 Review
Review

Affiliation:
1Merogenomics (Genomic Sequencing Consulting), Edmonton, AB T5J 3R8, Canada
ORCID: https://orcid.org/0009-0003-5755-3415
Affiliation:
2University of Lincoln, LN6 7TS Lincoln, UK
ORCID: https://orcid.org/0009-0008-0109-3495
Affiliation:
3Department of Biological Science, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
4Therapeutic and Protective Proteins Laboratory, Protein Research Department, Genetic Engineering and Biotechnology Research Institute, City of Scientific Research and Technology Applications, New Borg El-Arab 21934, Egypt
ORCID: https://orcid.org/0000-0001-8246-0075
Affiliation:
5Department of Molecular Medicine and USF Health Byrd Alzheimer’s Research Institute, Morsani College of Medicine, University of South Florida, Tampa, FL 33612, USA
Email: vuversky@usf.edu
ORCID: https://orcid.org/0000-0002-4037-5857
Affiliation:
6Autlan Regional Hospital, Health Secretariat, Autlán, JAL 48900, Mexico
7Biology Laboratory, Autlan Regional Preparatory School, University of Guadalajara, Autlán, JAL 48900, Mexico
Email: alberto.rubio@sems.udg.mx
ORCID: https://orcid.org/0000-0003-2978-5107
Explor Immunol. 2024;4:267–284 DOI: https://doi.org/10.37349/ei.2024.00140
Received: December 07, 2023 Accepted: March 20, 2024 Published: April 25, 2024
Academic Editor: Dominique J. Charron, Hospital Saint Louis, France
The article belongs to the special issue Old and New Paradigms in Viral Vaccinology
Repeated inoculation with messenger RNA (mRNA) vaccines elicits immunoglobulin G4 (IgG4) antibody production. Such an increase in the concentration of specific and non-specific IgG4 antibodies allows the growth of some types of cancer by blocking the activation of effector immune cells. This work proposes the hypothesis that cancer growth may be indirectly promoted by increased concentrations of non-specific IgG4 antibodies by the following mechanisms: 1) IgG4 antibodies can bind to anti-tumor IgG1 antibodies and block their interaction with receptors located on effector cells, thus preventing the destruction of cancer cells, 2) IgG4 can interact with fragment crystallizable gamma receptor IIb (FcγRIIB) inhibitory receptors, thus reducing effector functions of innate immune cells, and 3) targeting of specific epitopes by IgG4 could be oncogenic by inducing the production of a microenvironment that can promote cancer development. This article reviews the supporting literature and suggests several experimental protocols to evaluate this hypothesis in the context of repeated inoculation with mRNA vaccines. Additionally, this work proposes some management options aimed at reducing the unfavorable molecular consequences that could mediate cancer development when encountering high concentrations of IgG4 antibodies.
Timely vaccination is considered an essential tool to cope with pandemic crises [1, 2]. The first two messenger RNA (mRNA) vaccines authorized for administration to humans during the current pandemic were BNT162b2 from Pfizer/BioNTech and mRNA-1273 from Moderna [3]. Some studies have found that humoral responses evaluated after the initial two dose protocol were less effective than antibody responses observed following the third inoculation in terms of their neutralizing power towards a wide variety of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) variants of concern [4–6]. Additionally, it was shown that the strength of antibody-antigen-binding was enhanced after receiving an mRNA booster [7]. This discovery was somewhat defined by persistent B cell development and prolonged germinal center activation [7]. After administering the first two mRNA injections, scientists have shown that the antibody subclasses immunoglobulin G1 (IgG1) and IgG3 instantly predominated the IgG response [8]. Nevertheless, six months after the second BNT162b2 injection, a third mRNA dose enhanced spike-specific antibodies [8].
However, after the second mRNA vaccine injection, an unexpected long-term side effect has been observed worldwide: a switch in the isotype of IgG antibodies took place. Before the emergence of these observations in the general mRNA vaccinated population, this phenomenon appeared to have been documented in single individuals and described as a rare vaccine side effect; either producing IgG4-related disease (RD) [9–13] or experiencing a relapse of IgG4-RD symptoms [14]. The phenomenon of rising IgG4 antibodies post mRNA vaccination has now been documented in studies involving human participants [8, 15–28] and at least one animal study [29]. It appears that the rate of increase of IgG4 antibodies can surpass all other IgG antibodies developed towards the spike protein, rising consistently from an average of 0.04% after the second immunization to 19.27% after the third one [8]. This was echoed by another study, where the median level of IgG4 antibodies directed against the spike protein was 21.2% of all IgG antibodies [19]. In stark contrast, this phenomenon has not been reported in non-vaccinated individuals [16, 17, 23, 25, 27].
