 Review
Review

Affiliation:
1Independent researcher, Biochem123 Education, NW7 4AU London, UK
Email: abrownbscmsc@gmail.com
ORCID: http://orcid.org/0000-0001-5238-6943
Affiliation:
1Independent researcher, Biochem123 Education, NW7 4AU London, UK
ORCID: http://orcid.org/0000-0002-8200-0885
Affiliation:
2Think Vaccines LLC, Houston, TX 77005, USA
ORCID: http://orcid.org/0000-0003-2466-4920
Affiliation:
3Independent researcher, 31195 Lamspringe, Germany
ORCID: http://orcid.org/0000-0001-7638-3181
Explor Immunol. 2024;4:238–266 DOI: https://doi.org/10.37349/ei.2024.00139
Received: June 17, 2023 Accepted: January 15, 2024 Published: April 25, 2024
Academic Editor: Dominique J. Charron, Hospital Saint Louis, France
The article belongs to the special issue Old and New Paradigms in Viral Vaccinology
Ebola virus (EBOV) is a zoonotic virus comprising of six known different species, designated within the family Filoviridae and genus Ebolavirus. The first recorded outbreak of an EBOV disease (EVD) was in Yambuku, Zaire EBOV (ZEBOV) in 1976, followed by the Sudan EBOV (SUDV) later that year. Outbreaks have been increasing throughout the 21st century, and mortality rates can reach up to 90%. Such extraordinary virulence is evidenced by a few pathogens, similar to the Marburg virus (MARV) that originated in Uganda and was first detected in Germany in 1967. The virulent nature of filovirus disease has established these related viruses as a formidable global concern. There are currently four types of Ebolaviridae species known to infect humans, with two more recently identified in other animals that are genomically different concerning cellular pathogenesis or aetiology of disease. Recent advances in understanding the pathogenesis of filovirus disease infections have been remarkable, yet the immunological response to filovirus infection remains unknown. Scientific analysis of cellular mechanisms can provide insight into virulence factors utilised by other pathogenic viruses that also cause febrile illness with occasional haemorrhagic fever in humans. In this review, a brief summary of EBOV protein structure and functional cellular effects is covered. The role of innate and adaptive immune cells known since 1976 is considered with the relevance and implications of immunological proteins measured by cluster of differentiation (CD) molecule, alongside cytokine, chemokine, and other biologically relevant pathways, and through genetic research. A thorough understanding of immunological correlates affecting host responses to EBOV will facilitate clinical and applied research knowledge, contributing to protection against potential public health threats.
Ebola virus (EBOV) is the causal virion of a severe viral-induced pathology and disease affecting human hosts through cellular infection and across specific tissues, via transmission through host cell receptors or membranes and intracellular propagation. Total cases of EBOV disease (EVD) have occurred with more than 34,626 infections since the virus was first isolated and characterised [1, 2]. Past outbreak mortality was at least 15,266 individuals, which is indicative of an infection fatality rate (IFR) of around 50% (range: 48–74%), and a reproduction number (R0) was estimated (range: 1.51–2.53) during 2013–2014 [1, 2]. Consideration of the global incidence of disease or pathology of any description in all age groups is required to clarify research undertaken so far [3]. Despite developments occurring before and during the tragic outbreaks in the Republic of Guinea and surrounding areas before and during 2013–2023, much remains unclear. Transmission of zoonotic viruses to humans can occur from animal species [4]. In 2017, the International Code of Virus Classification and Nomenclature, ratified by the International Committee on Taxonomy of Viruses (ICTV), was published with updates (see Supplementary materials). Subsequently, another species of the Ebolavirus genus, named Bombali virus (BOMV), was defined in 2018 and observed so far without observed pathogenicity in humans at the date of writing (see Figure 1) [5].
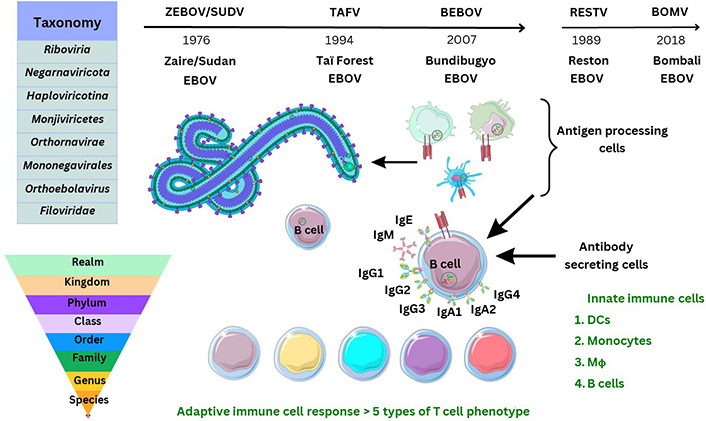
Historical discovery and taxonomy of Ebolaviridae. TAFV: Taï Forest EBOV; BEBOV: Bundibugyo EBOV; RESTV: Reston EBOV; IgG1: immunoglobulin G1; DCs: dendritic cells; Mϕ: macrophages. The Figure was partly created with Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported license. Virus schematic was adapted from ViralZone, SIB Swiss Institute of Bioinformatics (https://viralzone.expasy.org/207), licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License
EBOV is categorised as a biosafety level 4 (BSL-4) pathogen, only researched in designated laboratories (see Supplementary materials). Diagnostics are discussed elsewhere in other articles, subject to criteria from the World Health Organization (WHO) protocol and criteria “ASSURED”, to meet the benchmark for usage as point of care (PoC) products to test for a pathogen [6, 7].
From the research that has been conducted, the EBOV virion particle size is estimated at a length of 1–2 μm, with a diameter in the region of 80–100 nm [8]. The EBOV virion is composed of a negative (–ve) sense single-stranded ribonucleic acid (RNA; ssRNA) genome, of around 18–19 kilobases (kb) in size encoding for at least seven predominant proteins [8]. Several enzymes are necessary for the cleavage of host or virion particles for transcription including polymerases, transferases, but also methyl-transferases [8]. The interaction between EBOV viral proteins (VPs) within a host cell is still being further clarified [8]. In general, this family of viruses utilise the host genome as a complementary strand to synthesise messenger RNA (mRNA) via the RNA-dependent RNA polymerase (RdRp) within a host cell to replicate. Viral, bacterial, and fungal pathogens utilise different mechanisms for adhesion, permeation, propagation, and replication within a host. Other (–ve) ssRNA viruses include the rabies lyssavirus (RABV), hantavirus, mumps virus (MuV), measles virus (MeV), and Rift Valley fever (RVF) phlebovirus which also infect human hosts.
In this article, the structure and function of EBOV virion particles are discussed in brief. Immune cells, known as white blood cells or leukocytes, are evoked by viral antigens and respond differently during different viral infections. These are defined by cluster of differentiation (CD) proteins as observed during EBOV infection, specifically as membrane-bound proteins expressed by specific leukocytes (see Supplementary materials). Some CD proteins can also be shed by immune cell membranes, as soluble serum proteins, released upon intracellular or extracellular pattern recognition receptor (PRR) stimulation by pathogenic antigens. Furthermore, the humoral antibody responses measured by serology, alongside the adaptive T cell response are summarised further.
The significance of this evaluation is that herein, current leukocyte behaviour upon EBOV infection is further documented based on prior research. The relevance and expression of CD molecules, within specific immune cell phenotypes is outlined. Relevant immune cell interactions measured by either soluble or membrane-bound CD molecules occur accompanied by cytokine secretion and chemokine changes during leukocyte development, maturation, and response to any infection. The leukocytes considered include those that usually process viral antigens, which are denoted as antigen-presenting cells (APCs), including DCs, monocytes, and Mϕ. Following this neutrophil, B cell and T cell phenotypes measured in serology and research are considered with natural killer (NK) cells throughout 21st century laboratory analysis and further development.
Therefore, this review explores complete knowledge thus far on the interplay between innate/adaptive immune system cells during EBOV infection. This includes autocrine/paracrine cellular synthesised cytokines including interleukins (ILs), interferons (IFNs), and chemokines; denoted by C-C or C-X-C with the receptor or ligand. CD molecules during and after EBOV infection are considered in this evaluation expressed by specific immune cell phenotypes, as outlined prior [8].
A comprehensive PubMed literature review encompassing the period from the discovery of the EBOV, in 1976, until December 31, 2023, using the search terms “Ebola”, “Filovirus”, “Cluster of Differentiation”, “Immunology”, “Innate”, and “Adaptive” or in various combinations was performed. Relevant articles to the manuscript title were included out of the 11,940 listed.
