
Abstract
Dendritic cells (DCs) play an important role in the formation of the immune response, and they are involved in the pathogenesis of autoimmune diseases. Targeting DCs has thus emerged as a new therapeutic modality in the management of inflammatory and autoimmune diseases. DCs can be manipulated ex vivo and then injected back into humans to suppress the immune response. They can also be manipulated in vivo by delivering specific molecules into the DCs. Co-stimulatory molecules that shape DCs interaction with T cells can also be targeted to suppress immunity. This review tackles the latest advances in each of the 3 presented approaches.
Keywords
Autoimmune diseases, dendritic cells, immune tolerance, co-stimulatory molecules, immunotherapyIntroduction
Autoimmune diseases affect around 5–10% of the world’s population [1], with autoimmune thyroid disease and type 1 diabetes (T1D) being the most common. There are nearly 100 distinct autoimmune diseases, which can be either organ-specific such as primary biliary cirrhosis, or systemic, involving multiple organs such as systemic lupus erythematosus (SLE) [2]. Incidence is influenced by demographic factors like age, gender, ethnicity, and others. They can occur at any age, but each disease has its characteristic age of onset, with females being usually more affected than males [2].
Due to the evolution of research, significant advances in the understanding of human autoimmunity have been achieved, allowing improvements in the classification, diagnosis, and, most importantly, management of these autoimmune diseases.
The cornerstone for the treatment of autoimmune diseases has long been immunosuppressants (e.g., corticosteroids), until the advent of immunomodulatory biologic drugs, which block inflammatory mediators, including proinflammatory cytokines. At the frontier of these biological drugs are tumor necrosis factor (TNF)-α blockers, used in rheumatoid arthritis (RA), psoriasis, ulcerative colitis, and Crohn’s disease for example [1]. They have been considered the “standard of care” due to their effectiveness in disease control and risk reduction [1].
Despite the success of these immunomodulatory therapies, there still exist significant unmet clinical demands. First, currently approved therapies are immunomodulatory, symptom-relievers, rather than direct curatives for loss of immune tolerance [1]. Second, due to their action on the reduction of systemic inflammation, they increase susceptibility to opportunistic infections or malignancies and have critical side effects [3–5]. Third, these agents are of no efficacy in the case of refractory autoimmune diseases (e.g., multiple sclerosis) and in patients who have grown unresponsive [6, 7].
The new treatment paradigm is based on targeting known immune tolerance mechanisms, such as increasing regulatory T cells (Tregs) and inducing anergy or elimination of auto-reactive immune cells. These comprise coinhibitory checkpoint agonists and co-stimulatory checkpoint antagonists to expand Tregs and/or suppress pathogenic effector cells [1]. Moreover, a review presented by Lopez-Santalla et al. [8] addressed a new type of treatment, which is cell therapy.
Autoimmunity is indeed a challenge; nevertheless, the prognosis for patients with these diseases has and will continue to remarkably improve as new approaches are being investigated.
Physiology of the immune system and key co-stimulatory molecules
Brief review of the human immune system
The human immune system is the major defense against foreign pathogens and intrinsic aberrant malignancies while concurrently preventing deleterious responses against the self [9]. It comprises two important complementary and not mutually exclusive components, innate and adaptive immunity, where different molecules, cells, and organs are involved in the protection from external and internal damage [10].
Innate immunity cells, including phagocytes, dendritic cells (DCs), mast cells, basophils, eosinophils, and natural killer (NK) cells, respond to molecules exhibited by pathogens, called pathogen-associated molecular patterns, such as lipopolysaccharides of the bacterial cell wall and RNA of viruses, after recognizing them via pattern recognition receptors on their surface. Following this recognition, the now-activated immune cells produce cytokines and chemokines, recruiting more immune cells to sites of infection and ultimately resulting in inflammation.
Cells of adaptive immunity include antigen (Ag)-specific T cells, which are activated by Ag-presenting cells (APCs), and B cells, which differentiate into antibody (Ab)-producing plasma cells. APCs express a group of proteins known as the major histocompatibility complex (MHC), classified as MHC class I (MHCI), which presents endogenous (intracellular) peptides to cytotoxic T lymphocytes (CTLs), and MHCII, which presents exogenous (extracellular) peptides to helper T (Th) cells.
T cells, on the other hand, express a wide range of unique T cell receptors (TCRs), which can bind to specific foreign peptides expressed on MHC. They also possess cluster of differentiation 4 (CD4) or CD8 proteins. Mature APCs can activate T cells resulting in the release of cytokines that influence the activity of many cell types, i.e., potentiating the action of macrophages and allowing differentiation into CD8+ CTLs, CD4+ Th cells, and memory cells.
B cells, however, can recognize Ag directly, without the need for APCs, through unique Abs expressed on their surface. They present processed pathogen peptides on MHCII to TCR of CD4+ cells; meanwhile, CD40 protein on the B cell surface also binds to CD40 ligand (CD40L) of T cell. This interaction will result in the differentiation of B cells into plasma cells and memory cells.
On the other hand, a major malfunction of the immune system occurs when it recognizes self-Ags as foreigners, inducing a cascade of molecular and cellular events that destroy human tissues. This happens due to inadequate immune tolerance, and thus autoimmune diseases emerge [11]. Contemporary theories suggest that the development of an autoimmune disease requires both genetic predisposition and environmental stimuli (nutrition, microbiota, microorganisms, xenobiotics, etc.) that trigger the immune pathways leading to tissue destruction [1].
