effective, because it is readily inhibited by autoantibodies. The lack of efficiency of replacement therapies with extracted or recombinant human proteins, to supply the missing factor, is of special relevance for components present in blood at very low concentrations, in the μg/mL range or below: The patient’s antibody concentration can neutralize many times (up to hundreds or thousands fold) the physiological factor concentration. For example, FVIII is present in plasma at a concentration of only 0.100 μg/mL in normal individuals, but < 0.001 μg/mL in severe hemophiliacs A. When inhibitors are present, they are able to neutralize dozens to hundreds fold the FVIII normal concentration, and only alternative therapies with FEIBA can be used. Treating autoimmune episodes remains a big challenge as few therapeutic strategies are available, and they must be managed case by case. rFVIIa or blood exchange as the last-chance solution can be used in acute situations, and corticoids or high immunoglobulin infusions can be options to reduce autoantibody concentration and better control their burden.
At any time that the inducing trigger is duly characterized, the first action is to withdraw the cause to stop immunostimulation and to limit the development of autoantibodies along with their deleterious effects. However, due to autoantibodies and B lymphocytes’ half-lives, reducing and suppressing their pathogenic effects can require several weeks to various months. Therapies started for treating the symptoms are not always capable of overcoming all the deleterious effects of autoantibodies. Laboratory testing helps to better manage patients by following their temporal evolution.
For a correct diagnosis and an efficient management of patients with autoantibodies, it is essential to first characterize the antibody specificity, its pathogenic effect, and to identify the root cause of the autoimmune complication. Laboratory testing provides then a major support for patients’ care.
The major laboratory techniques used for detecting autoantibodies associated with hemostasis dysfunctions (either bleeding or thrombotic) are presented in Table 4.
Main laboratory exploration methods for autoantibodies associated with hemostasis disorders
| Laboratory testing | Assay principle | Measurement type |
|---|---|---|
| Functional assays | Inhibition of activity | Measurement of factor activity; mixing studies. |
| Titration of inhibitor titer (Bethesda method) | Incubation of a normal sample with serial dilutions of patient sample and testing residual activity | Dilution inhibiting 50% of activity (corresponds to 1 unit) |
| Flow cytometry | Detection of circulating autoantibodies through their binding to Plt or blood cell GPs | Detection of bound antibodies to specific GPs |
| ELISA | Autoantigen capture-based assays | Detection of autoantibody by ELISA |
| MAIPA | Immunocapture of Plt GP and reactive autoantibodies | Identification of specific anti-GP autoantibodies |
| Coombs test | Detection of cell-bound antibodies | Detection of global anti-cell autoantibodies |
Assay methods include: i) functional assays, to evaluate the physiological activity inhibited by autoantibodies; ii) antibody titration methods, to estimate the autoantibody concentration through its capacity to inhibit a biological activity, or to neutralize antigen; iii) immunological methods, which investigate autoantibodies binding to the targeted autoantigens, especially when they can be characterized and available; iv) cell-based methods for autoantibody binding. Antibody titer is measured using serial dilutions of sample containing it, which is mixed with normal plasma, or purified autoantigen at a defined concentration, and evaluating the excess of activity or antigen. The highest dilution which neutralizes a defined activity or antigen amount allows measuring the titer.
Laboratory monitoring of autoantibodies is of special relevance for surveillance of patients’ evolution and adjustment of treatments when possible. These investigations concern autoantibodies’ interference in physiological functions through the direct inhibition of physiologically active sites, accelerated protein clearance by non-inhibitory autoantibodies, or the specific measurement of autoantibodies with capture immune-assays designed autoantigens. When present, autoantibodies induce different consequences, depending on their target autoantigen, their concentration and affinity, and the immune complexes binding onto the various body’s structures and organs.