Several investigations have now demonstrated that this response is only produced by the mRNA-based vaccines (Pfizer/BioNTech or Moderna); individuals who received adenoviral vector-based or protein-based vaccines did not generate such a rise in IgG4 concentrations [8, 15, 16, 18–20, 26]. Heterologous use of SARS-CoV-2 vaccines (mixing mRNA vs. adenoviral vector vaccines) appears to lead to marginal increases in IgG4 [15, 18, 24] or none at all [8, 20]. The difference in these observations could be due to the timing of measurement, as another emergent trend is that the longer the time lapse between the last mRNA vaccination and sample measurement, the more likely IgG4 antibodies are to be detected.
The most frequent observation of IgG4 antibodies is post-booster shot (third mRNA injection) [8, 16–20, 22–25, 27, 28]. Subsequent injections maintain or further increase the antibody levels [23–25, 28]. Finally, a breakthrough infection can help induce IgG4 antibody production. Once again, infections after the third injection result in more pronounced outcomes than after the second mRNA injection [8, 16–18, 20, 23]. Thus, it appears that two factors influence the levels of IgG4: 1) time span after the second mRNA injection and 2) continued exposure to spike protein antigen, whether through boosting or infection, with data available up to a total of seven antigen exposures [28]. The likelihood of IgG4 production could also be dependent on the type of mRNA vaccine used, current data hints that Moderna injections are more likely to elicit greater IgG4 antibody levels than Pfizer/BioNTech vaccine [15, 26].
This observation also adds support to the notion that a significant contributing factor to IgG4 development is the over-presentation of the antigen. Since Moderna vaccines delivered a greater amount of genetic material coding for the spike protein than the Pfizer/BioNTech injections, they had a greater capacity to elicit the production of a greater amount of spike protein antigen [15, 26]. Hence Moderna injections could have a greater likelihood of inducing IgG4 antibody production. Another important factor could be the antigen delivery method. The accumulating evidence is mounting indicating that significant spike-specific IgG4 antibody concentrations are induced by mRNA vaccines only [8, 15, 16, 18–20, 26]. Contrarily, a strong IgG4 spike-specific response was not generated by those individuals who have gotten mRNA vaccines after being infected with SARS-CoV-2 [15, 16, 18, 25, 30], and also those who received viral vector-based vaccines [8, 15, 16, 18–20], or protein-based vaccines [26], did not generate a strong IgG4 spike-specific response.
It has been demonstrated that IgG2 and IgG4 antibodies have a reduced capacity to activate fragment crystallizable gamma receptor (FcγR)-dependent effector activities [31]. Therefore, Irrgang et al. [8] performed an antibody-dependent cellular phagocytosis (ADCP) test using the human leukemia monocytic THP-1 cell line. They verified that anti-spike IgG3 and IgG1 are more powerful in eliciting phagocytosis than IgG4 and IgG2. They also found that the interaction of IgG2 and IgG4 resulted in lower activation of the FcγRIIA, which is an essential regulator of ADCP. After the third inoculation, there was a decrease in antibody-dependent phagocytic responses and complement accumulation in blood serum, along with a rise in the percentage of anti-spike IgG4 antibodies [8].
On the other hand, Selva et al. [18] documented activation of the FcγRIIA in fewer individuals who had three mRNA injections as compared to those who recovered from coronavirus disease 2019 (COVID-19) prior to taking two mRNA immunizations. In addition, they also showed that immunized only individuals without prior infection had lower activation of the FcγRIIIA in comparison to those who contracted SARS-CoV-2 before immunization [18]. A comparable pattern has also been discovered in macaque animal models [29], where three immunizations with Moderna mRNA vaccine, or mRNA shots followed by a Novavax protein-based vaccine booster resulted in very low levels of ADCP and an antibody-dependent neutrophil phagocytosis (ADNP) with further concomitant reduction of these processes in some of the tested animals. In contrast to this, antibody-dependent complement deposition (ADCD) appeared to be normal with only limited reduction over time. Farkash et al. [21] showed that reduced complement binding occurred as early as 5 weeks after the second mRNA injection, while effector functions via FcγR were not affected. However, functional studies such as ADCP/antibody-dependent cellular cytotoxicity (ADCC) assays were not carried out. Finally, Kalkeri et al. [26] demonstrated reduced capacity to induce ADCP, ADCC, and ADCD with Novavax immunization after three mRNA vaccine injections in comparison to after three Novavax inoculations. Together, these studies suggest that fragment crystallizable (Fc)-mediated effector functions are reduced concomitant with an increase of IgG4 antibodies.
With elevated IgG4 concentrations, the cancer risk is not just theoretical. In patients with hepatocellular carcinoma, it was demonstrated that IgG4 concentrations were elevated in those individuals with tumor recurrence and poor clinical outcomes; elevated serum IgG4: IgG ratio can serve as a novel prognostic predictor for hepatocellular carcinoma patients undergoing resection [32].