Other reviews summarise current knowledge of proteins known to have been sequenced from the EBOV genome in great depth [8]. The method of transmission of EBOV may be considered to be through bodily fluid and similarly discussed in other reports with sequencing ongoing [9]. At least seven known EBOV proteins are characterised between the 3’ to 5’ EBOV genomic (–ve) ssRNA strand. These include a helical nucleoprotein (NP), and glycoprotein (GP), together with four VPs, VP35, VP40, VP30, and VP24, and a polymerase enzyme protein required for replication [8]. During 1998, the structure of EBOV GP was further elucidated after prior outbreaks, with a second GP (GP2) appearing to form a trimer with GP1 more important for receptor binding/viral entry [8, 10]. This requires a comparatively conserved furin enzyme for protein cleavage, differing between Filoviridae, with soluble GP (sGP) traversing organelles including the Golgi apparatus and endoplasmic reticulum (ER) [8, 10]. Particle entry of EBOV occurs by forming a ribonucleoprotein (RNP) complex with VP35/VP30 interacting as a dimer, and then VP24 followed by the matrix protein, VP40 [8, 11]. Recently in 2022, reports suggest that VP35 forms a dimer with the polymerase enzyme protein and may underpin the transcription process of the NP, as well as replication thereafter [8, 11]. It is necessary to correlate the individual size, structure, and function of protein composition, as immune cell epitope recognition relies on antigen presentation of degraded viral peptides. The EBOV NP protein is estimated at 739 amino-acids (aa) in length, VP35 is 340 aa, VP40 is 326 aa, GP a total of 1,378 aa, VP30 288 aa, with VP24 251 aa, and the longer polymerase enzyme protein of length 2,212 aa at the 5’ end of the EBOV genome [8].
Characterisation of the (–ve) ssRNA EBOV GP occurred before 2009 and elucidated that GP1/GP2, together with a small sGP (ssGP) were required [8]. The ssGP is described as a di-sulphide linked homodimer and N-glycosylated protein, produced through transcriptional RNA editing that can occur of GP1/GP2 with the addition of seven uridine residues in GP1 and GP2 together with an adenine forming nucleotide [8, 12]. It was previously considered that three biological processes contribute to EBOV virion particle cellular entry. Two methods of virion particle entry were described before 2016, which were either clathrin-dependent and/or caveolin-dependent EBOV virion particle cellular uptake [13, 14]. Lately, between 2010 and 2011, this was clarified as remaining a grey area, although logical suggestions were that EBOV utilises micropinocytosis as the main process modifying actin cytoskeletal elements for cellular vesicular entry [13, 14]. Thereafter, virion transport occurs intracellularly where GP1 undergoes proteolysis by one of two cathepsins (B/L) eventually leading to GP2-dependent host/virion endosome entry [8]. Further detail is shown below (see Figure 2).
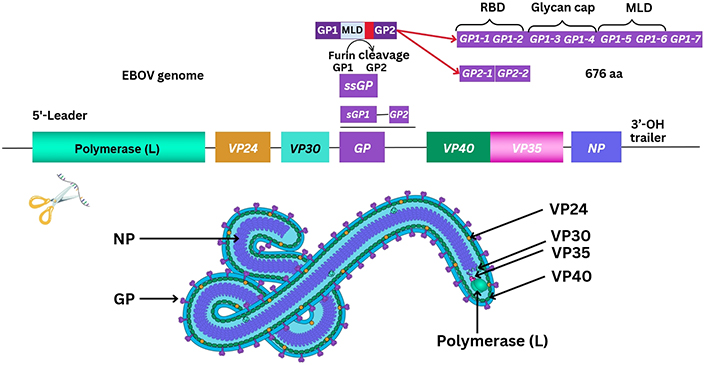
Graphical outline of the arrangement of the EBOV genome. RBD: receptor binding domain; OH: hydroxyl group; MLD: mucin-like domain
Note. Adapted from ViralZone, SIB Swiss Institute of Bioinformatics: (https://viralzone.expasy.org/207), licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License.
Therefore, cellular EBOV virion-like particles (VLPs) produce variable and complex functional proteins within host cells through transcription of viral genome elements, affecting the host homeostatic cellular compartment with much remaining unknown. For example, EBOV VP35 has aa residues considered to inactivate protein kinase R (PKR), whilst interacting with a protein activator of IFN-induced PKR (PACT) [8, 15]. Predominant functions of VP40 include those of maintaining virion particle structure with significance as a matrix protein; while VP30 role is that of transcription, activation as well as initiation [8]. In comparison, VP24 was first mentioned in literature in 1985, and recently seemingly interacts with the non-phosphorylated signal transducer activator of transcription 1 (STAT1), but also two types of karyopherin-α (KPNA; KPNA1/5) downstream reducing the nuclear accumulation of phosphorylated STAT1 [8]. Therefore, it can be considered that a key cellular pathway producing type I IFN (α/β) release is disrupted that trains the immune system response. Many authors concur that the mechanisms of viral EBOV cellular responses are unclear to date [16, 17]. Intriguingly, utilising co-immunoprecipitation (co-IP) assays in 2021, it was discovered that the EBOV VP24 protein may interact with emerin (encoded by the EMD gene), lamin A (LMNA), and LMNB [18]. It was highlighted and seen that each protein was required to stabilise nuclear membranes that are disrupted [18–20].
Prior articles investigating EBOV GP mutations, specifically within A82V, are relevant and may or may not enhance cellular virion particle entry in vitro of the EBOV GP [21]. Nevertheless, it was highlighted in vitro that an aa substitution where alanine was changed to valine, as both contain neutral non-polar groups and a methyl addition, that an increase in EBOV cellular infectivity was observed [21]. Moreover, a change between threonine to isoleucine at residue 544 of the GP increased virion particle kinetics during viral host plasma membrane (PM) fusion utilising the intracellular Niemann-Pick type C1 (NPC1) receptor within endosomes [21]. The crystallised protein structure of NPC1 was observed with clarity that a pocket exists within the cholesterol transport pathway where low-density lipoproteins (LDLs) are processed [22]. Thereafter, in vivo, NPC1 was detected in deficiency studies, where EBOV was non-contagious, with EBOV GP appearing to form a complex with the NPC1 luminal domain [22]. One enzyme, a serine cathepsin protease (cathepsin L), was considered to be more important in proteolytic cleavage, enhanced by the virion EBOV GP around or within the endosome [22]. To replicate, the RNA polymerase transcribes EBOV RNA from the 3’ trailer to the 5’ leader sequence intracellularly (see Figure 2). This occurs through capping and adenylation utilising the RNA polymerase protein, but also through host RNA editing to express full-length GP [23]. EBOV replication through endosomal pathways remains under research investigation [23]. The exact mechanism EBOV employs to form intracellular late endosomes remains unknown. Recently, VP40 has been ascribed to be crucial in membrane fusion during endosomal disassembly within cells [24].
Viral entry occurs by attachment to host cell membrane receptors or disruption of the cell/PM using either receptors, ion channels, adhesion mechanisms, or attachment factors for permeation. It is considered that the EBOV GP employs a C-type lectin, DC-specific intercellular adhesion molecule-3 (ICAM-3) grabbing non-integrin (DC-SIGN or CD209) receptor, expressed by DCs and Mϕ, as well as other host cells that facilitate virion particle entry [25]. The other corresponding receptor of DC-SIGN is CD50 (ICAM-3), a receptor constitutively expressed by leukocytes. Data on http://www.proteinatlas.org/ shows DC-SIGN is differentially expressed by host cells but at increased levels throughout the gastrointestinal (GI) tract, bladder, and heart muscle, as well as within the lymphoid organs that are the thymus, spleen, and lymph nodes. It is also present in smaller quantities within the brain (e.g., cortex, hypothalamus, etc.), but specifically within the choroid plexus. The current understanding of DC-SIGN evolved from 2000 research [25]. It was considered both DC-SIGN, together with a homologous protein, liver/lymph node-specific ICAM-3 grabbing non-integrin (L-SIGN, CD209L), have mannose-binding motifs, are calcium-dependent, and bind with high affinity to mannose N-glycans present in pathogens [25]. Between 2001 and 2003, it was implicated that this receptor played a role in Mycobacterium tuberculosis infection of DCs. Potentially DC-SIGN is utilised along with L-SIGN on T cells, forming an immunological synapse crucial for priming naive T cells (TN) [26, 27]. In addition, L-SIGN is suggested to have high avidity for EBOV GP with both binding to DC-SIGN [28].
In 2006, emerging in vitro research indicated a synthetic peptide that could affect (inhibit) two types of Filoviridae (Marburg virus; MARV as well as EBOV) through the activation of neutrophil cells [29]. Activation of neutrophils during EBOV infection is intertwined with the pro-inflammatory response. This occurs through triggering receptors expressed on myeloid cells-1 (TREM-1, known as CD354), regulating viral-induced increases in intracellular calcium as well as reducing the secretion of the cytokine tumour necrosis factor-α (TNF-α) [29, 30]. During an immune response, circulating neutrophils are central to both endothelial cell (EC) adhesion, and platelet aggregation, as well as the activation of other lymphocytes with the secretion of serine proteases (e.g., cathepsin G, elastase, etc.). Of note, these neutrophil cells are sensitive to IL-6 due to their high expression of the IL-6 receptor α (IL-6Rα, CD126) which may associate with gp130 (IL-6Rβ) receptor as a complex subunit domain during an inflammatory response [30–32]. It was indicated then that TREM-1 could be shed from the PM whilst increasing phosphorylation of decay-activating proteins (e.g., DNAX-activating protein-10; DAP-10/DAP-12), possessing a conserved immunoreceptor tyrosine-based activation motif (ITAM) [33, 34]. Since discovery, the DAP-12 protein has been described as a signalling adaptor molecule containing aspartic acid within the PM forming stable non-covalent homodimers that may interact with several NK cell receptors like NK2GC/E (CD94), but also CD158 and other immune cells [33]. Gene transcripts and proteins of DAP-12 have been described as being in abundance in plasmacytoid DCs (pDCs), monocytes, Mϕ, as well as NK cells [33]. Lesser amounts are described as present in known T cells expressing T cell receptors (TCRs), well known as CD4+ and CD8+ T lymphocytes, with comparatively less research on DAP-12 signalling in 2024 [33]. Signalling regulation occurs through stress-activated PKs (known as c-Jun N-terminal kinases; JNKs), but also spleen tyrosine kinase (Syk) in myeloid cells [33]. Further to this, a homologous zeta-chain-associated protein kinase 70 kDa (ZAP70) was identified as a crucial molecular co-partner of CD3 TCR signalling expressed during T cell maturation [35]. Syk is not expressed in T cells, but ZAP70 is expressed in NK cell and T lymphocyte cell populations with research ongoing [33, 35–37]. The importance of ZAP70, a cytoplasmic tyrosine kinase, can be considered, as ITAM tyrosine residues can be doubly phosphorylated; thereafter resulting in signalling through ZAP70 with high affinity through an Src homology 2 (-SH2) structure domain within the locality of the TCR/major histocompatibility complex (MHC) class II complex required for antigen/peptide dependent TCR signalling [33, 35–37].