DCs: key cells governing immune responses
DCs are professional APCs that efficiently link innate and adaptive immunity and modulate T cell responses. They constantly capture, process, and present Ags to T cells, influenced by the surrounding milieu. Subsequently, T cells either express stimulatory molecules to activate protective immunity against pathogens/cancer cells or inhibitory ones to tolerate self-Ags [12]. For this, understanding the role of distinct co-stimulatory pathways on DC function is indispensable.
DCs are said to be immature during homeostatic conditions, with minimal expression of co-stimulatory molecules and proinflammatory cytokines. Environmental cues such as pathogen- or danger-associated molecular pattern molecules direct their differentiation into mature DCs, with upregulated expression of co-stimulatory molecules and proinflammatory cytokines.
DC maturation leads to an upregulated expression of homing receptors [e.g., chemokine receptors (CCRs)], augmenting DC migration to lymph nodes, and the development of cellular extensions to expand the contact surface area with T cells. Additionally, there is an upregulation of MHC groups and an enhanced release of cytokines for more potent T cell recruitment [12]. On the other hand, interleukin-10 receptors (IL-10Rs) are downregulated to resist the immunosuppressive signals of interleukin-10 (IL-10) [13, 14].
Activation of naïve T cells by DCs relies on 3 signals (Figure 1): MHC presentation of the processed Ag to specific TCR, the action of multiple co-stimulatory molecules, and the role of the released cytokines in the milieu (e.g., IL-12, IL-6, TNF-α). The integration of all these signals directs T cell differentiation to an inflammatory versus tolerogenic profile [15].
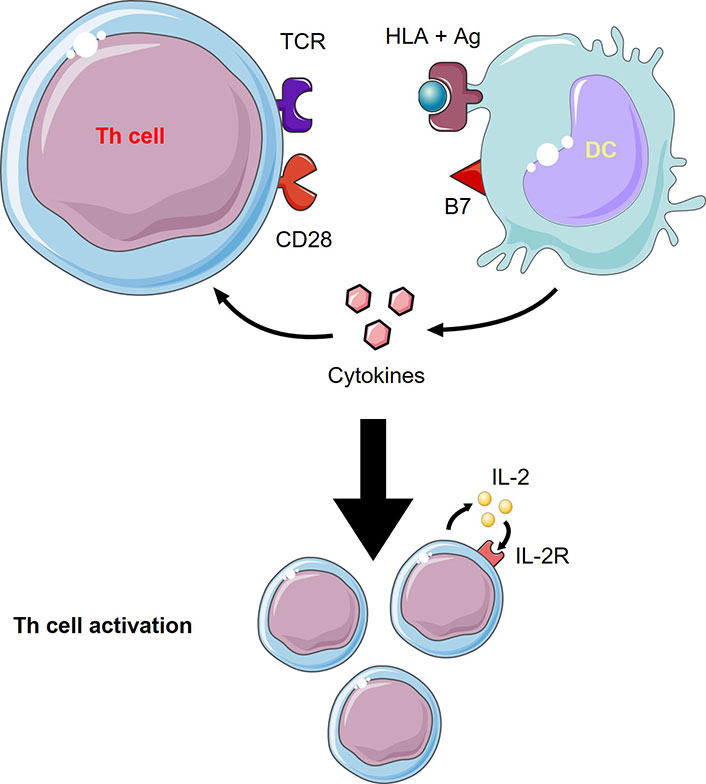
T cells activation by DCs. Th cell activation from DCs needs 3 signals which are 1) TCR recognition of the human leukocyte Ag (HLA) + Ag complex, 2) co-stimulatory molecule, B7, interaction with CD28, and 3) cytokines released from DCs to act on Th cells. Th cell activation involves proliferation through autocrine IL-2 signaling. This figure was created by Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported License
DCs also participate in mediating self-tolerance and thus the prevention of autoimmune diseases. This is due to their ability to eliminate autoreactive T cells and their support of the Treg population. In the absence of immune aggression, and thus the absence of proper co-stimulation and cytokines necessary for maturation of DCs and T cells, immature DCs presenting self-Ags will not yield any response and would rather result in autoreactive T cell apoptosis and anergy [12, 16]. Also, immunosuppressive molecules [e.g., IL-10, transforming growth factor-β (TGF-β)] act on DCs that present self-Ags on MHCII resulting in the expansion of the Treg pool [17].
Hence, it can be conferred that DCs may perform different actions under different conditions. Eliciting a tolerogenic vs. an immunogenic T cell response relies heavily, yet not solely, upon the nature of the available co-stimulation.
Co-stimulatory molecules
B7
B7, the most important co-stimulator of T cell activation [18], is a membrane protein of APCs. It comprises two closely related units: CD80 (B7.1) and CD86 (B7.2); the latter being constitutively expressed in intermediate amounts on immature DCs [19].
B7 can interact with 2 specific entities: CD28 for the initiation of T cell response and CTL-associated Ag 4 (CTLA-4) for its termination. Upon binding to CD28, B7 induces the production of IL-2 in CD4+ T cells, allowing their proliferation and differentiation into Th cells (Th1, Th2, and Th17) [20], while inhibiting Treg production [21]. CTLA-4 molecules are expressed on Tregs and activated T cells. As the immune response reaches its end, they are upregulated on the T cell surface, occupying B7 with higher affinity than CD28, and thus blocking the activation signal. Thus, B7 plays a role in launching and subsiding T cell response.
CD40
CD40, a member of the TNF receptor family, is constitutively expressed by mature DCs and other APCs and is upregulated during inflammation [22–24]. It interacts with CD40L (CD145) on the surface of activated T cells to allow T cell priming and differentiation [23, 25].