Elevated IgG4/IgG circulating levels in melanoma patients also correlated with worse progression-free survival and overall survival. Of the studied tissue samples, infiltration by IgG4 plasma cells was seen in 43% of melanomas, 21% of involved lymph nodes, and 30% of metastases [33]. Later more advanced stages of melanoma were more likely to exhibit higher serum levels of IgG4 (55% for stage 3 vs. 19% for stage 1) [34]. Patients with esophageal cancer also have been reported to exhibit increased serum levels of IgG4 and infiltrating IgG4 plasma cells and higher levels correlated with more aggressive cancer growth and worse prognosis [35].
Increased IgG4+ cell levels were reported in gastric cancer, once again, with higher levels linked to poorer prognosis [36]. The authors also noted IgG4+ cell levels correlating to gross tumor appearance, tumor depth, lymph node metastasis, venous invasion, and lymphatic invasion [36]. Similarly, higher IgG4 levels in the serum and higher IgG4/IgE ratios were observed in patients with metastatic colorectal cancer than non-metastatic patients [37]. Tissue samples from these patients indicated that IgG4 was found very near to macrophages and that IgG4s were inducing these macrophages to express cell surface and secretory markers characteristic of an immune-suppressive state that could promote a pro-tumorigenic microenvironment. The macrophages also exhibited reduced effector functions [37].
There is presently no targeted treatment available for triple negative breast cancer (TNBC), a highly malignant form of the disease. Although they have been detected in the tissues of TNBC patients, tumor-infiltrating B cells are unknown in terms of their intended function [38]. Scientists wanted to know if there were any IgG4+ B cells and what processes led to the change in isotype in TNBC. According to research, interactions between tumor-infiltrating B cells and TNBC activate long-term inflammatory mediators like interleukin-10 (IL-10) and IL-4, which cause class switching to an IgG4+ subtype and potentially inhibit antibody-mediated immunological responses. IgG4+ B cell counts could be used as a biomarker for an adverse clinical outcome [38].
Immune modulation towards the type 2 helper T (Th2) cells immunity bias was reported in glioblastoma patients as measured by increased serum levels of IgG4, M2 monocyte cells, IL-4, IL-10, and IL-13 [39]. Finally, high levels of serum IgG4 have also been reported in cholangiocarcinoma [40] and pancreatic cancer [41–43], while higher tissue IgG4 levels have been reported in papillary thyroid carcinoma [44], extrahepatic cholangiocarcinoma [45], and pancreatic cancer [46].
Most crucially, however, it is the “hyperprogressive disease” phenomenon that has been associated with some cancer treatments involving IgG4 antibodies such as nivolumab, which uses IgG4 with a stabilizing S228P mutation [35]. One case of rapidly growing renal tumor post mRNA vaccination that was accompanied by high levels of IgG4 has been described [12]. Furthermore, several examples of development, growth, worsening, and spontaneous regression of T cell lymphoma in humans have been documented following mRNA-based SARS-CoV-2 immunization [47–53].
The emerging hypothesis is that cancers can trigger IgG4 production for the purpose of evading immune recognition by inhibiting antibody-mediated anti-cancer responses [35, 40]. The hypothesis of how the immune checkpoint IgG4 inhibitors might lead to hyperprogressive disease is that inhibition of anti-cancer immune responses is mediated at the level of the Fc region of the IgG4 antibody, not by means of the antigen-binding fragment (Fab) [35, 54]. Furthermore, the undesired effect of IgG4 in the cancer microenvironment is exacerbated by glutathione antioxidant which aids in IgG4 Fc conformational changes favoring the hyperprogressive disease state [55]. In such conditions, hyperprogressive disease appears to be driven through IgG4 Fc interference with Fc portions of other cancer-targeting IgGs [35, 55] and via blocking access to Fc receptors of effector cells [54], thus reducing these important anti-cancer processes and leading to immune escape.
Proposed mechanisms by which IgG4 antibodies could inhibit anti-cancer responses.
The possible role of IgG4 Fc component in cancer development has been observed even prior to the currently proposed etiology of hyperprogressive state [56]. To investigate possible ways by which IgG4 may impact anti-tumor outcomes, Karagiannis et al. [56] created two antibodies with IgG1 and IgG4 Fc domains that shared the exact same variable regions that identified the melanoma-associated antigen chondroitin sulfate proteoglycan 4 (CSPG4). This was done to simulate a therapeutically relevant scenario in which IgG1 and IgG4 were detected separately or in combination in the presence of 1) cancer cells that expressed tumor antigens and 2) immune effector cells, such as patient monocytes and macrophages that expressed the FcγR. The two antibodies bound to melanoma cells, to patient monocytes, and to U937 monocytes that expressed the FcγR receptor [56]. The cancer-killing ability of anti-CSPG4 IgG1, formerly identified in ADCC and/or ADCP tests, was considerably diminished in the presence of anti-CSPG4 IgG4 when anti-CSPG4 IgG1 and anti-CSPG4 IgG4 were mixed with patient-derived monocytes and melanoma cells. This demonstrated that the anti-CSPG4 IgG4 antibody is not only unable to activate effector cell-driven malignant tumor elimination but can also inhibit tumor-specific IgG1’s ability to destroy tumor cells [56].