Before 2011, reports emerged investigating other receptors further clarifying unknown mechanisms of EBOV cellular PM entry. The T cell Ig-mucin-domain 1 (TIM-1, CD365) was of consideration, in vitro, that may facilitate EBOV cellular infection in overall Filoviridae research [38]. Mucosal epithelial expression of TIM-1 was suggested to be a potential receptor for EBOV cellular infection and route of entry [38]. Recently, TIM-1 has been suggested to facilitate late viral virion particle egress [38]. In 2017, T cell studies investigating the immune response further clarified that TIM-1 is a relevant attachment factor for EBOV together with a further three attachment factors which are L-SIGN, folate receptor-α, and Tyro3 receptor tyrosine kinases [39, 40]. Conundrums remain, as during 2019, TIM-1 would appear in vivo not to affect overall mortality, although as discussed further, EBOV stimulation does affect TCRs with TIM-1 affecting cytokine regulation [39, 40]. As research evolved, deletion of the MLD of EBOV GP1-5 has some effects unknown so far and may hold future promise with chemokines emerging that include C-X-C chemokine ligand 10 (CXCL10) and the monocyte chemoattractant protein-1 (MCP-1, known as C-C chemokine ligand 2; CCL2) with others of note in immunological responses detailed further below [40–42].
It is indicated that the EBOV VP40 protein requires phosphatidylinositol (PI) 4,5-bisphosphate [PI(4,5)P2] and phosphatidylserine (PS), forming oligomers at the PM surface also facilitating vesicular particle egress [43]. Recently, this was followed up to note VP40-membrane binding via electrostatic interactions with cationic VP40 residues [43]. Therefore, VP40 interaction with lipid residues was further clarified with phosphatidic acid (PA), as a key part of the formation of EBOV matrix proteins [44]. It is of note that PI is phosphorylated at the PM surface into PI(4,5)P2 and requires PA, but also phospholipase C (PLC), and can hydrolyse phospholipids; whilst PI(4,5)P2 regulates actin formation required within epithelial layers, an important EBOV method of transmission [45]. Other receptors remain unclear, although considered prior were fibronectin receptors (denoted by integrin subunit domains) present within the extracellular matrix (ECM; e.g., α5β1) [45]. As discussed, NPC1 protein is a 1,278 aa internal cellular receptor localised to endosomal and lysosomal membranes, and in 2015 was confirmed to be essential for EBOV replication and pathogenesis in vivo [46]. These were notable findings because NPC1 is uniformly expressed throughout the brain, but preferentially within pDCs and ECs of the vascular system, and possibly at decreased quantity within other immune cells. The unique receptor that EBOV utilises remains obscure, but viral EBOV GP is required for micropinocytosis and vesicular transport within cells [13, 14]. Furthermore, the ECM may contain unknown signalling peptides in cryptic pockets, which EBOV proteins interact with triggering structural conformational changes and allowing differential EBOV binding to cellular receptors [47].
In vitro research in 2011 elucidated that VP24 disrupted a nuclear p38 mitogen-activated protein kinase (MAPK) gene transcript (MAPK14) and its resultant enzyme synthesis [48]. It was inferred that MAPK p38-α disruption was one key factor in type I IFN-β inhibition, but also EBOV VP24 binds to KPNA1 as well as inhibiting phosphorylated STAT1 nuclear accumulation [48]. Therefore, as KPNAs usually transport proteins through nuclear pores, this could affect the resultant STAT1 signalling usually leading to IFN synthesis through IFN stimulatory genes (e.g., IFN-stimulated gene 15; ISG15) [48]. MAPKs are serine/threonine-PKs regulating the p38 cell cycle progression. Disruption to p38-α regulation between the growth and mitosis cell cycle (G2/M) could plausibly be considered; because p38 may regulate stability and translation of both cytokines, TNF-α as well as IL-6, affecting neutrophil migration and can be expressed within endothelial/epithelial cells [49]. Even more so, since the deletion of p38-α within DCs in vivo has consequential effects on DC function [50]. The p38-α deletion can result in changing viral antigen presentation to cytotoxic T (TC) cells expressing CD8 [50]. In effect, conventional DCs (cDCs) may have reduced antigen presentation ability accompanied by reduction of DC maturation cytokines, IL-12p40 and IL-12p70 [50]. Therefore, viral EBOV mRNA may in effect dysregulate cytokine production as well as nuclear cell cycle regulation and transcriptional regulation affecting antigen presentation to T cells remaining unknown to date (see Figure 3).
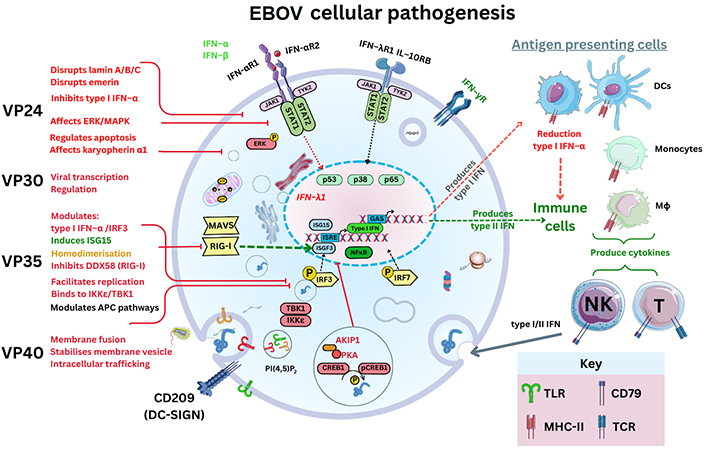
EBOV pathogenesis and the graphical outline of EBOV proteins and its known cellular targets. IFN-αR1: IFN-α receptor 1; MAVS: mitochondrial antiviral-signalling protein; RIG-I: retinoic acid-inducible gene I; IRF3: IFN regulatory factor 3; AKIP1: A kinase interacting protein 1; CREB1: cyclic AMP response element-binding protein; pCREB1: phosphorylated CREB1; ERK: extracellular signal-regulated kinase; JAK: Janus kinase; DDX58: DExD/H-box helicase 58 gene; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; TBK1: TNF receptor-associated factor (TRAF) family member associated NF-κB activator (TANK) binding kinase 1; ISRE: IFN-stimulated response element; ISGF3: ISG factor 3; IKKε: IκB kinase epsilon; TLR: Toll-like receptor; GAS: gamma IFN activation site. The Figure was partly created with Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported license
During 2006, early research articles implicated the GP relevance to Filoviridae pathogenesis, as development and results allowed more insight through genomic sequencing technology advancement [51]. At this time within MARV research, whole-genome sequencing identified the existence of a Ravn virus (RAVV) which was characterised [51]. Initial immunological research investigating Filoviridae suggested that an imbalance in helper T (TH) cell, as well as TC cell responses, may occur. The combined interaction of innate and adaptive regulation of the T cell response remains a topic requiring detail. It is known that optimal immune responses require B cell antibody production; but also, T cells alongside type I/II/III IFN synthesis require a homeostatic cytokine/chemokine balance to function in an autocrine/paracrine systemic manner. This in effect regulates antiviral, antibacterial, and antifungal activity through cytolytic actions and phagocytosis by APCs, TC cells, and NK cells. In a recent article, APCs were discussed encompassing DCs, monocytes, and Mϕ (M1ϕ and/or M2ϕ) by CD molecule expression [32]. These cells present and process pathogen-derived peptide epitopes to T cells utilising MHC class I/II molecules. DCs are considered to have a unique tolerogenic profile to viruses. This cell population has the highest expression of MHC class II molecules and presents EBOV antigens [52]. Monocytes are classified as non-classical, intermediate, or classical with varying functions including pathogen phagocytosis [32]. Mϕ categorisation is intertwined with cellular polarisation and the upregulation/downregulation of various CD markers [32]. Recently these have been referred to as M1ϕ or M2ϕ which can have pro-inflammatory or anti-inflammatory functions [32]. In 2017 it was designated that four EBOV proteins could potentially be key immunogens for innate and adaptive immunological responses, namely VP24, NP, GP, and sGP [53]. Below are shown many of the known EBOV protein functions currently with some of the recently observed gene transcripts and cytokines during infection and within laboratory research hitherto that affect IFN synthesis and coagulation regulation pathways (see Figure 4) [8, 54].
