 Review
Review

Affiliation:
1Department of Experimental Medicine and Biotechnology, Post Graduate Institute of Medical Education & Research, Chandigarh 160012, India
†These authors contributed equally to this work.
Affiliation:
1Department of Experimental Medicine and Biotechnology, Post Graduate Institute of Medical Education & Research, Chandigarh 160012, India
†These authors contributed equally to this work.
Affiliation:
1Department of Experimental Medicine and Biotechnology, Post Graduate Institute of Medical Education & Research, Chandigarh 160012, India
Affiliation:
2Department of Immunopathology, Post Graduate Institute of Medical Education & Research, Chandigarh 160012, India
Affiliation:
1Department of Experimental Medicine and Biotechnology, Post Graduate Institute of Medical Education & Research, Chandigarh 160012, India
Email: alkabhatia@ymail.com
ORCID: https://orcid.org/0000-0002-7976-0824
Explor Immunol. 2024;4:169–188 DOI: https://doi.org/10.37349/ei.2024.00135
Received: May 12, 2023 Accepted: January 19, 2024 Published: April 02, 2024
Academic Editor: Narinder K. Mehra, Indian Council of Medical Research (ICMR), India
Cancer stem cells (CSCs) are a unique population of tumor cells with stem cell-like properties. They are believed to be involved in drug resistance, potential therapy failure, tumor relapse after treatment, and ultimately reduced overall survival of cancer patients. One of the causal factors that may lead to CSC formation is chromosomal instability (CIN), a dynamic event leading to numerical and structural changes in the chromosomes. The CIN is also proposed to aid the maintenance of CSCs, contribute towards their heterogeneity, and facilitate their immune escape. However, the role of CIN in the modulation of the immune system in tumors remains contradictory. Studies have revealed that it can lead to both activation and suppression of the immune system. Previous literature suggests that the CIN, CSCs, and cancer immunity (3Cs), interact with and complement each other to create a pro-tumor environment. However, the mechanisms underlying such an interaction are poorly understood. So, in this review article, an attempt has been made to understand the nature of the interaction between the triad of CIN, CSC, and the immune response in tumors and some of the pathways governing the same. Understanding the above may be a positive step towards the complete cure for malignant diseases.
Cancer stem cells (CSCs) are a unique population of cells in the tumor that have stemness-like properties, cause drug resistance, potential therapy failure, tumor relapse after treatment, and ultimately decrease the overall survival of the patients [1]. Several theories have attempted to explain the origin of CSC, but none is entirely accepted. One of the factors implicated in the formation of CSCs is chromosomal instability (CIN) [2], which is a defect involving chromosome loss or rearrangement during cell division [3]. CIN may lead to an upregulation of oncogenes or a decrease in the level of tumor suppressor genes [4]. As the tumor grade increases, the rate of occurrence of CIN also increases, and this may lead to the conversion of either normal stem cells (SCs) to CSC or the conversion of non-stem cancer cells (NSCCs) to CSC [5]. Tumors can arise from transformed differentiated cells or transformed SCs [6]. The former can occur during stressful situations like infection, toxins, radiation, metabolic influences, and tissue regeneration, leading to mutagenesis [7]. Typically, complex carcinogenic events are triggered by overexpression of oncogenes, inactivation of tumor suppressors, and cell differentiation by acquiring SC-like characteristics [8].
On the other hand, the transformation of tissue-resident SCs or their progeny requires distinct but few genomic changes for proliferation independent of their niche. Such weak mutational changes have been demonstrated in some cancers, such as gastric cancer, suggesting their resident SC origin [9, 10]. Further, during tumorigenesis, the malignant cells may clonally evolve via the gradual acquisition of genetic changes to enable cancer progression, and some of these may change to CSCs in the process [11, 12]. However, whether the CSCs are present from the initial stages of cancer or formed at the later stages is not well-understood.
CSCs and SC share many common regulatory pathways, including Sonic Hedgehog (Hh), Wnt/β-catenin, and NOTCH, which are crucial for self-renewal [13]. Additional factors like PTEN and polycomb family members influence CSC growth regulation. The CSCs were classically described as a population capable of inducing tumors, especially when transplanted into severe combined immunodeficient mice [14]. Their vital role in tumor initiation and progression makes them ideal candidates for understanding cancer pathogenesis and developing novel therapies [13].
The role of CIN in the modulation of the immune system in tumors remains contradictory. Studies have reported that it can lead to the activation and suppression of the immune system [15]. Recent studies have pointed out how CIN helps CSCs modify themselves and the surrounding environment, thus enabling the malignant cells to evade the immune system [16]. So, in this review article, an attempt has been made to understand the cooperation and interaction between the CIN, CSCs, and cancer immunity (3Cs) in tumors. The review also highlights some of the mechanisms CSCs utilize to evade the immune response with the help of CIN, ultimately leading to the progression of the malignancy. Further, the potential importance of targeting them together in treating advanced-stage cancers is also briefly highlighted.
SCs are undifferentiated cell populations that can self-renew and differentiate into diverse mature progeny [17]. The origin of tumor development from SCs (embryonic rests) was described long back in the nineteenth century [5]. After that, Bonnet and Dick [18] revealed that, like the hematopoietic system, leukemia also has a rare population of cells with stemness-like properties, which he addressed as CSCs. Like leukemia, solid tumors were also observed to contain a similar cellular sub-population capable of self-renewal, differentiation to a variety of cell types, and harboring cancer-initiating properties. Nowadays, CSCs have been reported in many solid tumors, including breast, colon, brain, lung, pancreas, and glioblastoma [19, 20].