These assays evaluate the autoantibody impact on biological activity. The most frequently observed concerns prolongation of clotting methods. Antibodies targeted to phospholipid-binding proteins form complexes with electronegative phospholipids, either directly like β2GP1, or indirectly through divalent cations (especially Ca++, but also Mg++, Zn++, and Mn++), like vitamin K-dependent proteins or A5. They interfere in all phospholipid-dependent assays used for coagulation testing. Clotting methods are then prolonged or even unclottable. Electronegative phospholipids are exposed on many cell surfaces, like Plts, endothelial cells, and other blood cells, especially when activated, and they bind to phospholipid-dependent proteins through the same mechanisms. This binding targets the immune response onto these cells, focusing its deleterious effects. Antibody affinity and concentration determine the intensity of that harmful activity. There is this laboratory paradox for these autoantibodies, which induce anticoagulant activity in coagulation assays and are thrombogenic in-vivo. Pathogenicity concerns: thrombotic or thrombo-inflammatory complications, thromboembolic diseases, skin or limb extremities necrosis, vasculitis, myocarditis, etc. These autoantibodies are usually detected through their anticoagulant activity, reported as LA: This is a very heterogenous group, reacting with many different phospholipid-binding autoantigens [109–111]. Obviously, autoantigens presenting the highest phospholipid affinity in physiological conditions are the most frequently involved in autoantibodies’ development. Assays must then mimic physiological conditions whenever possible, as testing conditions highly impact autoantigen interaction with phospholipids, and therefore autoantibody detection. For example, whether calcium is present or not can induce opposite effects for the autoantibody interaction with its target autoantigen, as reported for β2GP1 capture assays [112]. Noteworthily, low-affinity autoantigens can express a much higher affinity for phospholipids when dimerized through the divalent autoantibody binding. Specific functional assays designed to detect LA activity in citrated plasma present different specificities and sensitivities [113]. This is why it is recommended to combine 2 different assay principles based on phospholipid dependence for a wide-spectrum diagnosis of LA. In practice, assays are performed at both low and high phospholipid concentrations: The low one allows to detect this anticoagulant activity, whilst the high one confirms LA. The 2 currently used methods are the APTT and the diluted Russel viper venom time (dRVVT). The International Society for Thrombosis and Hemostasis (ISTH) publishes and updates from-time-to-time guidelines for the optimization of LA and APA laboratory testing [114, 115].
Aside from these large-spectrum functional methods, specific immunoassays can be used when the autoantigen is well-characterized. Capture techniques are designed by using the purified autoantigen, either blood extracted or recombinant or its phospholipid complexes fixed on a solid surface. Usually, the ELISA is used. The coated ELISA plate captures autoantibodies, and then is measured with a tag-antibody directed to human IgGs, or eventually IgMs or IgAs, all isotypes being possibly present in autoimmune diseases. IgGs are often the most pathogenic, but in some cases, IgMs or IgAs can induce clinical complications and must be detected [114, 116, 117]. The autoantigen used for capturing autoantibodies must be in a presentation mimicking the in-vivo conditions, especially those present in pathology. Each assay needs then to be developed and validated for each class of autoantibodies measured. Concerning phospholipid-binding proteins, many have been reported as possible targets for autoantibodies. The highest incidence concerns β2GP1 which binds phospholipids in a calcium-independent way, and whose interaction is highly enhanced through its binding to anti-Β2GP1 autoantibodies. Others concern first PT, then A5, PS, PC, PZ, etc., all proteins, which bind to anionic phospholipids in a calcium-dependent manner.
A similar mechanism is involved in autoantigens’ interaction with electronegative polysaccharides (containing a high density of sulfate groups). The latter can interact with many soluble blood components or structures exposed on cell surfaces. Those with the highest binding affinity for polysaccharides are those involved the most frequently in the development of autoantibodies. Especially, the PF4 chemokine exhibits a strong affinity for heparin and electronegative polysaccharides, and its tetrameric structure exposes a ring of positive charges. In presence of stoichiometric concentrations with heparin, a long chain electronegative polysaccharide with many sulfate groups, and its structure is altered [89, 118]. New epitopes are exposed and can be immunogenic, inducing PF4-dependent autoantibodies. When present, these antibodies bind to HPF4 complexes. This observation allows to design specific assays for identification and estimation of heparin-dependent anti-PF4 autoantibodies, responsible for HIT. Capture autoantigen is then a stoichiometric HPF4, which presents a strong reactivity with autoantibodies involved in HIT. The IgG isotype, alone or combined with IgMs and/or IgAs, is involved in pathogenicity [119]. Many anti-HPF4 antibodies are asymptomatic. Their concentration and affinity for HPF4 are essential factors for pathogenicity. Other heparin-binding chemokines, although with a lower affinity, have been reported to be associated with clinical thrombogenic complications, like IL-8 or neutrophil-activating peptide 2 (NAP-2) [101]. Recently, PF4 has been identified to be the target autoantigen in VITT, but the mechanism differs from that involved in HIT. Conversely, to heparin-dependent antibodies, whose reactivity is the strongest to stoichiometric HPF4 complexes, those developed in VITT are mainly reactive with PF4 alone, and this binding is inhibited by low heparin concentrations [87, 88]. Actually, VITT autoantibodies bind to a defined PF4 domain located on the ring of electropositive charges on the PF4 tetramer, which is the heparin-binding structure [97]. Using HPF4 or PF4 capture immunoassays for the IgG epitope allows differentiating HIT and VITT autoantibodies: Those present in HIT bind preferentially to HPF4 complexes, whilst those developed in VITT exhibit a much higher reactivity with PF4 alone.