On one hand, it enhances adhesion, MHC expression, and B7 upregulation in DCs. The stimulation of CD40 by CD40L results in DC licensing, meaning its capacity for Ag cross-presentation to CD8+ T cells [26]. On the other, it induces positive signaling in T cells, activating the nuclear factor kappa B (NF-kB) pathway, leading to the production of cytokines like IL-12 and IL-6 [27, 28].
As for its role in activating humoral adaptive immunity, signaling through CD40/CD40L induces B cell proliferation through activation of the germinal center and refines the B cell response through promoting immunoglobulin (Ig) class switch, somatic hypermutation, and production of memory cells [29]. All this provides a more specific, efficient, and long-lived Ag clearance. The fact that X-liked hyper-IgM syndrome is associated with faulty CD40L expression in T cells has established the importance of this signaling pathway in regulating T cell and B cell responses [30].
OX40L
OX40L is constitutively expressed in APCs, endothelial cells, and smooth muscle cells [31–33]. It is upregulated on APCs under the action of CD40 signaling, TLR signaling, and inflammatory mediators like IL-18, interferon (IFN)-γ, TNF-α, and prostaglandin E2 (PGE2) [32, 34–38].
OX40L signaling is essential for T cell activation, survival, function, and memory formation. It promotes Th2 differentiation in the absence of IL-12 [39, 40], and Th1 polarization in its presence [41].
It is worth mentioning that OX40 stimulation of Treg represses forkhead box protein 3 (Foxp3)+, thereby leading to reduced Treg function [42, 43]. Thus, blocking OX40/OX40L signaling can modulate peripheral tolerance and serve as a handy tool in treating immune-mediated diseases like asthma, arteriosclerosis, diabetes, colitis, etc. [44, 45].
Inducible co-stimulator ligand
The inducible co-stimulator (ICOS or CD278) is a CD28 homolog, expressed by TCR-activated CD4+ T cells [46]. ICOS ligand [ICOSL, B7-homolog 2 (H2)] is expressed on APCs, upregulated in inflammation, and doesn’t bind CD28 or CTLA-4 [47].
ICOS/ICOSL pathway regulates T cell growth, proliferation, survival, and polarization by initiating intracellular cascades that prevent apoptosis and induce immunomodulatory cytokines [48–50].
On the other hand, several studies have revealed a potential role of ICOS in IL-10 and Treg induction [51–55]. ICOS serves a major role in immune tolerance as immature DCs are rich in ICOSL which outweighs their weak CD28 stimulation. As a result, activated T cell sensitivity to IL-10 produced by DCs will increase, resulting in immunosuppression and directing T cells toward the Treg pool [56]. This process is prevented by potent CD28 signaling during disease [21, 51].
Programmed cell death ligand 1
Programmed cell death 1 (PD-1) is a membrane protein of activated T cells. It possesses two ligands, programmed cell death ligand 1 (PD-L1) and PD-L2. PD-L1 is constitutively expressed on resting DC and further enhanced by pathogen-associated molecular pattern molecules or CD40-mediated signaling; it is the main ligand for PD-1 [57]. PD-1/PD-L1 interaction serves as a co-inhibitory signal that attenuates the immune response to prevent further tissue damage unless this effect is masked by a potent enough co-stimulating milieu.
Through PD-L1, tolerogenic DCs (tolDCs) can shut down self-reactive T cells, promoting peripheral tolerance [58, 59]. However, during ongoing immune processes, CD28 co-stimulation potently overcomes PD-1-mediated inhibitory effects [60].
During hematologic malignancies, such as leukemia and lymphoma, cancer cells display PD-1 abundantly to escape elimination by the immune system and ultimately lead to immunosuppression and effector T cell deficiency [61]. Clinical trials targeting the inhibition of PD-1 and PD-L1 by monoclonal Abs or small interfering RNA (siRNA) revealed promising results in terms of tumor growth regression and patient stability [62, 63]. On the other hand, many autoimmune diseases, such as T1D, multiple sclerosis, SLE, and RA, were associated with dysregulated PD-1 expression [64, 65]. Thus, targeting PD-1 could be of potential therapeutic benefit in such diseases.
CD137
CD137 (4-1BB) belongs to the TNF receptor superfamily and is expressed primarily by activated T cells within 24 hours of stimulation, peaking at days 2–3 [66]. Its ligand, CD137L (4-1BBL), is a member of the TNF superfamily and can be expressed by APCs.
CD137/CD137L engagement comprises a forward CD137 signal, which results in T cell co-stimulation, and a reverse CD137L signal, which activates APCs.
In T cells, it activates NF-kB signaling necessary for proliferation, potentiation, and memory [67], with a predilection towards CD8+ T cell expansion and Th1 polarization [68, 69].
In APCs, this stimulus induces TNF-α, IL-8, IL-12, and IL-6 and inhibits IL-10 release. It aids in DCs differentiation and cytokine release and promotes B cell proliferation and survival, but not class switching [70, 71].
In conclusion, the expression of co-stimulatory molecules on DC is essentially involved in controlling T cell differentiation and the resulting response of defense versus tolerance. No single co-stimulatory molecule pathway defines this outcome, but it is rather the integration of them all, interaction with soluble co-factors, and the differentiation state of interacting T cells that dictate the immune response.
DC abnormalities in autoimmune diseases
In the normal physiological steady state and in the absence of danger or pathogen signals, DCs are naturally tolerogenic. This means that they do not activate T cells to mount an immune response. Instead, they promote the development of Tregs. Naturally, tolDCs have low MHC and co-stimulatory molecules on their surface [16]. They also produce IL-10 and TGF-β, which are well-known for tolerance induction and immunomodulation [16]. All these properties, among other complex intricate interactions between DCs and T cells, lead to self-tolerance. On the other hand, the presence of proinflammatory molecules that get upregulated chronically leads to an imbalance in the DC profile, which in turn promotes the mounting of an immune response to self-Ags [72]. DC-T cell interactions through surface molecules, as well as secreted cytokines, play an important role in achieving tolerance versus disease [73]. It thus comes with no surprise that these cellular interactions are extremely targeted in therapies aiming to treat autoimmune diseases.