CSCs are relatively quiescent cells with the ability to initiate growth and maintain heterogeneity, ultimately promoting metastasis and developing therapeutic resistance in tumors [21]. According to the theory of Paget [22], metastasis involves the detachment of cancer cells from the site of the primary tumor and their spread to distant tissues and organs to establish the secondary tumors. However, this process is imperfect due to the challenging journey the tumor cells must undertake during their mobilization from the familiar microenvironment. CSCs with exceptional survival capabilities, heightened expression of transmembrane transporters, and tumorigenic potential are more capable of enduring stressful conditions in the bloodstream. Thus, they constitute the ideal candidates required for successful distant seeding of the tumor cells [23–25]. CSCs overexpress ATP-binding cassette membrane transporters that lead to high drug efflux, which further contributes to the ineffectiveness of different therapies, including common chemotherapeutic agents such as vinca alkaloids, anthracyclines, and taxanes [13, 26]. Additionally, the CSCs demonstrate an enhanced capability of DNA repair and a high ability to mitigate reactive oxygen species, which may further lead to apoptosis if DNA damage occurs [27]. The above may help them to counter the impact of anti-cancer drugs, which target essential processes in cancer cells like mitosis and DNA repair.
CSCs possess immune evasion capabilities and metabolic adaptations to survive under low nutrient conditions [28]. Further, they demonstrate a high frequency of epithelial-to-mesenchymal transition (EMT). A study on breast cancer highlighted the connection between EMT and SCs [29]. Induction of EMT was observed to elevate the SC gene (CD44+high/CD24–/low) profile in the above malignancy [30]. Also, the inhibitors targeting EMT have been noted to suppress SC-like traits. The interaction between EMT and CSCs is believed to play a vital role in relapse, drug resistance, and metastasis [30].
The CSCs express many surface proteins. In past studies, the same have been used as markers to identify and isolate them. The use of these biomarkers in determining the prognosis of tumors has gained broad interest [31]. However, though many of the markers have been listed, a specific marker for each CSC population has not been well-defined. In solid tumors, the CSCs are identified using a panel of cell surface markers like CD44, CD133, epithelial cell adhesion molecule (EpCAM), sex determining region Y-box2 (SOX2), octamer binding transcription factor 3/4 (OCT3/4), NANOG, etc. [28]. Biomarkers for the CSC population found in hematological cancers include CD33, CD44, CD123, C-type lectin-like molecule-1 (CLL1), T cell immunoglobulin and mucin domain-containing protein 3 (TIM3), etc. [10].
There have been several fascinating theories regarding the origin of CSCs. Whether they are formed from SCs or cancer cells has been continuously discussed. SCs are pluripotent, so it has been proposed that NSCC gives rise to CSC by upregulating pluripotency-related pathways like Wnt, JAGGED2/NOTCH, and β-catenin [13]. CSCs have been shown to display similar surface and intracellular markers to SCs. However, unlike SCs, the CSCs differentiate into disordered cell types without any role in the functional or structural restoration of the normal tissues. Thus, some authors prefer to label CSCs as SCs with a perturbed differentiation potential [32]. Some studies have suggested that the tumor cells may fuse with hematopoietic SCs, generating multinucleated CSCs with a high migration potential [33]. CSCs also have a high metabolic reprogramming ability, which can switch between glycolysis and oxidative phosphorylation [34].
The CSC microenvironment is a complex, heterogeneous milieu within which cells like stromal cells, epithelial cells, and immune cells, and a complex of extracellular macromolecules collectively foster the proliferation, sustenance, and differentiation of CSCs [35, 36]. Extracellular matrix (ECM) is a dynamic structure that undergoes a continuous remodeling process and is a vital component of the tumor microenvironment (TME) [37]. It comprises fibrous proteins, proteoglycans, polysaccharides, and glycoproteins (gp) necessary to sustain CSCs. ECM is a physical barrier to drug delivery to cancer cells [38]. ECM-CSC interaction via CSC receptors such as discoidin domain-containing receptors (DDRs; DDR1, DDR2), CD47, and CD44 enhance CSC nature. For example, CSCs bind to CD44 receptors through CD44 and facilitate the upregulation of stemness factors SOX2 and NANOG concomitant with the augmentations of multidrug resistance-1 expression. These molecular events contribute to heightened drug resistance in breast and ovarian CSCs [39]. ECM can switch NSCCs to CSCs by inducing EMT (Figure 1) [38].