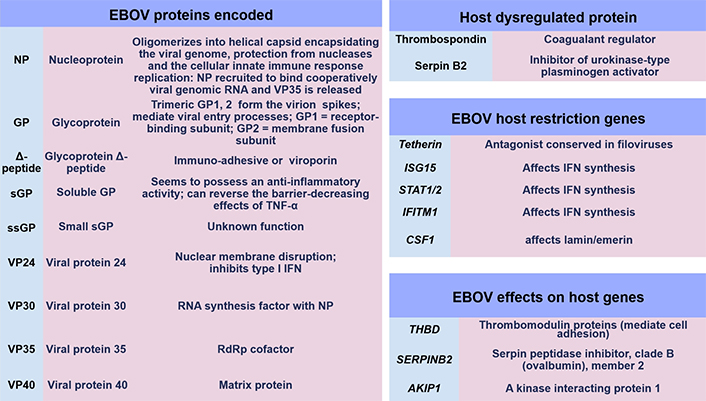
EBOV proteins and gene regulation factors. IFITM1: IFN-induced transmembrane protein 1; CSF1: colony stimulating factor 1; THBD: thrombomodulin protein; SERPINB2: serpin peptidase inhibitor, clade B (ovalbumin), member 2
Initial immunological laboratory observations are researched and measured by serum cytokine concentration changes during infection often referred to as a “cytokine storm” in an autocrine/paracrine-dependent manner in conjunction with chemokines and TLRs, with a 2015 CD classification update on all relevant immune cellular markers [55]. Studies of these can explain some of the cellular/molecular immune system synchronises during health and disease with much remaining unclear [56–58]. During 2003, the predominant APCs were further investigated with regards to Filoviridae infection. DC research has evolved since 2017 [59]. Accordingly, the profile of DCs remains a mystery in EBOV infection amongst other viral diseases that is dependent on a multitude of transcription and activation factors. These contribute towards the tolerogenic and anergic profile of DCs. Since 2017, DCs are broadly classified into non-classical pDC, but also a further three classical/conventional subtypes (cDC1, cDC2, and cDC3) [32].
In 2003 it was indicated that EBOV VP35 could infect DCs of the monocytic lineage affecting the T cell response [60]. It was then observed through in vitro culture that DCs maturing into a monocytic lineage secrete IL-1β and IL-6 together with IL-8 while expressing CCL5, also known as regulated on activation, normal T cell expressed and secreted (RANTES) protein, with abrogation of type I IFN-α that is usually secreted in high quantities [60, 61]. More recently, it was confirmed that DCs obtained from EBOV peripheral blood mononuclear cell (PBMC) monocyte-derived DCs (moDCs) could permit EBOV replication, but pDCs did not [60]. Importantly, the generic leukocyte T cell common antigen, CD45+, was used to determine the cellular lineages as either moDC (CD11c+CD16–CD14– human leukocyte antigen; HLA-DR+) or pDC (CD11c+CD14–CD16–CD123+HLA-DR+) expressing HLA-DRs that are required to present viral peptide antigens to T cells [60]. With research ongoing, it was confirmed that several key immune cell checkpoint gene transcripts (CD40, CD80, and CD83) were potentially affected in DCs by EBOV during the 2013–2016 outbreak with clarity that VP35 could unblock antigen processing pathways and IFN transduction [48, 62]. Uniquely, in 2015, it was seen in a crucial study that each of the type I/III IFN gene transcript changes in DCs [52]. This indicated that during EBOV infection, VP24 may counteract earlier VP35 antagonism of DC IFN transcription of genes between 8–24 h after infection and up to 5 days later [52, 63]. Type I IFNs (α or β) are usually required to activate and regulate both Mϕ, NK cells, and the T cell effector response [64]. The significance of this remains unclear.
Seminal in vitro reports before 2013 also explained some of the mysteries investigating monocyte cell roles during EBOV infection. It was indicated that monocytes could be permissive to infection after a delay of up to 48 h in effect allowing EBOV cellular replication [65]. Key differences were resolved with preferential mRNA expression in monocytes of three IFN regulatory gene transcripts (IFITM1, IFITM2, and IFITM3), that in effect regulate intracellular viral replication cycles through IFN synthesis [65]. NPC1 was distinctively expressed in pDCs at higher concentrations than within monocyte-derived lineages usually presenting viral antigenic epitopes to T cells [65]. It is therefore observed that EBOV can infect two APCs (Mϕs and DCs), with EBOV VP24 and VP35 having dual effects modulating the overall duration of leukocyte type I IFN synthesis and secretion of which the significance is obscure [48, 52, 62].
During cellular EBOV infection, early indications were that a change in secretion of type II IFN-γ might occur usually secreted by T cells and NK cells [60, 66]. This was accompanied by reduced IL-12 and IL-2 synthesis with increased IL-10 and increased T cell apoptosis within 12 h upon cellular stimulation [60, 66]. Synthesis of IL-12 is crucial, as transduction affects p35/p40 subunits, affecting DC responses in synergy with IL-23 and other cytokines via the heterodimer p70 formed with IL-12R (IL-12Rβ1/IL-12Rβ2), and a heterodimer shared with IL-23 [67, 68]. In 2015, it was examined that external stimulation could effectively inhibit EBOV replication within Mϕ in vivo. This was linked to the expression of four chemokines (CXCL9, CXCL10, CCL8, and CXCL11), and complement proteins (C1s and C1r), typically regulated by IFN-γ. Notably, three of these have a receptor (C-X-C chemokine receptor 3; CXCR3 or CD183) expressed by DCs [69–71]. Moreover, upregulation was noted of the Mϕ endocytic marker (CD163) shed from the PM localising with viral EBOV antigens around Mϕ and hepatocytes [72]. CD163 is also expressed by DCs during maturation. It is a haemoglobin scavenger receptor (HbSR) specific for both monocyte and M1ϕ/M2ϕ lineages and facilitates the uptake of haptoglobin-haemoglobin (Hp-Hb) complexes in the circulation where lysis of erythrocytes occurs during EBOV infection [72]. It was further noted in 2006 that lipopolysaccharide (LPS) stimulation of TLR4 also causes CD163 shedding in monocyte lineages with TLR2 and TLR5 present at the PM cell surface [73–75]. Recently in 2017, TLR4 studies in vivo and in vitro in the absence of T cells reveal that EBOV activates TLR4 with each of the CD11b+ and CD11c+ lineages (DCs, monocytes, M1ϕ, and neutrophils) circulating in draining lymph nodes (dLNs) [76]. Further to this observation, using GP-stimulated bone marrow-derived Mϕ (BMDM), it was observed in vivo that TNF-α, IL-1β, and IL-6 normalised within 24 h; moreover, CCL2 and CCL4 together with T cell-derived cytokines (IL-2, IL-4, IL-5, and IFN-γ) and IL-10 remained elevated [76]. Noteworthy was that CCL5 and IL-12 expression/synthesis remained unchanged [76]. However, in vivo studies in detail indicated Mϕ continue to upregulate two co-stimulatory molecules (CD40/CD80) and potentially could still signal to three respective T cell ligands, CD40 antigen ligand (CD40L), CD28, as well as TC lymphocyte-associated protein 4 (CTLA-4), which are required for adaptive T cell responses [76].
From 2016, further detailed analysis occurred in vitro comparing EBOV and MARV of consideration [77]. This laboratory utilised cellular transcriptome mapping, in vitro, to note the upregulation of an annexin A3 (ANXA3) gene transcript, with the potential protein encoded known to inhibit PLA2 enzymes [77, 78]. These were key observations as PLA2 is crucial and metabolised by cyclooxygenases (COXs) into anti-inflammatory mediators including leukotrienes (LTs), prostaglandins (PGs; e.g., PGE2), as well as eicosanoids. Also observed were IL-8 (also referred to as CXCL8) and IL-32 which could potentially affect T cell apoptosis [77]. Another cysteine-rich protein gene transcript, CYR61, was detected known to bind to heparin and is a growth factor (GF) affecting vascularisation [77, 78]. The dual specificity protein phosphatase 1 (DUSP1) gene transcript was highlighted as encoding a protein processing intrinsic phosphatase activity that can inhibit the ERK1/2 and MAPK pathway together with phosphothreonine and phosphotyrosine residues present in STAT proteins [77].
In longer-term studies of non-human primates (NHPs; n = 36), it is indicated that Mϕ expressing CD68+, together with leukocytes expressing CD45+, and also T cells (denoted by CD3+) could infiltrate heart ventricles and the choroid plexus [79]. Historically, and since 2021 the blood-brain barrier (BBB) in terms of how immune cells permeate across semi-permeable membranes is unclear [80].