DC-based therapies
Many new potential therapies for autoimmune diseases are based on DCs (Table 1). Using this approach, DCs are cultured ex vivo and manipulated in various ways to generate DCs that have tolerogenic properties. These DCs are then administered to the patients with the goal of reaching T cell anergy or Treg differentiation. TolDCs are characterized by low presentation of co-stimulatory molecules (CD80/CD86), high production of the anti-inflammatory cytokine, IL-10, and low secretion of pro-inflammatory cytokine [74].
Summary of clinical trials using therapies based on DCs
| Method used for tolDCs generation | Clinical trial phase | Target disease | Results | References |
|---|---|---|---|---|
| ATDCs treated with Dexa | Phase 1 | RA | Synovitis decreased Safe and well-tolerated | [78] |
| Semi-mature DCs treated with Dexa and vitA | Phase 1 | Crohn’s disease | Clinical improvement in 1/3 of patients Well-tolerate | [79] |
| ATDCs treated with vitD3 and pulsed with proinsulin peptide (C19-A3) | Phase 1 | T1D | Safe No systemic immunosuppression | [81] |
| DCs treated with Dexa and vitD3 and pulsed with auto-Ags collected from inflamed synovial joints | Phase 1 | RA | Clinical improvement No side effects No systemic immunomodulatory effect | [82] |
| Semi-mature ATDCs pulsed with 4 Ags related to RA | Phase 1 | RA | Good to moderate EULAR score Minimal side effects | [84] |
| DCs generated in the presence of NF-kB inhibitor and pulsed with RA Ags | Phase 1 | RA | Decreased DAS-28 score | [85] |
This table presents the various clinical trials using tolDCs to target autoimmune and inflammatory diseases. ATDCs: autologous tolDCs; EULAR: European Alliance of Associations for Rheumatology; DAS-28: disease activity score 28; Dexa: dexamethasone; vitA: vitamin A
There are several techniques to generate this immature tolerogenic profile of DCs. In most cases, monocytes are first extracted from the patients, transformed into DCs by using granulocyte-macrophage colony-stimulating factor (GM-CSF) and IL-4, and then modulated in a certain way for them to be tolerogenic [75]. Moreover, using low concentrations of GM-CSF by itself was proven to generate ATDCs [76]. The generated DCs may be pulsed with the auto-Ag to be targeted in order to produce a specific tolerogenic response for this particular Ag, or they may be injected in specific regions of the body where the disease mainly manifests.
A widely used technique for generating tolDCs is based on Dexa addition during DC differentiation from monocytes as it prevents the formation of fully mature DC [77]. This approach has been used in many clinical trials for RA, inflammatory bowel disease (IBD), Crohn’s disease, and multiple sclerosis. For RA, in a phase 1 clinical trial, ATDCs treated with Dexa were injected intra-articularly in the knees. After 3 months, synovitis in the knee joint has decreased, and this treatment proved to be safe and well-tolerated [78]. Furthermore, in a phase 1 clinical trial for Crohn’s disease, semi-mature DCs generated by adding IL-1β, IL-6, TNF-α, and PGE2, in addition to Dexa and vitA, were administered intraperitoneally. A clinical improvement was observed in one-third of patients, and the treatment was also generally well-tolerated. However, a significant increase in Tregs was observed in the circulation after 12 weeks with T cell hypo-responsiveness to polyclonal activation [79].
Moreover, 1,25-dihydroxyvitD3 [1,25(OH)2D3], the active form of VitD3, generates tolDCs when added to DCs in the differentiation phase by causing low Ag presentation, low co-stimulatory molecule presentation, and inhibition of IL-12 [80]. Thus, it comes as no surprise that this molecule has been implemented in clinical trials. In T1D, a phase 1 clinical trial using intradermal vitD3-treated ATDCs, pulsed with proinsulin peptide C19-A3, proved to be safe without producing systemic immunosuppression [81]. An interesting study used DC treated with Dexa applied on day 3 of DC differentiation and both VitD3 and Dexa applied on day 6 [82]. These DCs were then stimulated with monophosphoryl lipid A (MPLA), to become semi-mature, and pulsed with autologous Ags collected from the synovial fluid of inflamed joints. After intra-articular injection of the resulting tolDCs, patients who received the highest dose had improved clinical symptoms without side effects or systemic immunomodulatory consequences [82]. VitD3-treated DCs are also currently being studied for multiple sclerosis treatment [83].
Other than the previously mentioned studies about RA, there are two more worth mentioning. CreaVax-RA consists of autologous semi-mature DCs pulsed with 4 Ags related to RA [recombinant peptidylarginine deiminase 4 (PAD4), RA33, citrullinated-filaggrin, vimentin]. When CreaVax-RA is administered repetitively as a subcutaneous injection, a significant decrease in auto-Abs and IFN-γ-secreting T cells was noted, in addition to a good-to-moderate EULAR response. These clinical responses were achieved with minimal side effects [84]. Rheumavax, which is similar to CreaVax-RA in regard to pulsing with putative RA Ags and intradermal injection, uses however DCs generated in the presence of NF-kB inhibitor (BAY 11-7082). After one month from the injection, Rheumavax decreased DAS-28, with a reduction in pathogenic T cells and inflammatory chemokines secretion [85].