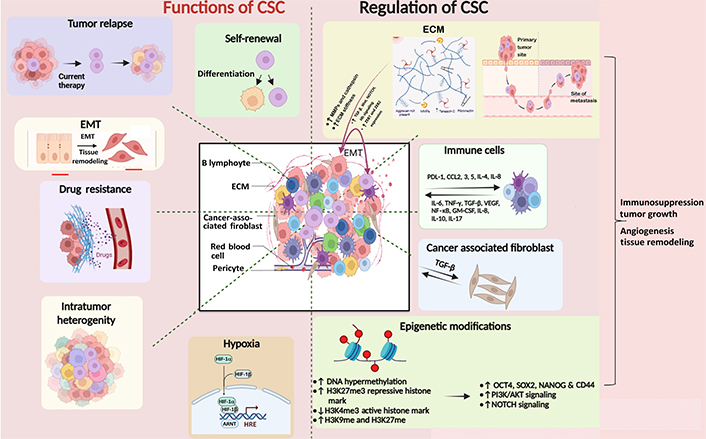
Diagram depicting the functions of CSC and their regulation by ECM, immune microenvironment, and epigenetic modifications. CSCs are regulated by other surrounding cells such as stromal cells, immune cells, cancer-associated fibroblasts, and epigenetic factors like histone modification and DNA methylation. They, in turn, cause relapse of the tumor by promoting EMT, drug resistance, intratumor heterogeneity, and hypoxia. The Figure was created with BioRender software (https://biorender.com/). TGF-β: transforming growth factor-beta; ZEB1: zinc finger E-box binding homeobox 1; PDL-1: programmed death ligand 1; CCL2: chemokine C-C motif ligand 2; IL-4: interleukin 4; TNF-γ: tumor necrosis factor-gamma; VEGF: vascular endothelial growth factor; NF-κB: nuclear factor kappa B; GM-CSF: granulocyte-macrophage colony-stimulating factor; H3K27me3: histone 3 lysine 27 trimethylation; PI3K: phosphoinositide 3-kinase; AKT: protein kinase B; HIF-1α: hypoxia inducible factor 1 alpha; HRE: hypoxia response element; ARNT: aryl hydrocarbon receptor nuclear translocator; MMPs: matrix metalloproteinases. ↑: upregulation; ↓: downregulation
This niche also provides physical support, signaling cues, and biochemical interactions that regulate CSC self-renewal and differentiation. The ECM components, such as fibronectin, collagen, and laminin, can activate signaling pathways crucial for CSC maintenance and survival, including integrin-mediated signaling, which regulates cell adhesion, migration, and survival [40, 41]. It provides a physical scaffold for cells to move through and influences the expression of genes related to the EMT. This process enhances cells’ ability to migrate and invade the surrounding tissues. Growth factors, such as fibroblast growth factor, TGF-β, and epidermal growth factor (EGF), are often present in the TME and can stimulate CSCs. These factors activate CSC-related signaling pathways, including the Wnt/β-catenin and NOTCH pathways. These pathways are critical for CSC self-renewal and differentiation [13]. ECM remodeling, often driven by enzymes like MMPs, can facilitate CSC invasion and metastasis. CSCs may use MMPs to degrade ECM barriers and invade the surrounding tissues [42]. Invasion through the ECM is a crucial step in metastasis, and CSCs are thought to play a significant role in this process [42].
Epigenetic factors are recognized to be crucial in the evolution of malignancies. Epigenetic modifications like histone modifications and DNA methylation are also known to influence the stemness potential and differentiation [43]. Methylation and acetylation of histone proteins are done by histone methyl and acetyltransferase enzymes, respectively [43]. Acetylation of histones makes DNA accessible to RNA polymerases for gene transcription, whereas methylation can activate histone 3 lysine 4 (H3K4) methylation [44] or repress gene transcription (H3K9 and H3K27) [45, 46]. Variable transcriptional outcomes result from irregular patterns of histone modifications. Cytosine-phosphate-guanine (CpG) islands are genomic regions containing large CpG nucleotides, which may extend to 3,000 base pair (bp). DNA methyl transferases (DNMTs) are crucial in checking gene transcription, which occurs when CpG islands are unmethylated and are impeded when they are methylated [47]. DNMT1, DNMT3B, and DNMT3A lead to methylation of CpG islands. The methylation of CpG islands plays a critical role in cell differentiation. OCT4-regulated embryonic SCs express enzymes like ten-eleven translocation 1 (TET1) and TET2 associated with DNA demethylation [48]. Polycomb repressive complexes 1 and 2 repress transcription by H3K27me3, thereby downregulating the genes related to differentiation, development, and choice of lineage [49]. Epigenetic reprogramming is associated with maintaining homeostasis in normal SCs. Its disbalance in CSCs is, however, related to their uncontrolled proliferation and self-renewal ability, thus resulting in tumor initiation and progression. Epigenetic modifications can be associated with the acquisition of hallmarks of malignant CSCs. Epigenetic traits of CSC include hypermethylation of CpG islands and silencing of tumor suppressor genes and pro-differentiation factors (Figure 1) [50]. Methylation enzymes like plasma membrane resident transporter 1 (PMRT1) are involved in SC transformation in leukemia [51], and SET and MYN-domain containing 3 (SMYD3) are observed to maintain self-renewal and tumorogenic potential in gastric cancer [52]. ECM factors like matrix softness can induce epigenetic alterations with enhanced self-renewal and survival of CSCs [53]. CD133 is a CSC marker, and its promoter’s hypomethylation influences its expression in gliomas [54]. EMT helps in the generation of cells with SC-like characters via epigenetic alterations. Histone modification is another epigenetic alteration that induces changes in the gene expression pattern of SCs, resulting in pro-tumorigenic structural and functional changes in the cell. CSCs are maintained by signaling pathways like Hh, NOTCH, Janus kinase (JAK)-signal transducer and activator of transcription (STAT), and Wnt-β-catenin signaling [55]. These pathways are involved in self-renewal in SC and are regulated by epigenetic mechanisms. Dickkopf Wnt signaling pathway inhibitor 1 promoter silencing involves decreased acetylation of H3K16 and H3K27me3 [23]. Histone acetylation is observed at the promoter region of NOTCH receptor-ligand JAGGED2, activating NOTCH signaling in myeloma cells [56]. Rhabdoid tumors show decreased activation of SNF5, a member of the chromatin remodeling complex, leading to Hh signaling activation [57]. The mixed lineage leukemia (MLL) gene encodes for a histone methyl transferase that influences many cellular processes [23]. MLL is found in several cancers and is involved in carcinogenesis. Leukemia SC with MLL-AF9 fusion protein can maintain the identity of progenitors from which they arose while maintaining a self-renewal-associated program [58]. CSC helps in propagation, growth, and metastasis in colorectal cancer (CRC), including the hypermethylation of several tumor suppressor gene promoters, including p16, retinoblastoma, secreted frizzled-related protein (SFRP), and mutL homolog 1 (MLH1) [23]. The driver mutation in CRC is the adenomatous polyposis coli (APC) mutation, which influences the activities of DNMTs [59]. Increased expression of DNMT1 is believed to downregulate the transcription of APC, a tumor suppressor gene in CRC [60]. It has been shown that the expression levels of DNMT1 within CSCs are critical for the development and progression of CRC, showing a clear link between epigenetic processes and the CSC-driven tumorigenesis process [61].