In the early 21st century between 2008 and 2009, reports appeared employing an isolated neutralising antibody (nAb) binding to EBOV GP to elucidate the crystalline structure of an antibody [81]. It was then specified as in vivo research continued that a mouse Ig2a monoclonal antibody could bind specifically to the glycosylated MLD domain of the EBOV GP (EQHHRRTDN, 405–413 aa) [82]. The overall structure of EBOV GP seemingly then was considered to be a GP1 trimer composed of base, head, and glycan cap forming a chalice with a trimer of GP2 subunits with a cradle around the GP1 EBOV domain [83]. Investigations began to indicate differential antibody responses against GP1/GP2 and the sGP cellular product [83]. Furthermore, the existence of a smaller Δ-peptide clarity remains unknown to date, with two theories. Initially, Δ-peptide could be immuno-adhesive or act as a viroporin [84–87]. To this effect, in 2011, observations were made in comparison with different Filoviridae between sGP and ssGP function, with hints of differences in the immunological function of the Δ-peptide resulting from a C-terminal cleavage product of sGP [87]. However, during longitudinal research studies of the 2013–2016 outbreak, indications were that a polyfunctional B cell and/or APC-induced TC phenotype expressing CD8 remained crucial (n = 206) for an effective immune response [88]. This was measured as cells being able to synthesise and secrete type II IFN-γ, alongside two other cytokines, TNF and IL-2, synthesised in 0.046% of total CD8+ T cells [88]. Observations of immunological relevance were examined within this case-control cohort (n = 206), where 96% of individuals possessed antibodies after EBOV infection with sub-characterisation confirming that 9% of blood samples were considered to possess nAbs [88]. It was observed in western blot analysis that antibodies could be polyfunctional against four of the seven EBOV virion-encoded proteins (GP, NP, VP35, and VP40) [88]. Therefore, this would indicate characterised viral-specific epitopes within EBOV particles that may generate antigen/epitope-specific innate and/or adaptive immunological responses [88]. In longer-term studies utilising the development of a cell-based reporter system, the affinity of EBOV-stimulated antibody binding via their respective fragment crystallizable receptors (FcRs) to effector cell surface receptors was examined. It was confirmed that EBOV infection may generate IgG1 antibodies that display the highest affinity for the FcγRI (CD64), rather than FcγRIII (CD16) and FcγRII (CD32), over a time frame of more than 10 years [89]. These three receptors are utilised differentially by antibody Fc regions to effect B cell antibody-dependent responses on a variety of host immune cells, like M1ϕ or M2ϕ.
Serology reports in 2020 demonstrated that EBOV protective antibodies can be synthesised in humans after infection with isotypes produced ten times higher in concentration that were mainly the IgG1 isotype compared to IgG2, IgG3, and IgG4 still produced [90]. In addition, IgM is produced which is historically seen to be a marker of natural infection in other viral pathologies. It is further indicated (n = 4) in a longer kinetics study that IgG specific for GP remained over 2 years and up to 12 years [91]. Concurrently, IgM was observed to decline, whilst IgA remained at increased concentrations alongside IgG responses to both EBOV NP and VP40 [91]. The characterisation of B cells producing antibodies then pinpointed antibody-secreting B cells expressing CD27hiCD38hi with less expression of other activation markers CD71+/CD20+ [91]. Notable at this point, was the removal of mucin and glycan caps of EBOV GP further increasing the binding affinity of serum IgG to cleaved viral GP (sGP) [92]. The research into antibody treatment peaked in the development of antibody drugs for the treatment of EBOV. The first cocktail of three monoclonal antibodies, designated Inmazeb™, was approved by the United States of America (USA) Food and Drug Administration (FDA) in October 2020 (see Supplementary materials).
A key laboratory report utilised a control group of non-pathogenic Filoviridae, RESTV, in comparison with pathogenic Filoviridae (EBOV and MARV) in both humans and macaques performed in 2006 [66]. It was noted, utilising annexin V and propidium iodide staining, that T cell apoptotic rates at least doubled indicating cell cycle changes and apoptotic cells, but not in the non-pathogenic control virus group (RESTV) [66]. Furthermore, employing in vitro viral peptide sequences stimulating PBMCs and measuring hypodiploid DNA content, a three-fold DNA synthesis reduction occurred during the T cell synthesis phase to each of the pathogenic Filoviridae (MARV/EBOV) [66]. It was considered that pathogenic Filoviridae could alter CD4/CD8 cell count ratios measured by TH1, TH2, and TC cell populations with reasons unknown as to cellular mechanisms. Indications may be evocative that this could be caused by nuclear membrane changes, cell communication, intracellular signalling, or single nucleotide polymorphism (SNP) affecting any of the proteins or immune cell populations. However, EBOV T cell research is evolving and continues with T cell phenotypes further researched including mucosal-associated invariant T (MAIT) cells [32].
It was noted in 2003 that EBOV may suppress TH1 cell phenotypes alongside IL-12p40 and type II IFN-γ production with inhibition of IL-2, TNF-α, as well as CCL2, but not CCL3, deploying monoclonal antibody reagents that are validated as specific [60]. The receptors for these cytokines and chemokine ligands were examined by flow cytometry using labelled monoclonal antibodies. This revealed their paracrine/exocrine functions during an EBOV infection. The reasons for this remain ambiguous. Further signals evidenced then were reduction in CD25 within T cell populations categorised into regulatory T (TREG) cells, amongst others known to express this soluble secreted PM TREG CD25 cell receptor [60]. Other CD receptors are predominantly membrane-bound GPs. Shortly after in 2011, independently, T cell phenotypes were further investigated and it was noted that a memory T (TMEM) cell subset could possess regenerative-like properties (denoted by CD45RO–CD45RA+CD62L+CD27+CD28+ expressing the C-C chemokine receptor 7, CCR7) and secreting IL-7 [93]. Importantly, it illustrated that T cells could be specific for viral as well as tumour-associated antigens (TAAs), whilst specifically expressing high concentrations of IL-2Rβ, CXCR3, and the leukocyte function associated-antigen-1 (LFA-1) with the CD95 cellular activation marker [93].
The role of T cell responses to EBOV in 2015 revealed polyfunctional TC (CD8+) cells could express CCR7, but not CD45RO, whilst phenotypical effector TMEM (TEM) cells developed EBOV-specific responses upregulating membrane activation proteins (CD28/CD95) in response [40, 94]. These were critical as CCR7 is expressed in the majority of the adaptive T cell population, whereas CD45RO/RA is used to define the activation status of TMEM leukocytes; while CD28 is a required co-stimulatory molecule for TC cell stimulation and survival. Other investigations of the exact mechanism of action of EBOV on T cells hint at a “superagonist-like” effect [40]. When EBOV-stimulated PBMCs were depleted of DCs/monocytes, it could be seen that activation markers CD25/CD69 were significantly upregulated at 48 h by T cells [40]. The reasons for EBOV stimulation and its effects on host cells remain unknown. Below is shown an infographic of the estimated overall immune system interactions based on prior research with much unknown (see Figure 5).
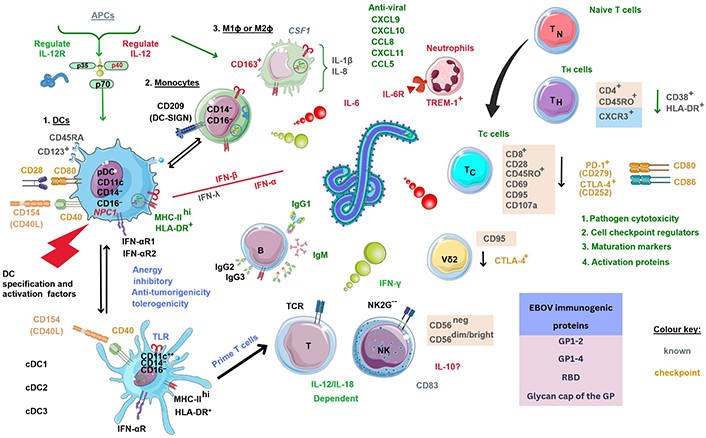
Estimated overall immune cellular response profile during EBOV infection based on known prior research. NK2G--: NK cell checkpoint; PD-1: programmed death-1. ?: no data. The Figure was partly created with Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported license. Virus schematic was adapted from ViralZone, SIB Swiss Institute of Bioinformatics (https://viralzone.expasy.org/207), licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License
With regards to the CD4+ T cell population, stimulation indicated preferential expression of CD45RO+ on T cells in survivors that were predominantly central TMEM (TCM) cells expressing CXCR3+ that were EBOV GP antigen-specific. The overall TH (CD4hi) cell population expressed another activation (CD38) marker [40, 95]. Simultaneously, in a 2018 report, it was noted in vivo that CD69 expression downregulation did not occur within myeloid-derived DCs (mDCs) in the spleen [95]. Though the earlier report did indicate T cells expressing both CD4+ and CD8+ did significantly downregulate CD69 at 12 h, it was recently confirmed that this occurred at 48 h, with CD25 upregulation by T cells [66]. In 2019, ANXA5 was suggested to potentially be a beneficial therapeutic during microvascular disease [96, 97]. ANX proteins are both soluble and hydrophilic and bind to oppositely charged phospholipids reversibly dependent on calcium. Such research would be interesting to see as the above ANX proteins can aggregate vesicles but also PS does appear to be implicated in EBOV host-cell interaction mostly unknown [96, 97].