There is a relationship between autoantibody concentration and the development of autoimmune pathology. Using quantitative or semi-quantitative assays could highly improve the immunoassays diagnostic value. The standard immunoassay procedures use a single 1:100 sample dilution (sometimes 1:50). This does not allow differentiating moderate from high or very high autoantibody concentrations. When present at a very large excess in the tested dilution, the assay absorbances correspond to the high concentration range of the ELISA method, and it is not possible to estimate the true concentration. Using successive and higher plasma dilutions allows for measuring better autoantibody concentrations. For HIT, and more especially VITT, we noticed that many of the most severe patients gave responses in the high assay range for plasma or serum dilutions up to 2:2,000 or even 1:5,000. This shows that when these patients are tested at the 1:100 dilution, the response obtained does not even discriminate on a scale from 1 to > 50. To calibrate immunoassays, we now internally use a recombinant monoclonal antibody to PF4 reactive HPF4 or affinity-purified anti-PF4 polyclonal antibodies. Immunoassays allow for detecting autoantibodies, but not their pathogenic effect. Functional assays must be used to confirm their deleterious impact. HIT functional assays are performed by testing patients’ sample capacity to activate/aggregate Plts from healthy donors at a low (0.1 IU/mL to 1.0 IU/mL) and a high heparin concentration (usually 100 IU/mL). The C14-serotonin release assay (SRA) is the most sensitive one and is often considered the gold standard, but PAT, heparin-induced aggregation assay (HIPA), and flow cytometry assay (FCA) have also been successfully used and are part of the panel available for characterization of symptomatic PF4-dependent autoantibodies [89]. When other chemokines are involved, using the purified protein or its recombinant form as capture antigen in the ELISA allows testing the correspondent autoantibody. If isotyping is required, specific tag antibodies reactive with IgG or IgM or IgA are then used.
For autoantibody testing, the right autoantigen available in a highly purified form and in a presentation mimicking its in-vivo structure must be used. In addition, the current challenge is to optimize sensitivity while keeping full specificity. Capture solid phase coated autoantigen, like in ELISA, must avoid non-specific binding from the tested specimen, especially when rheumatoid factor or heterophilic antibodies are present, or when “sticky samples” with circulating immune complexes are tested. When inflammation is present, high backgrounds can be generated [120, 121]. To achieve that goal, coated solid surfaces must be saturated with an animal serum (from the same species as that used for the tag antibody) to block possible interactions with antibodies. The same animal serum must be used for the sample diluent at a concentration of ≥ 10%, tenfold higher than that of the tested specimen (measured at the 1:100 dilution, i.e., 1%). Currently, we work with goat serum, which is from the same species as the detection antibody. In these conditions, one cannot react with the solid phase saturation serum or the sample diluent, and non-specific reactions are then suppressed or highly reduced. Another challenge is to define the cut-off value between the normal and pathological ranges and to differentiate the background from the specific antibody reaction [122]. In practice, a panel containing at least 120 normal individuals, male or female, out of any medication or disease, and as many pathological samples with autoantibodies as possible (ideally at least 30 or more), must be tested. The mean (M) value obtained for healthy individuals plus 2 standard deviations (SD) defines the highest value of the normal range, whilst M + 3 SD (or M + 5 SD depending on the local definition and assay specificity) defines the lowest value for the positive range. In between, from M + 2 SD to M + 3 SD (or M + 5 SD), there is a grey zone, which can concern borderline positive results or non-specific reactions. For diagnostic or clinical study purposes to rule out the presence of any autoantibody, the cut-off value for the negative range must be used, and complementary investigations must be performed for any reactivity above that limit.