CIN is the hallmark of most solid tumors and is believed to be an essential factor underlying cancer evolution. It is considered a well-known driver of tumor development and metastasis [62]. It is characterized by the frequent mis-segregation of chromosomes during cell division. The cells with CIN are proposed to have a mis-segregation rate as high as 20% (confer 1% in normal cells) of total cell divisions [63]. The chromosomal rearrangements in tumors may vary from very small to large, ultimately leading to its progression. However, CIN can be predominantly structural or numerical, and distinct types of CINs may exhibit different effects in the case of malignancy. As an example, most aneuploidies are thought to push the cancerous cells towards more aggressive phenotypes. Still, some kinds of CIN may drive the cells towards apoptosis due to extreme imbalance in gene dosage [64]. Malignant cells that have a medium level of CIN demonstrate a poorer prognosis than deficient and high levels [65], indicating that a narrow threshold level of CIN may be essential to facilitate cancer cell survival. High CIN can cause activation of the p53-dependent pathway if the same is active. The latter pushes the cells towards apoptosis and cell cycle arrest [66, 67]. CIN and somatic mutations serve as promoters of genetic heterogeneity and malignant phenotypes in cancer [68]. As malignancy progresses, the heterogeneous malignant cells undergo continual modifications due to CIN, and the novel clones that are fittest to survive are generated. The new karyotypes caused by CIN are responsible for phenotypically and functionally heterogeneous cancer cells co-existing within the same tumor (Figure 2) [15].
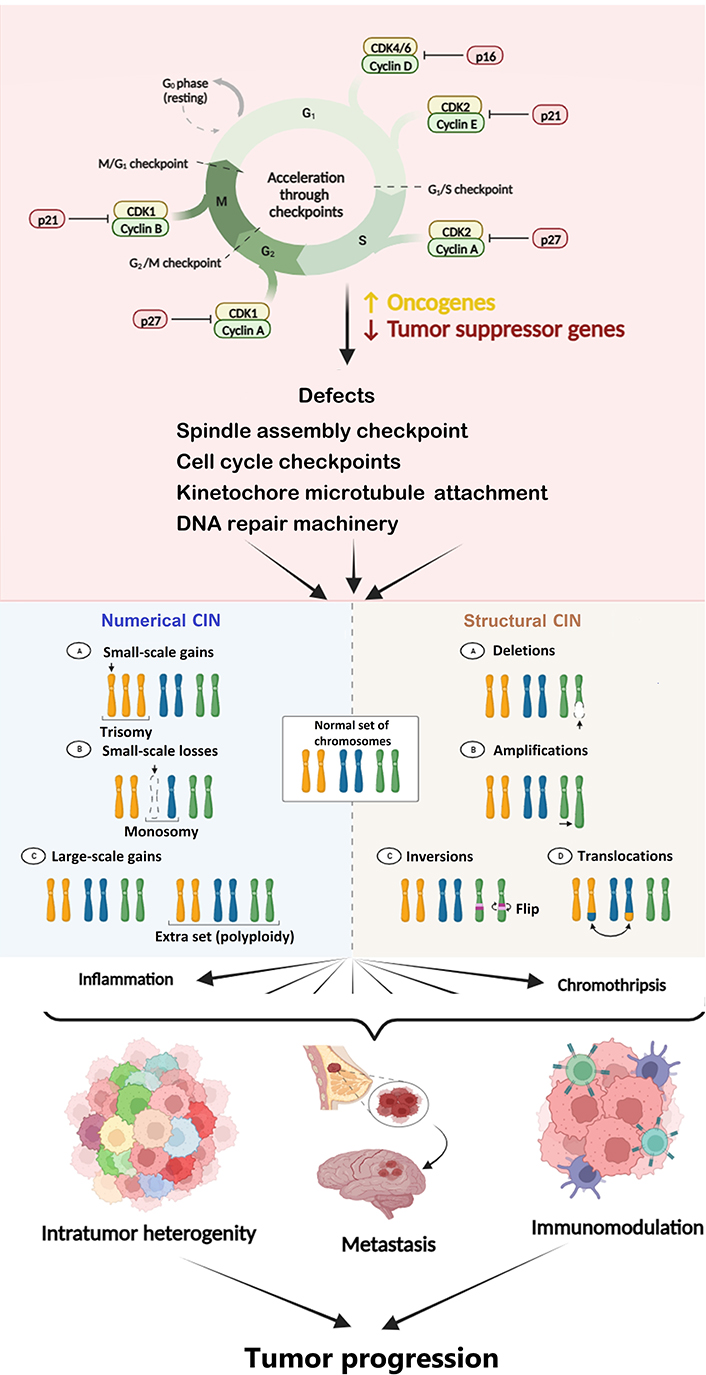
Diagram depicting the cause and consequences of CIN in cancer. CIN is a hallmark of cancer caused by defects in cell cycle and spindle assembly checkpoints, DNA repair machinery, etc. CIN leads to inflammation, endoplasmic reticulum (ER) stress, DNA damage, etc., which ultimately causes tumor progression. The Figure was created with BioRender software (https://biorender.com/). CDK1: cyclin-dependent kinase 1. ↑: upregulation or increase; ↓: downregulation or decrease
Mutations in the cell cycle genes lead to a significant fraction of chromosomes gained or lost during cell division and a change in the structure of the chromosome. Defects in mitotic spindle assembly checkpoint, loss of centriole, error in microtubule kinetochore rearrangement, sister chromatid cohesion defects, defects in DNA repair machinery, supernumerary centrosomes, mitotic plus-end assembly, and cell cycle checkpoints can all lead to CIN [65]. Several qualitative and quantitative approaches have been used for assessing CIN. The visual indicators of CIN include micronuclei, nuclear buds, chromatin bridges, and laggards [69]. These markers of CIN may have different fates, including degradation, chromothripsis, persistence, extrusion, and reincorporation inside the nucleus [70]. It is believed that tumor characteristics and their relationship with the surrounding immune and non-immune microenvironment play a critical role in defining the CIN level conducive to survival and growth.