Notable further investigations into EBOV host cell receptors showed deficiency of TIM-1 in vitro cell line cultures could regulate T cell secretion of crucial cytokines that were IFN-γ, IL-2, and TNF-α, as well as IL-12p40, together with the granulocyte-Mϕ (GM)-CSF [40]. Much remains unknown regarding the EBOV effect on T cells; however, it was evidenced that EBOV could both stimulate T cells and be internalised without replicating [98–100]. Investigations showed the EBOV NP protein could be found within T cells expressing the TCR complex differentially affected [98–100]. Specifically, the TCR CD3ε subunit domain could be found at increased frequency in EBOV-stimulated samples [98–100]. This was accompanied by downregulation and degradation of the TCR CD3ζ, with intracellular signalling maintained, whilst CD3ζ co-localised with a monomeric guanosine-5’-triphosphate (GTP) enzyme (a GTPase, Rab7), with EBOV GP in late endosomes [98–100]. The CD3/TCR complex predominantly utilises lymphocyte-specific protein tyrosine kinase (Lck), with phosphorylation of the TIM-1 cytoplasmic domain resulting in activation of PI3-kinase (PI3K), representing a known cell survival and proliferation pathway.
In other longitudinal kinetic studies (n = 2), where individuals underwent treatment for chronic EVD, the CD95 marker was observed as transiently upregulated on both TC/TH cells with the receptor, PD-1 transiently upregulated by TH cells indicating a cellular activation [101]. Antigen presentation proteins denoted by HLAs (HLA-DR/MHC class II) were present on CD8+ T cell PMs, but it was inferred that CD45RA and CCR7 may not be expressed on all T cell phenotypes [101].
During EBOV infection, each of the DCs, monocytes, Mϕ, as well as T cell phenotypes of the immune system are predominantly affected similarly to other viral pathologies [32, 102]. Recent observations are that EBOV sequesters IRF3 in viral inclusion bodies blocking required type I IFN induction [103, 104]. This usually occurs through IRF3 binding to the TRAF-associated proteins activating the NF-κB kinase, TANK, and TBK1, as well as IKKε [103, 104]. It is through kinetic studies outlined above that many of the delayed onset of immune cell upregulation of CD molecules could be regulated by type I/II/III IFN unknown so far. In conjunction with the tolerogenic profile of DCs, T, and NK cell responses usually stimulated by EBOV may be seemingly affected by subunit protein domains that are toxic with the appearance of agonist-like effects. However, the cytokine IL-8 is known to differentially affect CD4+/– T cell activation of TEM CD4+/– T cells, but not TN or TCM CD4+/– T cells [105]. In addition, IL-8 can upregulate IL-2 synthesis with no effect on IFN-γ and IL-4 production [105]. These observations remain key as IL-8 effects on T cell apoptosis remain unclear.
In 2017, authors indicated using digital cell quantification (DCQ) that NK cells showed an evidential increase after infection during recovery from EVD. In a comparison transcriptome analysis of NHP (n = 112), clear significance was noted between non-fatal and fatal outcomes with potential pro-inflammatory chemokine mRNA gene transcripts (CXCL10, CCL2, CCL8, and CXCL11) during severe EBOV infection [106]. Two of the potential encoded proteins (CXCL10/CXCL11) have an affinity to a known receptor, CXCR3, present in DCs, NK, and T cells [106]. Shortly after, in 2018, laboratory research investigated the role of Mϕ cells expressing soluble CD163 (sCD163), in comparison, given the similarities that severe EVD has to Mϕ activation syndrome (MAS) and haemophagocytic lymphohistiocytosis (HLH) [72]. At this juncture, it was hypothesised that Mϕ-CSF (M-CSF), two cytokines (IL-6 and IL-8), together with three chemokine ligands (CCL2, CCL3, and CCL4), alongside two modulatory cytokines IL-10/IL-1R antagonist (IL-1Ra) remained pertinent to immunological cell function and migration during EBOV infection [72]. The T cell marker expressed by NK cells denoted as an immune cell shed soluble IL-2R (sIL-2R and CD25+) is present on TREG cells and was also found at increased levels during infection [72]. As above, in vivo, TLR4 (CD284) is considered to be an activation marker instigated by both pathogenic/non-pathogenic viruses [72]. Authors noted that overall NK cell frequencies may be considered slightly reduced reflecting unknown NK cell activation and proliferation [72]. Furthermore, a decrease in CD56dim/bright NK cells, alongside increased frequencies of CD56– NK cells hold possibilities and may be attributed as positive regulators of immunological relevance unknown to date [72].
More recently in 2020, the role that NK cells perform illustrates that CD14+ monocytes may be considered the source of pro-inflammatory cytokines [107]. In vitro stimulation/inhibition studies using EBOV GP indicated that NK cell activation was dependent on IL-18 and IL-12 and could be enhanced by an antagonist that blocked the IL-10R [107]. Two NK cell activation markers, CD107a and CD25, were observed to be upregulated in response to EBOV GP, but without secretion of type II IFN-γ usually synthesised by both NK and T cells [108]. Simultaneously, the EBOV VP40 protein subunit domain was also found to stimulate IL-12/IL-18 production [109]. However, it was determined that the degranulation and cytolytic actions of NK cells were dependent on IL-12 to activate CD56+ NK cells [109]. These observations noted that NK cell function and maturation were crucial. IL-18 is considered to have pleiotropic functions, with IL-12 as a regulator of TH1 cell differentiation, along with IL-23 regulating TH17 cell secretion of IL-17 [109]. Dual regulatory effects can occur with the activation of TC cells and NK cells [110]. Activation markers like CD95 are used to examine NK cell maturation and observed to be upregulated without change during EBOV infection [109]; but concurrently more recent subtypes of immune cells have been characterised as lacking the chemokine CCR7, denoted as effector Vδ2 cells, that can be an antigen-stimulated source of TNF-α [111]. The other activation marker, CD69, was expressed on each of the observed NK cell phenotypes [111]. As above type II IFN-γ can inhibit EBOV infection and be produced by the above four cell types. Some of the unknown genetic mechanisms of IFN-γ synthesis became slightly clearer with predominant ISG gene transcripts researched and observed that are relevant. Initially, guanylate binding protein 5 (GBP5) upregulation was examined that is a factor that can inhibit type II IFN-γ in other viral infections like respiratory syncytial virus (RSV) [112]; but also, others were recognised that may affect other immune cell types regulating retinoic acid receptor responder protein 3 (RARRES3) and vesicle-associated membrane protein 5 (VAMP5) as yet unknown [70, 113].
Very recently, developments occurring showed the neutrophil chemoattractant IL-8, together with IL-1Ra, TNF-α, CCL2, and transforming GF alpha (TGF-α) as essential factors in beneficial immune responses [54]. Metabolic enzymes were depicted as significant including high-density lipoproteins (HDLs) that were apolipoproteins (E/L1/A4/C3/C4). Notably, CSF1, GM-CSF, CCL3, CX3CL1, and IL-18 binding protein (IL-18BP) were associated with high EBOV viral loads [54]. Additionally, VP40 was observed to be less stable than GP, and less detectable in samples with polymerase chain reaction (PCR) cycle threshold (Ct) values higher than 25 [54]. EBOV VP40 detection is negatively correlated with Ct value—a potential indicator of EVD severity, similar to the value gained from antibody titre serology [54]. These reports in combination illuminate that M-CSF is a key cytokine required during Mϕ development; but also IL-18 can independently mediate type II IFN-γ secretion from TH1 cells (through IL-18BP) and is usually produced by Mϕ that NK cells require for cytolytic function [54]. The role of IL-18BP in regulating type II IFN-γ during EBOV infection is worthy of exploring as it is produced by mononuclear cells, but also IL-18 does signal between IL-1R-associated kinase 1/4 (IRAK1/4), and could plausibly affect T cell and NK cell responses [114, 115]. The synergism of IL-18 activity with other cytokines does stimulate NK cell proliferation and cytotoxicity through the production of perforins and granzyme B remaining unsettled to date [116]. The duality of NK cell function remains vague as a multitude of activation and inhibitory receptors exist regulating NK cell-dependent cytotoxicity; however, activation/inhibition signalling was further researched in 2022 [28]. Jarahian et al. [28] performed in vitro research to observe the EBOV GP may stimulate NK cell cytotoxic activation receptors (NKp44/NKp46), selectin adhesion receptors (CD62L/CD62P), as well as inhibitory sialylated Siglec-7 (CD328) and Siglec-5 (CD170) receptor proteins [28]. These were significant findings because CD62L is constitutively expressed, required by leukocytes for cellular migration across EC layers, with CD62P similarly on platelets; while short-chain CD328 (also CD170) contains immunoreceptor tyrosine-based inhibitory motif (ITIM) cellular cytoplasmic domains and external domain receptors which bind to glycans containing sialic acid [28].