More techniques are also implemented, such as using DC treated with antisense oligonucleotides targeting co-stimulatory molecules (CD40, CD80, and CD86) decreasing their expression, or with rapamycin, which is an antibiotic with immunosuppressant properties [86, 87]. Furthermore, adding IL-10 during DC differentiation and maturation would result in a tolerogenic phenotype [88, 89]. In addition, experimental work in mouse models proposed that DC expressing Fas ligand would cause apoptosis of active T cells that express Fas, and thus, when these DC were injected in mouse models of collagen-induced arthritis (CIA), they resulted in disease amelioration [90]. In another murine model of T1D, autoimmune regulator (AIRE)-overexpressing DCs that express T1D Ags induced Ag-specific immune tolerance with lower blood glucose levels and less pancreatic inflammation. AIRE expression seems to downregulate the expression of co-stimulatory molecules (CD40, CD80/86) which would therefore cause tolerance to auto-Ags specifically [91].
In vivo manipulation of DCs
As generating DCs ex vivo, making them tolerogenic, and injecting them into the patient is quite complex and expensive, researchers thought of manipulating DCs in vivo by using nano-medicines and Ag-delivering Abs. These approaches, however, are quite new with limited translation into clinical studies.
Nanoparticles can be engineered to deliver certain drugs to the inside of a target cell-based on the nanoparticles’ size and chemical interactions [92]. This technique has been exploited with the aim of treating autoimmune diseases by developing nanoparticles that would target DCs. In pre-diabetic mice, T1D onset was prevented in 40% of mice treated with nanoparticles containing insulin B, GM-CSF, TGF-β1, and VitD3, which have been stated above to be tolerogenic [93]. This outcome was achieved because DCs would take up the potential auto-Ag, along with the tolerogenic molecules [93]. Thus the Ag would be presented in a tolerogenic environment, and cause T cell tolerance with an increase in Tregs [93]. In another approach, nanoparticles containing antisense oligonucleotides, targeting CD40, CD80, and CD86 co-stimulatory molecules, injected in an area that is drained by pancreatic lymph nodes, caused a reversal of diabetes in new-onset non-obese diabetic (NOD) mice [94]. The concept in this study is that even if the auto-Ag is not present within the nanoparticle, injecting nanoparticles with tolerogenic molecules into an area where DCs would naturally present the auto-Ag would also yield the same result [94].
Furthermore, mice with experimental autoimmune encephalomyelitis (EAE), which are models of multiple sclerosis, were also used in preclinical studies for nanoparticle therapy. Similar to the previously mentioned experiment with prediabetic mice, nanoparticles encapsulating the auto-Ag of EAE, proteolipid protein, with a tolerogenic agent, rapamycin, prevented EAE relapse [95]. IL-10 was also used as a tolerogenic molecule in nanoparticles for EAE [96]. Nanoparticles encapsulating the target Ag only, such as in the case of TAK-101 nanoparticles that encapsulate gliadin, also show promising results. In a phase 2 clinical trial, intravenous administration of TAK-101 nanoparticles in celiac disease patients on a gluten-free diet was associated with reduced gluten challenge-induced intestinal damage [97]. When TAK-101 nanoparticles deliver gliadin into the inside of DCs, this Ag will be presented in a tolerogenic environment as no inflammation will be provoked through this delivery mechanism [98].
Other than nanoparticles, Ag-delivering Abs also prove to be an efficient way of delivering molecules into target cells [99]. In theory, when Ags of interest are conjugated to Abs that would bind the target cell, these Ags would get internalized into this cell [99].
For example, naturally pro-tolDCs in the human body express DEC-205; thus, delivering anti-DEC-205 Abs conjugated to auto-Ags of interest would make DEC-205+ DCs present this Ag and promote tolerance. Anti-DEC-205 chimeric Abs fused with proteolipid protein helped reduce the severity of EAE in mouse models by inducing anergy in effector T cells, thus decreasing the number of pathogenic T cells in the CNS [100]. Several other studies have been carried out in a similar fashion for NOD, proteoglycan-induced arthritis, and experimental colitis mouse models for T1D, RA, and IBD respectively [101].
Co-stimulatory molecules-targeted therapies
Co-stimulatory molecules present on DCs and on other immune cells are being targeted by drugs for the aim of treating autoimmune diseases. This approach is not only used for autoimmune diseases as the new immune checkpoint inhibitors targeting these molecules showed robust results in cancer therapy as well [102]. Therapies developed for autoimmune diseases mainly target CD80/86 complex with CD28, CTLA-4, CD40, and CD40L, and some other less known molecules such as ICOS, CD137, OX40, etc.
CD80/86, CD28/CTLA-4
CD80/86 is an important co-stimulatory molecule, as aforementioned, since it provides “signal II” needed for launching an immune response. Without this signal, tolerance and anergy would rather develop; hence, it becomes clear why many therapies focus on blocking this molecule (Table 2 and Figure 2). Abatacept (CTLA-4-Ig), an Food and Drug Administration (FDA)-approved drug for the treatment of RA and psoriatic arthritis, is basically a fusion protein holding the extracellular portion of CTLA-4 fused with the modified fragment crystallizable (Fc) portion of human IgG1. CTLA-4 binds CD80/86 and blocks its interaction with CD28, mimicking a tolerogenic state in which CD80/86 molecules are less expressed. This drug proved to be efficacious and relatively safe in the long term [103]. The downsides to this medication are that it is costly and that it may predispose to infections as it targets all CD80/86 molecules and not only those related to the disease [103, 104]. A very similar drug is belatacept, which binds approximately 4-fold more strongly to CD86 and 2-fold more strongly to CD80 than abatacept [105]. Belatacept is FDA-approved for post-renal transplant immunosuppression as it does not cause the nephrotoxicity associated with calcineurin inhibitors, namely cyclosporine. Moreover, treatment with belatacept showed higher patient and graft survival after 7 years compared to cyclosporine. However, this treatment was associated with more early acute rejection [106]. Two additional fusion proteins XPro952348 and MEDI5256 bind CD80/86 molecules even more avidly than the previously mentioned drugs, with MEDI5256 having a preferentially way more stronger binding to CD80. These molecules also have a longer half-life due to changes in the Fc region of the fusion protein. Both newer drugs proved to be more efficacious and more potent than abatacept and belatacept in suppressing humoral responses following keyhole limpet hemocyanin (KLH) immunization [107, 108].