The relationship between CIN and cancer stemness is complicated and far from clear. However, some studies have suggested that CIN could be a potential originator of stemness in cancer [71]. Both cancer cells and SCs have an unstable genome, which provides CSCs with a cumulative reason for possessing genetic instability. Spectral karyotypic analysis of bone marrow mesenchymal SCs suggested that CSCs acquire genetic alterations initially absent in SCs.
In the case of normal SCs, whenever a certain level of mutation happens, it is repaired by the repair machinery. But in the case of cancer, since the mutational burden is high and due to CIN, the amplification rate of oncogenes and deletions of tumor suppressor genes is also high. Therefore, the CSCs with CIN also have a high mutation frequency and damaged DNA, which may not be repaired due to overburdened and defective repair machinery [71].
A study showed that when the level of CIN was increased in bone marrow-derived mesenchymal SCs, they converted into CSCs and gave rise to fibrosarcoma [72]. This malignant transformation was characterized by the accumulation of genetic aberrations, elevation in cellular Myc, and telomerase activity [72]. Similarly, on prolonged culturing, the neural SC was found to gather CIN, and the cell line obtained from it was observed to have CSC features. The above may be because SCs are most capable of accumulating genetic changes as they are immortal cells. For example, embryonic SCs have been shown to harbor numerical CIN involving sex chromosomes X and autosomes 12 and 17q [73]. The embryonic SCs have been reported to show uncoupling of apoptosis from mitosis, which may also be responsible for their predisposition to acquire CIN [74]. The CIN in these cells may be an inherent feature or acquired during their prolonged lifetime to aid their survival; however, it is debatable. Also, gene array analyses have indicated that CSCs resemble embryonic SCs more in showing uncoupling between mitotic checkpoint activation and apoptosis [74]. However, the genome of adult SC is relatively stable [75]. Therefore, from the ensuing discussion, it can be concluded that normal SCs via the acquisition of CIN may proceed towards CSC and malignancy [76]. On the other hand, the CSCs, which are long-lived, may keep increasing their CIN by umpteen number of cell divisions that they undergo during their lifetime.
CSCs are expected to possess CIN, which may contribute to intra-tumoral heterogeneity [77]. Some data points towards the presence of cancer progenitor cells, which give rise to cells with different levels of CIN. These genetic variants play a role in imparting metastatic potential and drug resistance [78]. The CSCs and CIN thus complement each other to drive therapy failure and metastasis.
From this section, it can be concluded that in normal cells, whenever a level of DNA damage occurs, it is repaired by the DNA repair machinery. But in the case of cancer, the latter is compromised, and the DNA damage overcomes the repair machinery due to over- and under-functional oncogenes and tumor suppressor genes respectively. The above may lead to the formation of CSCs from either normal SCs or NSCCs. The heightened level of CIN in CSC itself may further help in enhancing tumor heterogeneity and thus contribute towards adverse phenomena like drug resistance, metastasis, and tumor progression.
The TME contains different types of immune cells like natural killer (NK), dendritic cells (DCs), macrophages, and T cells of innate and adaptive immune systems, respectively [79]. The immune system exerts both stimulatory and inhibitory effects on cancers, and the balance of these has profound implications for tumor growth. Cancer immunoediting is defined as the continuous remodeling of the tumor immunogenicity by the immune microenvironment surrounding it [80, 81]. Therefore, the cells involved in cancer immunoediting include the immune cells as well as non-immune cells like the stromal and endothelial cells.
Recent studies have suggested that CSCs may play a significant role in immunoediting [82]. The concept of immunoediting is defined by elimination, equilibrium, and escape phases (3Es) [83]. The first is the elimination phase, defined as the immune system’s ability to recognize and remove cancer cells. CSCs remain relatively secluded in the niches that are the functionally diverse regions within the TME. The niches sustain CSCs, preserve their phenotypic plasticity, help in their protection from the immune system, and contribute to their metastatic potential. Quiescence and low immunogenicity may help CSCs remain protected from a highly functional and active immune system in the elimination phase [36].
In the equilibrium phase, cancerous cells’ growth is limited but not entirely removed. CSCs, due to their higher resistance to immune-mediated killing, may play a crucial part as they can constitute the hidden but continuous source of a few tumor cells that may be present in this phase. In the escape phase, the CSCs may remove all their restrictions and divide asymmetrically. These divisions may give rise to the progenitor cells with a high proliferation rate on one hand and daughter CSCs replenishing their pool on the other hand [84]. By increasing the heterogeneity in the CSC population or the progenitor cells downstream in the hierarchy, CIN may further aid them to escape immune destruction [18, 85, 86]. Also, it has been proposed that CIN may contribute to decreased immunogenicity of CSCs, thus facilitating their escape from the immune system. Overall, a higher CIN will result in more heterogeneity and a lower proportion of the immunogenic antigens of a particular type.