These observations were also supported by the work of Jordakieva et al. [37]. The authors also generated two cetuximab antibodies with either IgG1 or IgG4 Fc domains but targeting the same epidermal growth factor receptor (EGFR) antigen. Antibodies with IgG4 Fc domains were less capable of killing EGFR-expressing tumor cells via ADCP than antibodies with IgG1 Fc domains.
According to a recent study, IgG4 antibodies (independently of their antigen specificity) impaired the typical immune responses of ADCP, complement-dependent cytotoxicity (CDC), and ADCC against cancer cells in vivo and in vitro [35]. Such responses were mediated by the Fc region of the IgG4 antibodies reacting with the Fc region of IgG1 antibodies that have been attached to cancer antigens. Without knowing their antigen specificity, researchers investigated IgG4 obtained from intravenous IgG, genetically modified IgG4 purchased from several commercial sources, and IgG4 taken from the serum of cancer patients and healthy individuals. They all induced a similar inhibiting action on such immune responses [35].
These findings demonstrated that IgG4 antibodies are essential components in local immune escape by inhibiting immunological responses regulated by cancer-specific IgG1 antibodies (Figure 1).
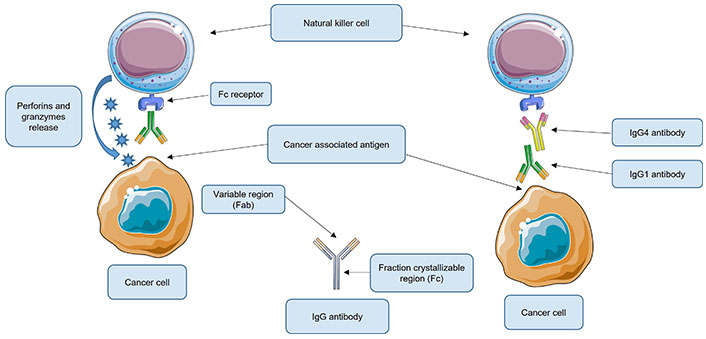
Roles of immunoglobulin G4 (IgG4) antibodies in local immune escape. Under normal conditions, the IgG1 antibody recognizes the cancer-associated antigen. IgG1 then binds through the fragment crystallizable (Fc) to its receptor, located on the natural killer (NK) cells (shown) or macrophages (not shown). These are activated and NK cells release perforins and granzymes that destroy the cancer cell while macrophages phagocytose the cancer cell. However, when present, IgG4 could bind to the Fc region of the IgG1 antibody, thus inhibiting its union with its respective receptor located on the NK or macrophage cell. As a consequence, these effector cells are not activated and therefore cannot destroy the cancer cell. Fab: antigen-binding fragment. This Figure was created using Servier Medical Art (https://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License
To learn more about the mechanism that these antibodies regulate, scientists conducted in vivo research utilizing mouse models with functional immune systems [35]. In one model, IgG4 antibodies that were not cancer-specific were injected into the subcutaneous injection location for breast cancer cells. Cancer cell growth was drastically enhanced in this group of mice (Figure 2). They generated a much bigger cancerous growth by 21 days when compared to other cohorts (injections of inert phosphate buffered saline or IgG1 without IgG4), and demonstrated that the IgG4 antibodies (regardless of Fab region specificity) can inhibit regional immune reaction and thus foster cancer growth in vivo via immune evasion [35]. Using such preclinical models, other researchers later confirmed the additive role of glutathione driving the IgG4 effect on cancer progression [55].
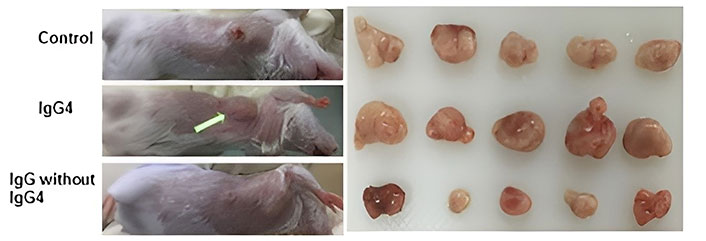
Immunoglobulin G4 (IgG4) antibodies enhance breast cancer cell proliferation. In the animal model, IgG4 antibodies that are not cancer-specific enhanced breast cancer cell proliferation. When immune-competent mice were locally injected with mouse breast cancer cells, large tumor masses developed after 21 days. When IgG4 antibodies that are not cancer-specific were locally co-administered, tumor masses doubled in size compared to animals who received equivalent injections of IgG1 without IgG4 or phosphate buffered saline (control group)
Note. Adapted with permission from “An immune evasion mechanism with IgG4 playing an essential role in cancer and implication for immunotherapy,” by Wang H, Xu Q, Zhao C, Zhu Z, Zhu X, Zhou J, et al. J Immunother Cancer. 2020;8:e000661 (https://jitc.bmj.com/content/8/2/e000661). CC BY-NC.