Summary of Abs targeting CD80/86 interaction with CD28
| Ab | Properties | Clinical application | Desired effects | Downsides | References |
|---|---|---|---|---|---|
| Abatacept | Fusion protein of the extracellular domain of CTLA-4 is linked to modified Fc portion | FDA-approved for: RA Psoriatic arthritis | Blocks CD80/86 interaction with CD28 Efficacious and relatively safe | Predisposes to infections Costly | [103, 104] |
| Belatacept | Similar to abatacept in structure 4-fold higher affinity to CD86 2-fold higher affinity to CD80 | FDA-approved for: Immunosuppression after renal transplant | Higher graft survival compared to cyclosporine Does not cause nephrotoxicity associated with cyclosporine | Higher early acute rejection | [105, 106] |
| XPro952349 & MEDI5256 | Similar to abatacept in structure Higher avidity to CD80/86 than abatacept and belatacept | - | Decreased humoral response to KLH immunization | - | [107, 108] |
| TAB08 | Agonistic anti-CD28 Ab | Phase 2 clinical trial for RA | At low doses, minimal activation of CD28 preferentially expands Tregs and increases serum IL-10 | Causes cytokine release syndrome if administered with a higher dose | [109–111] |
| FR104 | Pegylated Fab with antagonistic CD28 activity | - | Decreased humoral response to KLH immunization in healthy human volunteers Improved GVHD-free survival in primate models | Increased infectious complications | [112, 113] |
This table shows the various molecules generated to target co-stimulatory molecules (CD80/86) interaction with CD28. Inhibiting this interaction leads to T cell anergy and an inhibited immune response. -: no data. Fab: fragment Ag-binding; GVHD: graft-versus-host disease; TAB08: theralizumab
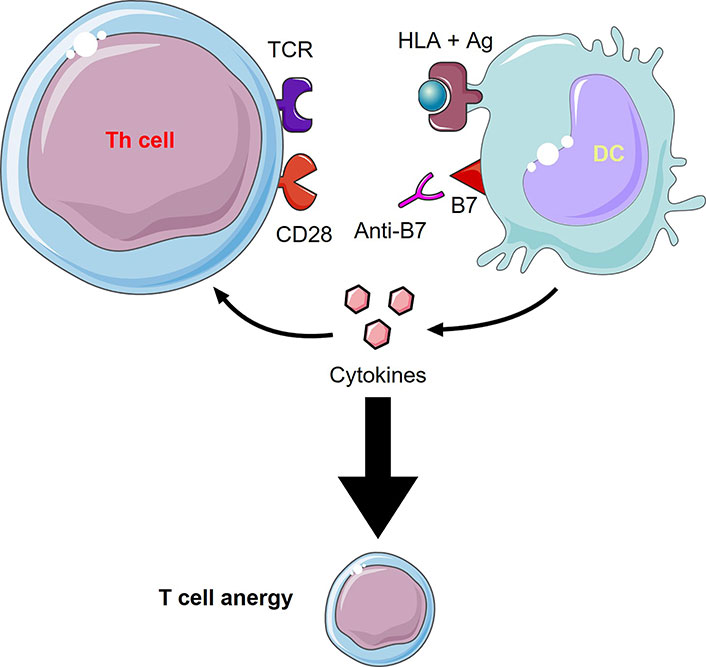
Anti-B7 molecules mechanism of action. Inhibiting the interaction between the co-stimulatory molecule, B7, with CD28 through an anti-B7 molecule (e.g., abatacept, belatacept) leads to T cell anergy and an inhibited immune response. This figure was created by Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported License
Other than CTLA-4-Ig, agonistic anti-CD28 Ab (TAB08) serves an intriguing finding, where at low doses of this Ab, minimal activation of CD28 would preferentially expand Tregs and thus help with tolerance [109]. However, careful dose titration is required since a previous trial of the same Ab but with a higher dose resulted in cytokine release syndrome [110]. With the appropriate dosing though, TAB08 increases serum IL-10 without causing cytokine storm. This Ab has entered phase 2 clinical trials in RA as it showed a clinical response with an acceptable level of adverse effects [111]. On the other hand, antagonistic anti-CD28 Ab also has a role in treating autoimmune diseases. FR104, a pegylated Fab with antagonistic CD28 properties, hampered humoral response to KLH challenge in human healthy volunteers [112]. Moreover, this Ab served better than CTLA-4-Ig in improving GVHD-free survival in non-human primate models, yet it has infection-related complications [113].