Thus, the threshold required to fire the anti-tumor effector cells may be difficult to reach [87]. CSCs may also modify the microenvironment around themselves, secrete some immune inhibitory factors, and increase the expression of surface inhibitory molecules like PDL-1 [88, 89]. The CSCs can secrete a variety of molecules, such as TGF-β, IL-6, IL-10, IL-4, and prostaglandin E2 [90], and also show improper antigen presentation that helps in their protection from both innate and adaptive immune systems [80, 91]. A lower level of CSC immunogenicity as compared to NSCC has been reported in previous studies [89]. The representatives of CIN, like micronuclei, may rupture to release DNA into the cytoplasm, which is recognized by nuclear DNA sensors like cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS) to initiate a stimulator of interferon (IFN) genes (STING)-mediated type-1 IFN response. The same may lead to inflammation [92]. However, prolonged inflammation via this pathway may further enhance CIN and promote tumorigenesis. Also, the cGAS-STING pathway may be downregulated in some viral-induced tumors or by some molecules like excision repair cross complementation group 1 (ERCC1) in tumors [93]. Nevertheless, the cGAS-STING pathway is proposed to play a vital role in cancer immune responses, which may be governed by factors like the type of tumor and strength and duration of its activation [94]. Moreover, the CIN-activated cGAS-STING pathway may impact CSCs. A study by Liu et al. [95] reported the potentiation of cancer stemness by intrinsic activation of cGAS-STING via increased STAT3 (Figure 3).
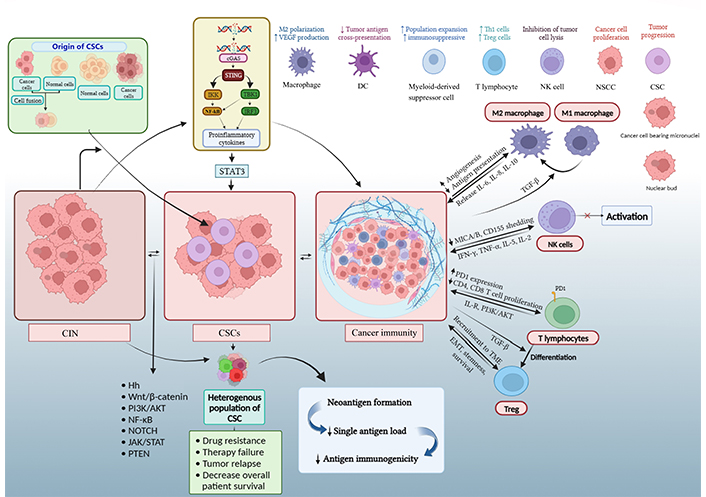
Diagrammatic representation of complex mechanisms underlying CSC, CIN, and immune response in cancer. An increase in CIN beyond a certain level by dysfunctional cell cycle and DNA repair genes or oncogenes and tumor suppressor genes are believed to contribute to malignant transformation and progression. CIN in NSCC or SC can lead to the generation of CSC. Also, CIN (through micronuclei) and stemness can produce a pro-inflammatory immune response. The CSCs can utilize CIN to enhance their heterogeneity and modulate the immune and other neighboring cells to favor self-survival [16, 96, 97]. The Figure was created with BioRender software (https://biorender.com/). IKK: Ikappa B kinase; TBK1: tank-binding kinase 1; IRF3: IFN regulatory factor 3; Th1: T helper 1; Treg: T regulatory; MICA: major histocompatibility complex (MHC) class I chain-related protein A; PD1: programmed cell death protein 1; IL-R: IL receptor. ↑: upregulation or increase; ↓: downregulation or decrease
T cells exert an immune response after recognizing antigens presented by antigen-presenting cells and non-immune cells. MHC I presents an antigen that is identified by CD8+ T cell receptor (TCR) [98]. Full activation of T cells requires TCR signaling and secondary signaling through costimulatory molecules. Though CSCs have low expression of ligands for secondary signals due to clonal heterogeneity caused by CIN (B7.1 and B7.2), they contain higher degrees of co-inhibitory molecular ligands like PDL-1 [99]. CSCs cannot constrain the cytotoxicity of activated and differentiated cytotoxic T lymphocytes (CTLs) but can prevent or inhibit the differentiation of CD8+ naive T cells into CTLs. In their comprehensive bioinformatics analysis, Davoli and colleagues [100] observed that the tumor samples that contain high CIN have low expression of CD8+ T cells. So, we can say that a level of CIN is required for the CSCs to evade the immune system. As CSCs proliferate, genetic variation occurs, and they select a population with decreased expression of the MHC I molecule. The low expression of MHC I may help them to survive and escape from the CTLs [101]. CSCs have been demonstrated to increase the expression of CTL-associated antigen 4 (CTLA-4). Then, the CTLA-4 binds with the CD80/86 expressed on antigen presenting cells and inhibits the function of T cells [102].
The complex interaction between immune cells and CSCs also involves Treg cells. Tregs are divided into two groups, one formed in the thymus (natural) and the second one that differentiates in the periphery (induced Treg; iTreg) [103]. With the help of Treg cells, CSCs evade the immune system. The role of cancer-associated fibroblasts and tumor-associated macrophages (TAMs) in the recruitment of Treg cells to the TME has been highlighted by several studies [104]. The CSCs produce TGF-β, which increases the levels of transcription factor forkhead box protein 3 (FoxP3) and helps differentiate Treg to iTreg with consequent suppression of tumor immunity [105]. The increasing number of Tregs in tumors prevents the activation of CD8+ T cells and the instantaneous secretion of inflammatory cytokines, such as TNF, IFN-γ, and CCL2, which are needed to activate the adaptive immune system [106]. By increasing the stemness, CIN may complement the immunosuppressive effects of CSCs mentioned above.