It has even been suggested that melanoma tumor cells could directly attempt to influence IgG4 production by tumor-derived B cells [56]. The rise in IgG4 was accompanied by Th2-biased inflammatory cytokines including IL-4 as expected in the class switch towards IgG4. The authors postulated that tumor cells might induce the production of IgG4 antibodies as a mechanism to avoid immune detection [56].
Another possible way that IgG4 antibodies could reduce the effector functions of other cancer-targeting antibodies is by interacting with FcγRIIB receptors [57]. FcγRs are either activating or inhibitory in their function. Currently, FcγRIIB receptors are the only known inhibitory ones. IgG4 antibodies happen to bind to FcγRIIB receptors with either similar or higher affinity than the other subclasses of IgG antibodies [57]. Therefore, IgG4’s ability to interact with FcγRIIB receptors could allow them to reduce the desired effector functions of other subclasses of antibodies, even if such antibodies are still binding to activating receptors. This also suggests that IgG4 antibodies could exert their effects even when presenting at lower concentrations than other antibodies.
This precise mechanism of action has also been proposed for the development of hyperprogressive disease. Lo Russo et al. [54] showed that all biopsies of patients with hyperprogressive disease showed tumor infiltration by M2-like CD163+CD33+ programmed death-ligand 1 (PD-L1)+ epithelioid macrophages that tended to cluster together. These results were recapitulated in a mouse model treated with nivolumab [a monoclonal antibody immune checkpoint inhibitor that binds to programmed cell death protein 1 (PD-1)]. Crucially, the Fab component of nivolumab without the Fc domain no longer induced hyperprogressive-like disease in mice. These results suggest that hyperprogressive disease is also partially triggered by nivolumab interaction with FcγRIIB on M2-like clustered macrophages [54]. Other group later also showed that the M2-like phenotype is at least partially induced by the cancer cells themselves via exosomes [58].
It is hypothesized that IgG4 antibodies can also impair anti-tumor responses by activating FcγRIIB receptors located on macrophages or natural killer (NK) cells (Figure 3).
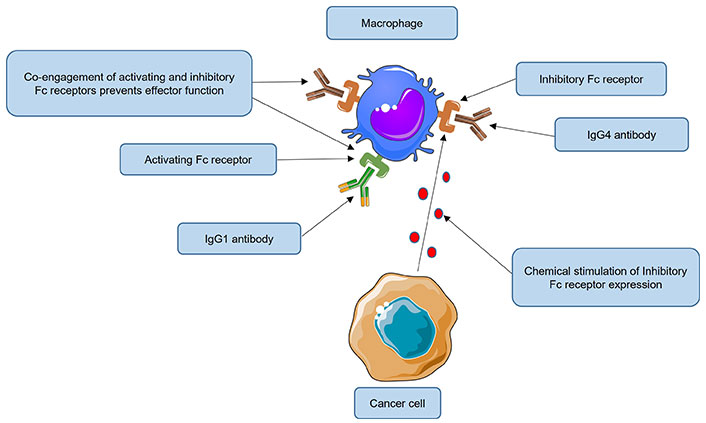
Impairment of anti-tumor responses by immunoglobulin G4 (IgG4) antibodies engaging inhibitory FcγRIIB receptors. In normal circumstances, macrophages can recognize and eliminate cancer cells by phagocytosis. Tumors nevertheless continue to grow despite their presence. Some cancers have evolved immune evasion mechanisms, for example, by inducing over-expression of the fragment crystallizable gamma receptor IIb (FcγRIIB) receptor on macrophages. High levels of IgG4 could activate this inhibitory receptor (when the same cell is co-engaged with an activating FcγR receptor), inducing a state of anergy or even apoptosis of effector cells. It is hypothesized that IgG4 antibodies, by engaging FcγRIIB receptors located on macrophages can also impair anti-tumor responses, thus reducing their phagocytic functions. Fc: fragment crystallizable. This Figure was created using Servier Medical Art (https://smart.servier.com), licensed under a Creative Commons Attribution 3.0 Unported License
Macrophages are extremely effective phagocytes that are capable of devouring pathogens, debris, and tumor cells [59]. A subpopulation of macrophages known as tumor-associated macrophages (TAMs) is prevalent in the tumor microenvironment [60]. Their significant contribution to tumor growth [61], resistance to treatments [62], and tumor metastasis [63] has established their clinical relevance. The general consensus is that M2 polarized TAMs have an anti-inflammatory phenotype, promote an immunosuppressive milieu, and generate anti-inflammatory chemokines and cytokines to promote tumor growth [64, 65]. With a pro-inflammatory phenotype, M1 polarized TAMs prevent tumor growth by sustaining an environment that is unfavorable to the tumor through the production of pro-inflammatory cytokines. Both M1 and M2 polarized TAMs can phagocytose cancer cells [66], with the former being far superior [67].