CD40, CD40L
CD40 and its ligand also play a significant role in boosting the immune response, and thus they provide a good therapy target (Table 3). Dapirolizumab pegol (CDP7657) is a Fab that antagonizes CD40L [114]. This Ab showed an improvement in clinical outcomes of SLE and was generally well-tolerated [115]. However, in a phase 2b clinical trial, this medication failed to reach the needed primary endpoint of dose-response [116]. The use of the Fab only, instead of the whole Ab, was because a previous humanized Ab with an Fc segment, Ruplizumab, caused thromboembolic events due to platelets aggregation dependent on Fc interactions [117]. Ruplizumab clinical evaluation was thus stopped even though it had an interesting clinical response in lupus nephritis by reducing hematuria in all involved patients [118]. Another important CD40L targeting molecule for RA reduced disease activity and was shown to be safe without serious adverse events [119]. This molecule is called VIB4920 and is composed of 2 Tn3 proteins, which are protein scaffolds with Ig-like folds engineered to bind CD40L [120].
Summary of Abs targeting CD40/CD40L interaction
| Ab | Properties | Clinical trials* | Results | References |
|---|---|---|---|---|
| Dapirolizumab pegol (CDP7657) | Anti-CD40L Fab | Phase 2 trial for SLE | Improved clinical outcomes of the disease Well-tolerated Did not reach the primary endpoint of dose-response in phase 2b | [114–116] |
| VIB4920 | CD40L binding protein composed of 2 Tn3 proteins fused to serum albumin | Phase 1b for RA | Decreased disease activity Safe without serious adverse effects | [119, 120] |
| Iscalimab (CFZ533) | Fully human antagonistic anti-CD40 monoclonal IgG1 Abs | Phase 2 for: Kidney transplant-SS | Safer than tacrolimus in regards to nephrotoxicity and infectious complications but less efficacious in preventing transplant rejection Efficacious for SS | [121–124] |
| BI-655064 | Humanized antagonistic anti-CD40 monoclonal IgG1 Ab | Phase 2 for RA | Good add-on to methotrexate Improved clinical outcomes Reduced RF auto-Ab titers | [126] |
This table presents information on the different molecules made to inhibit the CD40/CD40L interaction. Inhibiting this interaction greatly affects the immune response and thus it has been used in clinical trials against autoimmune diseases and transplant rejection. *: this list is not comprehensive. SS: Sjogren’s syndrome; RF: rheumatoid factor
CD40L binds molecules other than CD40; thus, blocking CD40L would disrupt all of its other possible interactions. Therapies targeting CD40 instead of CD40L were therefore developed. Iscalimab (CFZ533) is an anti-CD40 IgG1 Ab with a modified Fc portion not capable of performing effector functions [121]. This Ab was tested in renal transplant and SS, among others. Although it was shown to be safer than tacrolimus regarding nephrotoxicity and infectious complications, it was less efficacious in preventing transplant rejection [122, 123]. For SS, however, this biologic drug proved efficacy in a phase 2 clinical trial [124]. Some patients with certain CD40 variants may respond better to this drug, especially those having increased CD40 expression before receiving the treatment [125]. This opens up the possibility of applying precision medicine with this new drug. A similar drug called BI-655064 was tested on RA, and it turned out to be a good add-on to methotrexate, with better clinical results and lower RF auto-Ab titers compared to placebo + methotrexate [126].
ICOS, ICOSL
ICOS signaling is important for Th-dependent Ab production; thus, blocking this molecule would reduce the production of more refined Abs with isotype switching. In the phase 1 clinical trial, the human α-ICOSL Ab, AMG 557, diminished Ig class switching in patients with SLE challenged with KLH nasally [127].
CD137, CD137L
CD137, another co-stimulatory molecule found on T cells, is important for Th1 polarization. Reverse CD137L signaling in the APC enhances its inflammatory function [69]. Human clinical trials targeting this molecule are lacking; however, studies made on mice showed interesting results [128]. Agonistic CD137 Ab can prevent CD137L reverse signaling to the APC, suggesting some immunoregulatory effects, while still enhancing the Th1 response. Both effects help reduce Th2 autoimmune responses since Th1 cells inhibit Th2 cells and APCs would have a more regulatory profile [128, 129]. Antagonistic CD137 Ab, however, would be of benefit to autoimmune diseases mediated by Th1 [69, 128]. Thus both agonistic and antagonistic Abs may be potential treatments for autoimmune diseases based on their Th cell profile.
OX40, OX40L
OX 40 and OX40L are co-stimulatory molecules found in an inflammatory environment on activated T cells and DCs, respectively [130, 131]. Blocking this molecule with anti-OX40 Ab (GBR 830) was tested among atopic dermatitis patients in a phase 2 clinical trial. It showed promising results with significant improvement in clinical manifestations of the disease as well as reduced numbers of OX40+ T cells and OX40L+ DCs in the affected skin regions [132].
PD-1, PD-L1
The famous PD-1 molecule that is blocked in cancer therapy can be activated for the treatment of autoimmune diseases. The use of the fusion protein PD-L1-Fc would bind PD-1 and activate it. This molecule decreased disease activity in mouse models of RA and IBD [133, 134]. Phase 1 clinical trials using PD-1 agonist Ab (CC-90006) for the treatment of psoriasis are completed but official results are still pending [135]. In addition, interesting new techniques are currently being employed to promote PD-1 activity [136]. Immune Mobilising Monoclonal TCRs Against Autoimmunity (IMMTAAI) are composed of an agonistic anti-PD-1 coupled to a specific monoclonal TCR molecule. This complex would activate PD-1 only in sites where DCs present the specific target peptide on their MHC that binds to this monoclonal TCR. Therefore, this therapy would result in localized immune suppression [136]. It’s not unlikely that this technique may be used for other purposes also in the near future.
Genetic engineering of DCs
In addition to the previously mentioned approaches, gene therapy has also gained popularity in generating tolDCs. There are many techniques involved in gene therapy, such as using viruses and liposomes for gene transfer or using RNA interference (RNAi) to silence particular genes.