The interface between CSC and NK cells is associated with the upregulation of activating cytotoxicity receptors like NKp44 and NKp30 on NK cells and higher-level presentation of their ligands on CSCs [107]. The same is also expected as the CSCs show decreased MHC I expression and hence are susceptible to targeting by NK cells. Further, high expression of activation ligands like FS-7-associated surface antigen (Fas), MICA/B, and death receptor 5 (DR5) makes CSCs more vulnerable to NK cell-mediated cytotoxicity. However, CSCs also have a higher level of CIN that helps them to escape from NK cell-mediated killing, possibly by increasing the expression of inhibitory NK cell receptor ligands [89, 108]. Wang et al. [109] reported that CSCs evade killing by NK cells by shedding MICA and MICB ligands and via recruitment of Tregs. Further, through the loss or gain of chromosomes, CIN can generate variants of CSCs negative for the immune activation gene, thus enabling further evasion of NK cell-mediated immunity [110]. In addition, previous work has shown that as the CIN increases, there is improper functioning of type I IFN signaling that causes improper priming and cytotoxic activity of NK cell-related cytokines like IL-15 and IL-12 [15, 111].
Naturally activated macrophages, recognized as M1-polarized macrophages, are stimulated by cytokines like IFN-γ, generating an increase in levels of reactive oxygen and nitrogen species that cause the secretion of pro-inflammatory cytokines, like IL-23, IL-12, chemokine C-X-C motif ligand 10 (CXCL10), and CXCL19 in the microenvironment of CSCs [112]. The above promotes Th17, Th1, and NK cell phenotypes. Macrophages can abolish the cancerous cells and activate the tumor elimination environment; however, malignancy development causes a phenotypic shift from M1 to M2 macrophages [113]. Abundance of Th2 cytokines (such as IL-10, IL-13, and IL-4) and CSCs activate the M2 macrophages, also known as TAMs [114]. TAMs, an essential component of TME, regulate the functions of CSCs in many aspects. Together with tumor-associated neutrophils, the TAMs protect CSCs from constant immune surveillance in the TME [115]. TAMs help advance established malignancy by stimulating CSC survival and proliferation as well as lymphangiogenesis and angiogenesis and modulating the activated T cell responses [80]. Many mechanisms have been proposed by which the TAMs can affect CSCs. Milk fat globule EGF 8 (MFG-E8) is a growth factor that signals through integrin-αvβ3 and αvβ5 and facilitates processes, such as apoptotic cell phagocytosis and angiogenesis. High expression of αv-integrins has been reported on CSCs (confer NSCCs), which can interact with MFG-E8, thus resulting in tumor progression and development of drug resistance [116]. Another study showed that macrophages enhance the invasion of glioma CSCs through the TGF-β1 signaling pathway [117]. Further, TAMs have also been noted to promote breast CSC self-renewal in mice through EGF secreted by activation of STAT3/SOX2 signaling pathway. In hepatocellular carcinoma cells, the TAM-secreted IL-8 activates JAK2/STAT/Snail pathway to promote EMT [13]. Not many studies have elaborated upon the effects of CIN on macrophage behavior in tumors. However, one recent report has indicated that CIN can help to increase the anti-tumor effects of macrophages [118]. Therefore, the exact influence of interaction between CSCs, macrophages, and the CIN in the TME remains ununderstood.
Modern studies have highlighted the importance of the above 3Cs in carcinogenesis, however, in isolation. The level of CIN is determined by microscopy-based identification of morphological markers like micronuclei or hybridization techniques like fluorescence in situ hybridization or comparative genome hybridization. Similarly, past studies have used designated CD markers to identify stemness or the nature of immune response in cancer [119]. Choosing the most relevant panel of markers and assays for gauzing the 3Cs in the tumor needs to be undertaken in future studies. Further, techniques like flow cytometry or real-time polymerase chain reaction (PCR) can then be explored to assess these markers in the routine clinical setting for diagnosis and to decide upon the precision therapies from the existing or novel drugs. For example, drugs targeting multiple aspects of CSCs, whether they are the surface molecules or the signaling factors related to them, are being extensively investigated.
Similarly, the knowledge that CIN beyond a certain level is lethal to cancer cells is being used to develop novel therapies (Table 1). The immune response dynamics in cancer have also led to a torrent of immune-based therapeutics like antibodies, chimeric antigen receptor (CAR)-T cells, cytokines, immune checkpoint inhibitors, etc. However, despite so much progress, the treatment of advanced cancers remains problematic. At present, different types of approaches, ranging from gene therapy to small interfering RNAs (siRNAs) to novel compounds, are being used with or without conventional chemotherapy to achieve complete elimination of cancer. CAR-T and CAR-NK cells designed against the molecular markers overexpressed in CSC, like EpCAM, CD133, CD44 isoform variant 6, etc., are being investigated to treat certain cancers (phase I clinical trial) [120]. So, searching for a therapeutic strategy addressing the 3Cs together may be the elixir of life for difficult-to-target cancer patients. The above may involve a single compound or combination of drugs simultaneously targeting stemness, CIN, and disadvantageous immune factors in one go.