It is suggested that increased IgG4 levels could activate FcγRIIB receptors on macrophages and induce macrophage polarization from M1 to M2 and/or M2-like phenotype, thus impairing ADCP and promoting immune evasion.
There is also the possibility that IgG4 antibodies could promote cancer development precisely because they are specific against the spike protein. One such possibility is that IgG4 antibodies could target auto-antigens important for cancer suppression.
Binding such antigens by IgG4 antibodies essentially would prevent them from executing their tasks that are supposed to minimize cancer development. Spike protein is known to exhibit mimicry or geometrical shapes of native human proteins [68–71]. If the mimicry happens to be of a protein with tumor-suppressor activity, producing antibodies against such components of the spike protein could also have dangerous consequences. For example, Kanduc [68] showed that the spike protein can share a pentapeptide sequence with a staggering number of 294 tumor-suppressor proteins. The ramifications of such an overlap of immune targeting are still yet to be understood. Other biological targets based on spike protein mimicry could be pro-oncogenic, and one such possible target might be galectin-3 (Gal-3).
Research from 2019 by Perugino et al. [72] links Gal-3-associated residues to IgG4 class switching. They used mass spectrometry to analyze the Ig sequences of single-cell clones from patients with IgG4-RD and identified Gal-3 as the primary antigen recognized by both IgG4 and IgE. Twenty eight percent of patients in the IgG4-RD cohort demonstrated IgG4-specific anti-Gal-3 antibody responses, with minimal to nonexistent response from other IgG subclasses. IgG1, IgG2, and IgG3 isotypes did not exhibit any significant responses, and anti-Gal-3 autoantibodies were associated with increased concentrations of Gal-3, lymphadenopathy, as well as total IgG and IgG4 increase [72]. Perugino et al. [72] concluded that elevations in IgG4 and IgE observed clinically were due to the development of IgG4 and IgE anti-Gal-3 autoantibodies, at least in part. They suggest that this may be an attempt to interfere with Gal-3-induced myofibroblast proliferation—to mitigate the fibrotic process [72].
This discovery is significant for two reasons:
First, Gal-3 controls or is connected to a variety of fundamental cellular processes, such as adhesion, differentiation, angiogenesis, and apoptosis. As a consequence, Gal-3 is thought to have a role in many conditions and diseases [73], especially in some cancers. Its expression levels correlate with cancer aggressiveness, invasion, and metastasis [74]. One of the mechanisms is by stabilizing and extending the half-life of cancer-associated mRNAs [75]. Another mechanism is through the mediation of vascular endothelial growth factor induced angiogenesis [76], promoting cancer metastasis [77].
Second, as with other β-coronaviruses, the N-terminal domain of the S1 subunit (S1-NTD) of SARS-CoV-2 spike protein possesses a Gal fold that bears almost perfect structural homology to the galactose binding domain [78] of human Gal-3 (Figure 4).
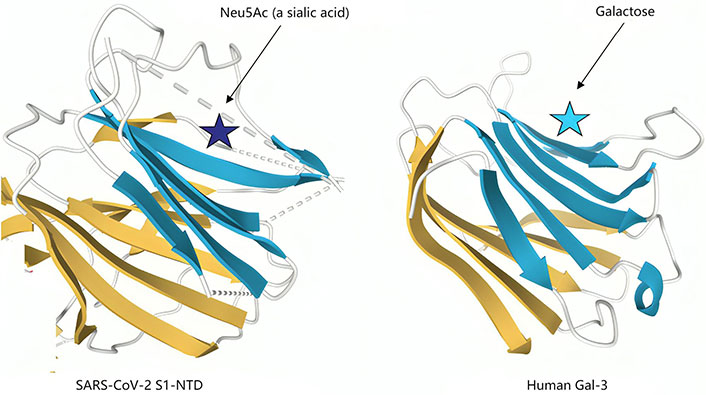
Structural similarities of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) N-terminal domain of the S1 subunit (S1-NTD) and human galectin-3 (Gal-3). The structural topologies of the SARS-CoV-2 S1-NTD (Left panel) and human Gal-3 (right panel) are shown as schematic illustrations, where β-strands are depicted as arrows and α-helices as cylinders. Neu5Ac: N-acetylneuraminic acid
Note. Adapted from “A potential role for Galectin-3 inhibitors in the treatment of COVID-19,” by Caniglia JL, Guda MR, Asuthkar S, Tsung AJ, Velpula KK. PeerJ. 2020;8:e9392 (https://peerj.com/articles/9392/). CC BY.
As Gals can recognize and bind reversibly to β-galactose residues, the evolutionary theory for this is that ancestral coronaviruses may have acquired the genes from a host Gal, which became the S1-NTD of their spikes. Selective pressure would have conserved the ability of spike protein Gal fold to recognize sugar receptors for cell entry [79, 80].