Recent developments during in vivo transcriptional research demonstrate that during EBOV infection (n = 9), four genes are markedly upregulated at all stages of infection (IRF7, S100A8, S100A9, and IFN-induced protein 44; IFI44) [117]. These correspond to calprotectin and IRF proteins potentially encoded [117]. A further seven mRNA transcripts were noted at early stages of infection, including CD19, IRF1, CXCR2, IL13RA1; notably, three remain crucial which include the PR domain zinc finger protein 1 (PRDM1, known as the B lymphocyte-induced maturation protein-1, BLIMP1), but also FCGR3, encoding FcγRIII (CD16), as well as the B cell antigen receptor complex-associated protein alpha chain (CD79A) [117]. These encode proteins that all may regulate B cells, antibody receptors, cytokines, receptors, and chemokine receptor synthesis [117]. Importantly, one key microRNA (miR) regulating post-transcriptional gene expression was highlighted and is under investigation in sepsis research [118]. Future research on the miR oligonucleotide, miR-122-5p, will be very interesting to see. Recently, miR-122-5p was observed to regulate coagulation, where mimics of miR-122-5p (n = 84) were observed to downregulate IL-1β, IL-6, CCL2, and TNF-α in sepsis models [118]. Therefore, these observations may have direct relevance to research in the future given that miR modulation of cytokine levels could potentially apply to different pathologies after extensive further research. More recently, nine EBOV GP epitopes were examined against HLA alleles in silico to predict that thirteen HLA-A and HLA-B alleles may represent optimal CD8+ T cell responses that could generate broad immunological responses. These include two predicted to be within regions of the EBOV GP1-1 and GP2-2 domains [119]. Below is shown interpretation of current immunological response factors so far based on research to date (see Figure 6).
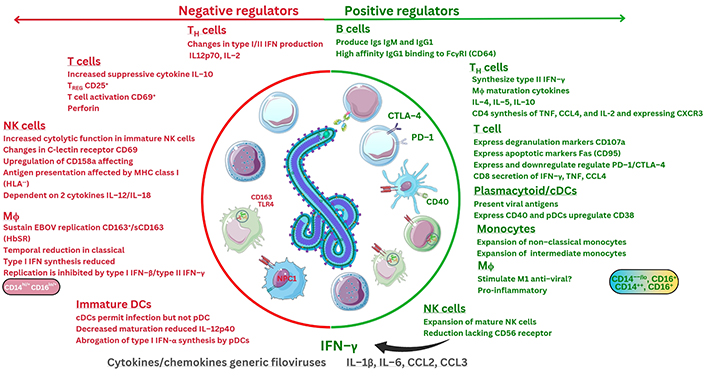
Interpretation of overall immunological factors during EBOV infection based on current research. Fas: apoptosis antigen 1, also known as CD95. ?: no data. The Figure was partly created with Servier Medical Art (https://smart.servier.com/), licensed under a Creative Commons Attribution 4.0 Unported license. Virus schematic was adapted from ViralZone, SIB Swiss Institute of Bioinformatics (https://viralzone.expasy.org/207), licensed under a Creative Commons Attribution 4.0 International (CC BY 4.0) License
Filovirus research utilises both in vitro and in vivo models [120]. In the 21st-century outbreaks since the 1976 discovery of both Zaire EBOV (ZEBOV) and Sudan EBOV (SUDV) strains have elucidated further detail regarding diagnostics and potential correlates of protection against such virulent pathogens (see Supplementary materials). Facilities necessary for BSL-4 pathogens require advanced training, high cost of reagents, and other materials, but also the development of future in vitro and in vivo, as well as organoid models to elucidate pathogen cellular mechanisms (see Supplementary materials) [120]. Developments are ongoing of future vaccines alongside clinical trial studies documented in other reviews (see Supplementary materials) [120]. In 2014, limitations of PCR testing were noted with a report outlining potential future options of lateral flow immunoassays (LFIs) as an accurate viral antigen diagnostic (see Supplementary materials) [121–123]. Other articles discuss monoclonal antibodies that have been identified (see Supplementary materials). Diagnostic and therapeutic monoclonal antibodies are expensive and require validation for both sensitivity and specificity affected by viral mutational evolution. Furthermore, SNPs can occur in any cellular host gene encoding any protein with many discovered recently. Therapeutic strategies used may vary and the differential role of TIM-1 (CD365) in T cell signalling is still being clarified [39, 40]. The role of STAT protein mutations and chemokines is still being discovered, with type II IFN-γ discovery some 50 years ago in 1965, with errors remaining unknown within populations globally [124]. Historically, errors in IFN-γ signalling are rarely documented in research or literature, apart from in mycobacterial infection, and remain a determinant of adaptive T cell function in pathogenic host immune cell responses. More recently, the 2003 discovery of type III IFN-λ, followed by type III IFN-λ4 in 2013 remains largely uncharted as to cellular signalling and transduction mechanisms [52]. Chemokines were only further characterised during 2000–2014, additionally highlighting atypical chemokine receptors (ACKRs) present within epithelial layers. In addition, TLR research during EBOV infection is comparatively recent; but also, human leukocyte alleles may vary between global populations encoding the MHC type I/II proteins required to present viral peptide antigens between immune cell phenotypes. However, much remains fuzzy with research ongoing, subject to ethical guidelines (see Supplementary materials).
Since 1976, both SUDV/ZEBOV strains were isolated and found to be causal to EVD. Technological advances and research remain of priority. From 2013, the Democratic Republic of Congo (DRC) and other countries experienced serious EBOV outbreaks (see Supplementary materials). The Ugandan Ministry of Health confirmed a recent EBOV outbreak on September 20, 2022, evidenced to be SUDV. This led to 164 affected individuals (142 confirmed), claiming the lives of 55 individuals with an IFR of 33.5% (see Supplementary materials). During any infection caused by viral, bacterial, or mycobacterial pathogens, the overall risk is defined by R0 as the transmission rate. This is a measure denoting pathogen transmission referred to as the reproduction number or how many people could be affected during contagious infectious disease outbreaks. The 2015 estimate for EBOV R0 is in the range of 1.37–2.53 [125–127]. Other reports indicate overall risk of EBOV infection is estimated to be around 45% within community settings [125–127]. Male/female survival rate differences following EBOV infection were indicated, with a slight increase rate of survival for EBOV-affected females [127]. The 2022/2023 Ugandan response through a detailed genomic sequencing report indicates 99.58% similarity of the September 2022 EBOV (SUDV, Mubende variant), with a prior May 2011 SUDV isolate (Nakisamata, JN638998.1) (see Supplementary materials) [128]. A difference of 10 aa is noted, calculated to occur at a rate of 2.23 × 10–5 substitutions per site occurring per year since EBOV characterisation [128].
Current indications are that the EBOV GP glycan cap and MLD of the 2022 SUDV analysed sequences differ by 1–2 aa (codon 711/821, within the EBOV NP or polymerase proteins respectively) [128, 129]. Significantly, enrichment of unprocessed glycans at two conserved sites, denoted by asparagine residues (positions 257/563) of the EBOV GP, are demonstrated to affect sensitivity to neutralisation by immune system cells [129]. C-Mannosylation and O-linked glycans outside the EBOV GP MLD have not as yet been fully investigated and require further research. Furthermore, mutations in the EBOV polymerase enzyme (N572S, Q986R, and F2061S) are suggested to correlate with regulating host EBOV viral load [130]. Viral mutations can have an effect on therapeutic monoclonal antibody interventions developed, but also organ disease phenotypes [131–134]. Given EBOV severity in disease, it is prudent to consider that adaptive immune systems can vary by age. For example, thymic T cell development is only mainly documented during and after cytomegalovirus (CMV) infection [135].
In 2022, it was clarified that EBOV could induce B cell, T cell, and NK cell apoptosis and be dependent/independent of caspase proteins with further research needed to ascertain such mechanisms [136]. Others indicate that EBOV is dependent on PS with caspase-dependent activated (XK-related; Xkr) scramblase proteins utilised further elucidating EBOV intracellular pathways [137]. In the 21st century, real-time PCR has become regularly used for sequencing viral genomes and EBOV has been further investigated in different age-ranges [138].
The significance, as shown in Figures 2–5 of this article, highlights various characterised proteins including RANTES, known as CCL5 [139]. This appears to correlate with EVD survival outcome; but also two soluble intercellular and vascular cell adhesion molecules (sVCAM and soluble ICAM; sICAM), and plasminogen activator inhibitor-1 (PAI-1), released by ECs can be considered detrimental factors [139]. Potential reasons for this correlation of expression by immune cells could be the effect on DC migration of monocytes expressing the corresponding receptor CCR5 changing migration into the dLNs [139]. Monocyte decreases are suggested in the periphery during EBOV infection alongside NK cell changes remaining unknown [106, 139]. Stimulation of NK and T cell phenotypes to effect pathogen cytolytic activity is required through antigen presentation by monocytes and DCs together affected by type I IFN. Specifically, in vitro, the EBOV GP was revealed to sterically be shielded from antigen presentation by MHC class I to the TCR [140, 141]. These complexities act through a multitude of activatory/inhibitory signals expressed by NK cells, e.g., NKG2D (CD159), NKp30 (CD337), usually effecting cytolytic pathogen clearance [140, 141]; however, with EBOV cellular infection, it has been observed that there could be temporal cytokine secretion reduction by NK cells alongside reduced degranulation [140, 141]. NK cell maturation is ascribed to be affected through STAT5, but also IL-15 stimulation is associated with memory NK cell survival [142]. As recently as 2021, IL-10 regulation of innate lymphoid cells, including NK cells could require further analysis [140–142]. The mechanisms that some EBOV proteins, like VP24, use to regulate CD38 and type I/III IFN synthesis are only now becoming slightly clearer [52, 143]. Specifically, some mutations within EBOV VP24 aa residues (137/140) are indicated to affect, through KPNA, antagonism of IFN transduction signals via the nuclear ISRE (see Figure 3) affecting the human immune cell response [121, 143, 144]. In NHPs, three predominant upregulated cytokines synthesised and compared to a base-line that were upregulated were IL-1Ra, IL-6, followed by IL-10 concurrently upregulated with sCD40L (CD154) downregulated at days 5–6 during severity [143–147].