A key factor for immunoassay specificity and reactivity remains the assay design and choice of reagents. The target autoantigen used for preparing the capture solid surface can be recombinant or extracted, but its purity grade is essential for avoiding non-specific interactions. Binding of “sticky samples” or containing immune complexes is avoided or reduced by introducing the saturation step, usually performed with animal serum, also used for the sample diluent [120]. We recommend employing a serum from the same species for the tagged antibodies, the saturation step, and the assay diluent to avoid any unwanted reaction. This animal serum can contain proteins homologous to human ones. Animal and human homologous proteins can share some epitopes, but from our experience, this rarely interferes in the assay specificity. When the autoantigen is unknown, running a control plate prepared in the absence of antigen or coated with an irrelevant antigen like bovine serum albumin (BSA) saturated allows for confirming specificity. If tested samples remain reactive with the control plate, presence of a cross-reactivity with the animal homologous protein is possible. The assay must then be performed by using a different plate saturation and sample diluent additive, like BSA, casein, hydrolyzed collagen peptides, or even poly-ethylene-glycol (PEG). The target autoantigen can then be confirmed. Another technical difficulty concerns antigen presentation. In some conditions, autoantibodies only react with a structure modification of the autoantigen inducing the exposure of neo-epitopes. This is the case for β2GP1 and ADAMTS-13 for example. Autoantigen can furthermore be altered when bound to the plate, but this often mimics its binding to in-vivo biological surfaces, exposing modifications responsible for the autoantibody generation. The laboratory challenge is then to reproduce the antigen presentation, react with autoantibodies, and avoid any assay failure due to autoantigen structural change when immobilized. Should this occur, complementary testing could be performed by using other techniques where the autoantigen presentation is preserved or by performing inhibition studies. In addition to ELISAs, automated immune assays become now available for the most frequently tested autoantibodies, like anti-β2GP1, APAs, or anti-HPF4. Assays use autoantigen bound to magnetic latex beads, and captured antibodies are detected by chemiluminescence. For autoantibodies to Plt antigens, the MAIPA technique can be used. Lastly, the global presence of antibodies on cell surface can be estimated using the Coombs method.
A very wide variety of autoantibodies can develop into various hemostasis proteins, either procoagulant, regulatory, or anticoagulant. They can be detected in many different physio-pathological circumstances, including infectious diseases, drug intake, metabolic disorders, malignant diseases, and tissue damages; and this list is non-limitative [123–125]. The major inducing cause concerns a structural modification of a self-component, which is then presented in an unusual way, and exposes cryptic epitopes or forms complexes with non-self-components, then inducing an extended immune response to the whole complex, through the epitope spreading mechanism. When autoantibodies to hemostasis proteins are generated, they can provoke bleeding episodes or thrombo-embolic diseases, depending on the physiological function of the concerned autoantigen. If they react with clotting factors, a bleeding tendency develops and can be very severe and life-threatening, especially when targeted factors are present at a very low concentration. Paradoxically, in a few cases, this apparent anticoagulant activity can evolve into thrombotic complications, when the immune response is targeted to blood or endothelial cells, which can bind the autoantigen complex with its autoantibody. Therefore, the autoantibody deleterious effect is double: i) inhibition of the biological function of the targeted protein; ii) focusing immune complexes to blood or endothelial cells, or other tissues, targeting the deleterious effect of the immune response onto those cells.