The similarity between the spike protein S1-NTD and Gal-3 was further underscored in 2021 by Ryzhikov et al. [81] who screened which glycan-binding proteins had the highest binding affinities for SARS-CoV-2 spike protein. The top 10 glycans were the typical ligands for human Gals. If Gal-3 levels can be implicated in cancer development, it is not unreasonable to ponder the possibility that indirectly the anti-Gal-3 antibodies themselves could be contributing to cancer development and that this could be achieved through induction of IgG4 antibodies against Gal-3. Therefore, just like the Gal fold of the spike protein mimics the function of Gal-3, the antibodies produced against S1-NTD could mimic the function of anti-Gal-3 antibodies. If targeting Gal-3 with antibodies could be oncogenic due to over-presentation of antigen leading to immunosuppressive IgG4 antibodies development, then the spike protein’s Gal fold area could run the risk of providing oncogenic epitopes in a similar fashion of antigen presentation.
Therefore, if IgG4 antibodies can be directed against Gal-3, potentially as an immunological response due to too much antigen presentation, could the same IgG4 response be observed in mRNA vaccinated? If the spike protein mimics Gal-3 with its Gal fold, then could the mRNA vaccination response mimic that of what is seen in IgG4-RD? The fact that S1-NTD is an important target for neutralizing antibodies, including those induced by mRNA vaccines, has been well established [82, 83]. Di Gaetano et al. [84] identified the crucial target residues in the S1-NTD pocket as E154, F157, Y160, and the “stabilizing loop” of glycans N122–N125. And Cerutti et al. [85] described how 7 N-terminal domain (NTD)-directed neutralizing antibodies targeted a single “supersite” formed by a mobile β-hairpin and several flexible loops, bordered by glycans N17, N74, N122, and N149.
The evidence that vaccinal antibodies included those specific to the Gal fold of S1-NTD includes a paper from 2022 by Caetano-Anollés et al. [86]. They hypothesize that increased infections associated with winter may be due to temperature and environmentally sensitive Gal homologues undergoing mutational bursts. These aid the virus in avoiding the host’s physiological reactions or adjusting them in a seasonal pattern.
They observed that the spike protein’s outer layer was where the majority of mutations were found, involving the ones in the Gal fold structure of the NTD [86]. Whilst the receptor-binding domain (RBD) is the immunodominant epitope, mutations in an “NTD supersite” of residues 14–20, 140–158, and 245–264 [87] can have a disproportionately large effect on antibody binding potential, leading to immune escape and vaccine failure, as seen with Omicron. Of these, residues 140–158 make up the Gal fold of the spike protein [88].
In summary, when developing an mRNA vaccine platform restricted to epitopes of the spike protein, one cannot avoid needing to induce antibodies to the S1-NTD, if neutralizing antibodies are to be used as the primary metric of efficacy. More investigation is warranted to confirm the involvement of the SARS-CoV-2 Gal fold in IgG4 class switching and to investigate if other motifs are involved, as well as to detail the specific signaling pathways invoked.
Numerous recently published studies from various geographical locations [8, 15–29] found that mRNA vaccines (two doses plus booster; where the booster could be an additional mRNA injection, natural infection, or possibly an injection of another type of COVID-19 vaccine) elicited a significant percentage of spike-specific IgG4 antibodies.
Together, these studies form a significant validation of each other. Contrarily, people who either 1) remained non-vaccinated; 2) received mRNA vaccines after contracting SARS-CoV-2; 3) received vector-based vaccines or protein-based COVID-19 vaccines; or 4) were on certain immunosuppressive drugs when being immunized with mRNA vaccines, all did not produce an IgG4 spike-specific response, and constituted a protected group from this unexpected side effect of mRNA injections.
The results of the Fc receptor competition studies demonstrated that a significant IgG4 concentration is necessary for competition with IgG1 in attaching to Fc receptors on macrophages and peripheral blood mononuclear cells (PBMCs) [35]. That is precisely what Irrgang et al. [8] found in their study: they observed that in a cohort of Pfizer/BioNTech vaccinees, there was a 1.4-fold increase in IgG1 total antibodies between the second and third immunization while nearly a 39-fold increase in IgG4 antibodies in same period. This clearly indicates an increase in the ratio of IgG4 to IgG1 antibodies. As repeated mRNA vaccination caused a decrease in the percentage of IgG1 antibodies relative to the percentage of IgG4 antibodies [8], the latter could contend with IgG1 in interacting with Fc receptors, thus blocking anti-cancer responses. In addition, IgG4 antibodies could interact with inhibitory Fc receptors, further attenuating the effect of IgG1 antibodies.
In science, the only way to confirm or refute a hypothesis is by performing experiments. This hypothesis may be incorrect, but it needs to be refuted experimentally. It is therefore encouraged that scientists experimentally evaluate this hypothesis. Useful information on how to perform such experiments is found in the previously discussed works [35, 54, 55] of IgG4-induced hyperprogressive disease. Several experimental protocols are proposed that could be undertaken to test this hypothesis:
In vitro assays:
Test anti-spike IgG4 isolated from mRNA vaccinated individuals (with or without added glutathione) for inhibition of cetuximab induced PBMC mediated phagocytosis of cancer cells and CDC.