DCs producing high levels of IL-10 have been genetically engineered using a lentiviral vector encoding the IL-10 gene. These cells, called DC-IL-10, have decreased allogeneic T cell responses in humanized mouse models and seem promising for use in clinical practice in the context of allogeneic transplantation [137]. This technique has also been exploited in generating Ag-specific tolerance in T cell-mediated diseases. Transferring immunodominant Ag-derived peptides along with the IL-10 gene to DC using lentiviral vectors helps in producing Ag-specific Treg [138]. These DC-IL-10/Ag cells prevented the development of T1D in preclinical disease models.
Other than IL-10, researchers have also tried to transfer other genes to DCs. Since NF-kB is very important for DC maturation, a gene coding for kinase-defective dominant negative form of IKK2 (dnIKK2), which blocks NF-kB activation, was transferred to DCs using an adenoviral vector. This way DCs will not be able to mature properly and would produce a regulatory profile of T cells. These produced Tregs also inhibit naive T cell responses to antigenic stimuli with mature DCs [139]. Moreover, IL-4 coding adenoviral vectors were used in the transduction of DCs producing DC/soluble IL-4 (sIL-4). These genetically modified DCs alter the immune response to the Th2 profile rather than Th1 and were proven to reduce the number of prediabetic NOD mice that developed diabetes [140].
Furthermore, RNAi is a widely used genetic engineering method for DCs. As previously mentioned, antisense oligonucleotides targeting CD40, CD80, and CD86 molecules produce tolDCs [86, 94]. In addition, silencing IL-12 expression with siRNA stimulates the production of Th2 cytokines, as well as IL-10. This therapy would help reduce Th1 dominant alloimmune or autoimmune responses [141]. Finally, immunoliposomes are also used in genetic engineering as non-viral vectors. For example, immunoliposomes can deliver to DCs a gene encoding for CTLA-4 protein fused to an endoplasmic reticulum retention signal. This fusion protein prevents CD80/86 molecules from reaching the cell surface by keeping them in the endoplasmic reticulum, thus promoting tolerance [142].
Conclusions
As previously mentioned, novel therapies with advanced strategies are being implemented for autoimmune diseases. These therapies are being improved based on intensive research studies ranging from preclinical and experimental works to clinical trials. Each therapy is designed to be efficient in inhibiting or reversing a specific disease process, thus targeting one single disease or a group of diseases with a similar pathophysiology. Moreover, the most appropriate dosing, routes of administration, and other pharmacokinetic properties are being established to have the best clinical results possible. These therapies, however, need time to be implemented into clinical practice as they are still new and need to be tested thoroughly before they get approved for the market. Nonetheless, it’s expected that in the near future, the management of autoimmune disease will evolve, and these molecular and cell-based therapies will be a major milestone in immunotherapy.
Furthermore, the novel immunotherapy agents match the modern shift in medicine towards precision and personalized treatments. As everyone has a specific set of genetic characteristics, the disease process in each patient will be slightly different from that in others. Thus, therapy should be tailored to the specific needs of each patient. Certain genetic mutations and pathological markers can be evaluated, and appropriate therapy will be administered. Novel immunotherapy can achieve this and oppose the disease process in each individual patient.
Many other treatment modalities not mentioned in this review are also being studied, such as mesenchymal stem/stromal cell-based therapies among others [10]. The hope is that autoimmune diseases and similar disease processes, such as allergies and transplant rejection, can be cured or prevented without the need for long term immunosuppressive therapy with devastating side effects.
Abbreviations
| Ab: | antibody |
| Ag: | antigen |
| APCs: | antigen-presenting cells |
| ATDCs: | autologous tolerogenic dendritic cells |
| CD4: | cluster of differentiation 4 |
| CD40L: | cluster of differentiation 40 ligand |
| CTLA-4: | cytotoxic T lymphocyte-associated antigen 4 |
| CTLs: | cytotoxic T lymphocytes |
| DCs: | dendritic cells |
| Dexa: | dexamethasone |
| EAE: | experimental autoimmune encephalomyelitis |
| Fab: | fragment antigen-binding |
| Fc: | fragment crystallizable |
| FDA: | Food and Drug Administration |
| GM-CSF: | granulocyte-macrophage colony-stimulating factor |
| IBD: | inflammatory bowel disease |
| ICOS: | inducible co-stimulator |
| ICOSL: | inducible co-stimulator ligand |
| Ig: | immunoglobulin |
| IL-10: | interleukin-10 |
| KLH: | keyhole limpet hemocyanin |
| MHC: | major histocompatibility complex |
| MHCI: | major histocompatibility complex class I |
| NF-kB: | nuclear factor kappa B |
| NOD: | non-obese diabetic |
| PD-1: | programmed cell death 1 |
| PD-L1: | programmed cell death ligand 1 |
| RA: | rheumatoid arthritis |
| SLE: | systemic lupus erythematosus |
| SS: | Sjogren’s syndrome |
| T1D: | type 1 diabetes |
| TAB08: | theralizumab |
| TCRs: | T cell receptors |
| TGF-β: | transforming growth factor-β |
| Th: | helper T |
| TNF: | tumor necrosis factor |
| tolDCs: | tolerogenic dendritic cells |
| Tregs: | regulatory T cells |
| vitA: | vitamin A |
Declarations
Author contributions
MEJ: Conceptualization, Project administration, Supervision, Writing—original draft. FS: Conceptualization, Writing—original draft, Writing—review & editing.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
Not applicable.
Copyright
© The Author(s) 2024.