Questions remain given EBOV similarities as outlined above with MAS, and sepsis, which are each systemic conditions of severity [72]. These observations will undoubtedly require further research in the future. Uniquely, Bruchez et al. [148] recently characterised a transcription factor (the MHC class II transactivator), seemingly with 2–3 times proprietary antiviral activity against two viruses (both EBOV and the severe acute respiratory syndrome coronavirus-2) [148]. This protein is induced by type II IFN whilst inducing virus resistance through a CD74/p41 isoform affecting a known host lysosomal cathepsin L protease required to allow endocytosis of VPs [148]. However, further research is required on the other immunologically relevant antibodies that include other subtypes of IgG (1–4), and IgA (1 and 2), alongside other NK cell receptors, mast cells, basophils, and eosinophils. Since the 1976 EBOV discovery, various studies are suggestive that there is a role for the ISG protein, cholesterol-25-hydroxylase (CH25H), that can inhibit EBOV with suggestions that this antagonises EBOV entry in the endosomal and lysosomal compartments where NPC1 is expressed [21, 149, 150].
During host immune response to pathogens, overall host innate and adaptive immune responses may remain consistent as viral genomes will differ upon transcription and translation between species. As a prerequisite, it is considered that both DNA and RNA viruses will evolve and/or mutate, with more or less severity regardless. Host immune cell proteins reliant on a myriad of signalling pathways that can vary with errors occurring during gene transcription and translation into proteins, many of which remain unknown. Previously CD4+ T cell upregulation was discussed during EBOV infection. This is important in determining the T cell response and contribution to disease phenotype regulation. T cell phenotypes also include MAIT, but some T cells may not express CD4 or CD8 characteristic receptors [32]. Whilst other T cell phenotypes secrete at least 6 IL-17 cytokine subtypes (IL-17A to IL-17F), acting through respective receptors requiring further clarity. The significant findings outlined in this article draft the progression in knowledge of CD protein indicators of immunological relevance, based on laboratory progress from many researchers and institutions across the world [32]. The upregulation of TH (CD4+) cells, alongside an optimal TC (CD8+) cell response, as illustrated in Figures 4 and 5, together with CCL5 expression can be considered further.
Therapeutic strategies are under further investigation for Filoviridae (EBOV/MARV) including antivirals, polyclonal/monoclonal antibodies, as well as proprietary small-interfering RNA (siRNA) molecules [151–155]. As of January 31, 2024, it is currently indicated that there are at least 115 clinical trials at various stages globally on the National Institute of Health (NIH) website either completed or in progress (see Supplementary materials). Of these, three vaccines are at various stages of development and are viral-vector modified with two monoclonal antibody therapeutics recently recommended by the WHO (see Supplementary materials) [151–157]. During this time, other announcements indicate that a novel orally administered monoclonal antibody, opaganib (ABC294640), has been developed in partnership between Redhill Biopharma Ltd. with the United States Army Medical Research Institute of Infectious Diseases (USAMRIID). On October 3, 2023, early prospects were that this sphingosine kinase-2 (SPHK2) inhibitor may deliver a statistically significant increase in survival time in vivo, with research ongoing (see Supplementary materials). Similarly, recent reports are also appearing evaluating other therapeutics. Further safety evaluations are occurring of a ZEBOV vaccine, followed by a combined modified vaccinia Ankara-Bavarian Nordic (MVA-BN) vaccine (n = 192) [158, 159]. The second is based upon a combined filovirus GP antigen, designed to counter EBOV, SUDV, and MARV [158]. In 2024, the antiviral activity of natural compounds (n = 974) is evaluating potential future options of tubeimosides I, II, and III as EBOV cellular fusion protein inhibitors to block binding to NPC1 [160].
EBOV aside from severe disease can be causal of long-term conditions [63]. Some have observed this occurs with EBOV infection adding to the requirement for further research. There are currently 371 CD molecules classified that immune cells could express with variable cellular function. Additionally, 10 TLRs classified within this nomenclature remain unclear as to relevance within how or why the EBOV evokes such differential immunological characteristics. Based on the above, B cell antigen presentation together with T cell phenotypes remain crucial to elucidate future dynamics of EBOV host immune responses and other pathologies. Single-cell RNA (scRNA) transcriptome studies since around 2009 allow further clarity and insights into gene expression factors, some of which are described above [161]. Recent developments in 2023 further clarify the role of NPC1, as a key receptor, together with potential future inhibitors of cellular EBOV infection [160, 162]; however, further information will be required elucidating the role of IL-8 and DAP-12 signalling within T cell phenotypes and B cell antigen presentation [162–169].
Characterisation of the EBOV genome is ongoing. In this holistic overview, the relevant cellular proteins are highlighted which are critical factors of the human host immune cell response. The authors therefore hope that the comprehensive information presented will be useful for clinicians, academics, and researchers alike in the future.
aa: amino-acids
ANXA3: annexin A3
APCs: antigen-presenting cells
CCL2: C-C chemokine ligand 2
CCR7: C-C Chemokine receptor 7
CD: cluster of differentiation
cDCs: conventional dendritic cells
CSF1: colony stimulating factor 1
CXCL10: C-X-C chemokine ligand 10
CXCR3: C-X-C chemokine receptor 3
DAP-10: DNAX-activating protein-10
DCs: dendritic cells
DC-SIGN: dendritic cell-specific intercellular adhesion molecule-3 grabbing non-integrin
EBOV: Ebola virus
EC: endothelial cell
EVD: Ebola virus disease
FcRs: fragment crystallizable receptors
GP: glycoprotein
HLA: human leukocyte antigen
ICAM-3: intercellular adhesion molecule-3
IFITM1: interferon-induced transmembrane protein 1
IFNs: interferons
IgG1: immunoglobulin G1
IL-18BP: interleukin-18 binding protein
IL-1Ra: interleukin-1 receptor antagonist
IL-6Rα: interleukin-6 receptor α
ILs: interleukins
IRF3: interferon regulatory factor 3
ISG15: interferon-stimulated gene 15
KPNA: karyopherin-α
L-SIGN: liver/lymph node-specific intercellular adhesion molecule-3 grabbing non-integrin
MAPK: mitogen-activated protein kinase
MARV: Marburg virus
MHC: major histocompatibility complex
miR: micro ribonucleic acid
MLD: mucin-like domain
mRNA: messenger ribonucleic acid
Mϕ: macrophages
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NHPs: non-human primates
NK: natural killer
NP: nucleoprotein
NPC1: Niemann-Pick type C1
PBMC: peripheral blood mononuclear cell
PCR: polymerase chain reaction
pDCs: plasmacytoid dendritic cells
PI(4,5)P2: phosphatidylinositol 4,5-bisphosphate
PI: phosphatidylinositol
PKR: protein kinase R
PLC: phospholipase C
PM: plasma membrane
PS: phosphatidylserine
RESTV: Reston Ebola virus
RNA: ribonucleic acid
sGP: soluble glycoprotein
ssGP: small soluble glycoprotein
ssRNA: single-stranded ribonucleic acid
STAT1: signal transducer activator of transcription 1
SUDV: Sudan Ebola virus
TC: cytotoxic T
TCRs: T cell receptors
TH: helper T
TIM-1: T cell immunoglobulin-mucin-domain 1
TLR: Toll-like receptor
TMEM: memory T
TNF-α: tumour necrosis factor-α
TREG: regulatory T
–ve: negative
VPs: viral proteins
ZAP70: zeta-chain-associated protein kinase 70 kDa
ZEBOV: Zaire Ebola virus
The supplementary material for this article is available at: https://www.explorationpub.com/uploads/Article/file/1003139_sup_1.pdf.
The authors are grateful to Mandana Akhavan, Theodore Carp, and Wenbo Li for data curation.
BB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing—original draft, Writing—review & editing. CI: Data curation, Formal analysis, Methodology, Investigation, Writing—review & editing, Software, Supervision. ECC and IF: Data curation, Formal analysis, Investigation, Methodology, Writing—review & editing, Resources, Visualization, Software, Supervision.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
All datasets analyzed for this study are included in the manuscript and the supplementary files.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Marc H.V. Van Regenmortel
Christine Jacomet
Vladimir N. Uversky
Chittaranjan Baruah ... Bhabesh Deka
Brent Brown ... Enrique Chacon-Cruz
Om Saswat Sahoo ... Subhradip Karmakar
Mikolaj Raszek ... Alberto Rubio-Casillas
Ankit Kumar ... Vijay Mishra
Yulia Desheva ... Irina Isakova-Sivak