Autoimmune diseases can affect many different body organs, cells, or proteins, and can be induced by a high variety of causes, especially in patients with a genetic susceptibility [72]. The clinical manifestations depend on the autoantibody concentration and affinity, the damages caused to the targeted body’s structures, and the impact on their biological function. In most cases, autoantibodies are transient and progressively disappear when the inducing cause ceases or is suppressed, but they can become chronic with a life-long impact on quality of life in some cases. They can then induce disabilities or lead to fatal issues. Even if major progresses have been undertaken in autoimmune disease management, these complications remain an important issue for global health. Autoantibodies involved in hemostasis provoke either hemorrhagic diseases or thrombotic complications, and they concern a small but specific group of autoimmune burdens. They are characterized when the targeted components are well-identified, which occurs now frequently. They can induce the rapid onset of severe life-threatening pathological conditions, through acute bleeding or thrombotic episodes. However, development of such autoantibodies remains rare, except when they are secondary to other pathologies, like infections, iatrogenic complications, or associated with cancer or metabolic diseases. These autoimmune diseases are often transient when provoked by an infection, a drug, or a chemical contact, although in a few cases, chronicity might develop. The targeted body’s proteins are usually soluble or exposed on blood cells or endothelial surfaces, and they are continuously synthesized. Therefore, when autoantibodies are no longer present, normal conditions are restored, and patients heal. When the cause is metabolic or tumoral, the autoimmune trigger remains for a longer time or becomes chronic, which can induce a long-lasting autoimmune disease. Similarly, when the targeted organ is damaged, with no or little regeneration capabilities, the autoimmune complication also becomes a life-long pathological condition.
For investigating hemostasis autoimmune pathologies, laboratory tools are essential. Thanks to the progress of biotechnology and life sciences, more specific assays can be designed, even if their access remains often restricted because of the low occurrence of many of these complications. Assays are often research tools only available on some sites, except for a few concerning pathologies with a higher incidence like LA, APA, and HIT. Rendering the assays quantitatively as recently proposed for HIT [126] could offer a more appropriate approach for bridging autoantibody concentration and clinical outcomes or evolutions.
In conclusion, although rare, the development of autoantibodies to coagulation proteins must be suspected in presence of an acquired hemostasis factor deficiency or the occurrence of bleeding or thrombotic events in the absence of well-identified triggers. Confirmation of diagnosis is mandatory as soon as possible, and laboratory methods must be used to identify the autoantigen. Clinicians then know how difficult it is to use replacement therapies for the missing factor because the presence of autoantibodies renders that approach inefficient, especially for factors present at a very low blood concentration. Remaining options then include the use of a bypassing activity, when available, and/or to reduce the antibody concentration by depressing the immune system, like with corticoids or immunoglobulins. New promising approaches introduce the use of specific humanized monoclonal antibodies. Management of these autoimmune complications remains always a challenge due to the rapid onset of life-threatening events, which are hemorrhages or thrombosis. Treating the symptom using anticoagulants is effective in controlling thrombotic episodes and their recurrence, however, the situation is more complex for bleeding events, and plasma exchange can remain a possibility.
A5: annexin 5
ACE2: angiotensin converting enzyme 2
ADAMTS-13: A disintegrin and metalloprotease with thrombospondin type I repeats-13
anti-FVIII: anti-factor VIII
anti-GP1b-IX: anti-glycoprotein 1b-IX
anti-HPF4: anti-heparin-platelet factor 4 complexes
anti-PT: anti-prothrombin
APA: anti-phospholipid antibody
APS: anti-phospholipid syndrome
APTT: activated partial thromboplastin time
AT: antithrombin
DIC: disseminated intravascular coagulation
ELISA: enzyme-linked immuno-sorbent assay
FEIBA: factor VIII bypassing activity
HIT: heparin-induced thrombocytopenia
IgG: immunoglobulin G
IL-8: interleukin-8
IU: international units
LA: lupus anticoagulant
M: mean
MAIPA: monoclonal antibody-specific immobilization of platelet antigens
PAT: platelet aggregometry test
PF4: platelet factor 4
Plts: platelets
PS: protein S
rFVIIa: recombinant activated factor VII
SD: standard deviations
TM: thrombomodulin
VITT: vaccine-induced thrombotic thrombocytopenia
vWF: von Willebrand factor
JA: Conceptualization, Writing—original draft, Writing—review & editing.
JA is consultant for HYPHEN BioMed SAS.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 3546
Download: 55
Times Cited: 0
Masahiro Ieko ... Akitada Ichinose
Gary W. Moore
Osamu Kumano ... Jean Amiral
Fariza A. Cheldieva ... Tatiana M. Reshetnyak
Tiffany Pascreau ... Marc Vasse