Known and investigational anti-cancer drugs with potential to target 3Cs
| Drugs | Effect on CIN | Effect on cancer immunity | CSCs | References |
|---|---|---|---|---|
| Chemotherapeutics drugs | ||||
| 5-Fluorouracil | ↓ DNA and RNA synthesis | ↑ Tumor-infiltrating CTLs | Suppress CSC in the PANC-1 cell line by inhibiting p38 | [121, 122] |
| Cyclophosphamide | ↓ DNA replication process | ↑ NK and T cell activity; ↓ Treg activity | CSCs found to be resistant to cyclophosphamide | [123, 124] |
| Doxorubicin | Perturbation of replication and transcription | ↓ Activation of the immune system | Co-delivery of all-trans-retinoic acid and doxorubicin inhibit CSCs | [125–127] |
| Gemcitabine plus cisplatin | ↓ DNA and RNA synthesis | ↓ Circulating Treg cells | Cisplatin and plasma-activated medium cause death of CSC | [128, 129] |
| Paclitaxel | ↓ Mitosis | ↑ CD68+ macrophage, NK, and CTL tumor invasion | Paclitaxel and sorafenib combination suppressed self-renewal of CSCs | [130–132] |
| Immunotherapeutics drugs | ||||
| Vemurafenib | Inhibit BRAFV600E | ↑ gp100, MART1, and other antigen expressions ↓ Immune-suppressive cytokine secretion | ↓ CSC traits | [133, 134] |
| Temsirolimus, rapamycin, and other mTOR inhibitors | Targets mTOR pathway | ↑ CD8+ T-cell activation, IFN production, and differentiation into memory cells; ↓ IDO expression by Treg homeostasis. ↑ Treg response to antigen | Can lead to preferential apoptosis of CSCs | [135, 136] |
| PI3K-AKT inhibitors | ↓ PI3K-AKT mediated signaling in malignant cells | ↑ The ability of CTLs and NK cells to lyse tumors using perforin-granzyme; ↓ pro-survival signaling, and pro-tumor inflammation | Rottlerin-induced apoptosis of CSC by inhibiting the PI3K-AKT-mTOR pathway | [137–139] |
| Imatinib | Targets multiple tumor-linked tyrosine kinases, like ABL and cKIT | Targets IDO, ↓ efficiency, and number of Treg cells; ↑ DC-NK cell cross-talk | Conflicting data | [140, 141] |
| CSC targeting drugs | ||||
| Paclitaxel + reparixin | ↓ Mitosis | NK and CTL tumor invasion | Targets CSC in triple-negative breast cancer | [142] |
| GDC-0449 + conventional therapy | ↓ Hh signaling in pancreatic cancer | Not reported yet | Targets CSC (phase II) | [142] |
| CAR-T EpCAM | Targets EMT | Improve infiltration of T-cells with Wnt inhibitor | Targets CSC (phase I/II) | [10, 143] |
IDO: indolamine 2,3-dioxygenase; MART1: melanoma antigen recognized by T cells; mTOR: mammalian target of rapamycin; cKIT: type III receptor tyrosine kinase. ↓: inhibition or decrease; ↑: activation or increase
The ensuing discussion proves beyond doubt that the 3Cs mentioned above constitute the crucial three pillars of carcinogenesis. Although 3Cs have been observed to influence each other in one or multiple ways, the pathways and molecules governing their interactions are complex and incompletely understood. The association between the 3Cs constitutes a vicious cycle that drives the cancerous process forward. Understanding the role of these, especially in the context of each other, may help in identifying a panel of biomarkers for early detection and therapeutic targeting of malignancies. Recent studies have drawn attention to the importance of modulation of CIN-induced cGAS-STING pathway and subsequent immune response in cancer treatment. Similarly, PI3K-AKT inhibitors may be used to target all three events together. CAR-based immunotherapy is being investigated to target surface markers of CSCs to kill them specifically. Thus, the effective targeting of the triad of CIN, CSCs, and associated hostile immune microenvironment together carries plenty of potential for improving the therapeutic outcome of highly aggressive, metastatic, and drug-resistant tumors with poorly defined molecular targets in the future.
3Cs: chromosomal instability, cancer stem cells, and cancer immunity
AKT: protein kinase B
CAR: chimeric antigen receptor
cGAS: cyclic guanosine monophosphate-adenosine monophosphate synthase
CIN: chromosomal instability
CpG: cytosine-phosphate-guanine
CRC: colorectal cancer
CSCs: cancer stem cells
CTLs: cytotoxic T lymphocytes
DDRs: discoidin domain-containing receptors
DNMTs: DNA methyl transferases
ECM: extracellular matrix
EGF: epidermal growth factor
EMT: epithelial-to-mesenchymal transition
EpCAM: epithelial cell adhesion molecule
H3K27me3: histone 3 lysine 27 trimethylation
H3K4: histone 3 lysine 4
Hh: Hedgehog
IFN: interferon
IL-4: interleukin 4
MHC: major histocompatibility complex
MICA: major histocompatibility complex class I chain-related protein A
MLL: mixed lineage leukemia
MMPs: matrix metalloproteinases
NK: natural killer
NSCCs: non-stem cancer cells
PDL-1: programmed death ligand 1
PI3K: phosphoinositide 3-kinase
SCs: stem cells
SOX2: sex determining region Y-box2
STAT: signal transducer and activator of transcription
STING: stimulator of interferon genes
TAMs: tumor-associated macrophages
TGF-β: transforming growth factor-beta
Th1: T helper 1
TME: tumor microenvironment
Treg: T regulatory
LK and SM equally contributed to: Writing—original draft, Writing—review & editing. RY: Writing—original draft. AB and YK: Conceptualization, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