Extract IgG1 and anti-spike IgG4 antibodies from the serum of mRNA vaccinated cancer patients and test IgG1 reactivity to cancer tissue sections of the same patients, followed by using biotin labeled anti-spike IgG4 antibodies to detect reactivity with cancer tissue bound IgG1s.
Extract PBMCs, IgG1, and anti-spike IgG4 from the serum of mRNA vaccinated cancer patients and test for anti-spike IgG4 concentration-dependent binding competition of IgG1 to PBMC and ADCC cytotoxicity.
Test anti-spike IgG4 isolated from mRNA vaccinated individuals for cross-reactivity with self-antigens.
Animal tests:
Inoculate immune-competent mice subcutaneously with cancer cells or a skin tumor-inducing carcinogen and subsequently inject in the same location with anti-spike IgG4 isolated from mRNA vaccinated individuals. This is compared to a sham (with or without competing IgG), and 21 days later measured for tumor number, incidence, volume, area/tumor as well as mouse weight. Nivolumab can be used as a positive control.
Athymic nude or severe combined immunodeficiency (SCID) mice implanted with human cancer cells or patient-derived xenografts are treated either intraperitoneally or peritumorally with IgG4 anti-spike antibodies isolated from mRNA vaccinated individuals, and about 21 days later measured for tumor parameters.
Patient tests:
Compare serum IgG4 and IgG4: IgG total levels in non-vaccinated cancer patients vs. mRNA vaccinated cancer patients vs. mRNA vaccinated non-cancer patients by stages of cancer.
Tumor tissue of non-vaccinated vs. mRNA vaccinated is compared for the M2/M1 macrophage ratio (ratio of CD163+ cells/CD86+ cells) and CD163+CD33+PD-L1+ tumor-infiltrating macrophages showing epithelioid morphology with the tendency to form dense clusters.
Immunohistochemistry for IgG4 antibodies or IgG4+ lymphocytes in cancer tissue of non-vaccinated vs. mRNA vaccinated patients in comparison with tissue more distant to the cancer mass and normal mucosa tissue.
One condition of particular interest in the context of emerging mRNA vaccination side effects of increased IgG4 levels is the IgG4-RD, which also frequently presents with increased IgG4 levels. It has been proposed that longstanding IgG4-RD may progress to malignancy [89]. A meta-analysis and systematic review of individuals with IgG4-RD found that standardized incidence ratios were significantly increased, i.e., 4-fold for pancreatic cancer and 69-fold for lymphoma [90]. It is suggested that after the launch of the widespread COVID-19 immunization program involving mRNA injections, lymphoma is unquestionably the sentinel condition to be attentively observed at the population level.
In the event that the mRNA injections lead to clinical complications similar to those observed with IgG4-RD, including promoting cancer development reminiscent of hyperprogressive disease, how should such a situation be monitored, and what can we learn from published literature about available options countering the effects of high levels of IgG4? In light of the potential mechanisms of pathological outcomes outlined here, the important question is what are the levels of IgG4 antibodies that should alert the healthcare professionals for potential clinical oversight of affected mRNA gene therapy vaccinated individuals?
While it remains to be determined at what serum concentrations IgG4 levels are pathological in nature in hyperprogressive disease patients, it should be noted that the IgG4 levels observed in cancers, while often exceeding the minimum required toward IgG4-RD diagnosis, can be substantially lower than what can be observed with IgG4-RD patients [91].
Thus, it could be assumed that the IgG4 antibody levels seen in IgG4-RD would be an appropriate starting point to commence monitoring the developing situation. The IgG4 serum levels are one of the most frequent markers investigated, with levels from 1.35 mg/mL to 2.48 mg/mL of IgG4 proposed by different authors [92] with 1.35 mg/mL value now recognized as part of the diagnostic criteria of IgG4-RD [93]. It should be noted, however, that different concentration values can be obtained depending on the reagents used for IgG4 measurements [94]. Another proposed measurement criterion is to consider the IgG4/IgG ratio with levels above 10% being indicative of the condition with even higher specificity [92].
It is proposed that physicians start taking note of IgG4 levels in all their mRNA vaccinated patients. A rise in such levels might be alert to start screening for a potential genetic predisposition to hyperprogressive disease as has previously been observed [32–37]; affected genes [such as Kirsten rat sarcoma viral oncogene homologue (KRAS), serine/threonine kinase 11 (STK11, also known as LKB1), mouse double minute 2 (MDM2)/MDM4, and EGFR] appear to play a role in the configuration of oxidative stress observed in the tumor microenvironment, including via glutathione metabolism [95]. Such individuals perhaps could benefit from increased monitoring frequency for cancer development. Investigations also need to be conducted to determine which epitopes on the spike protein likely mediate IgG4 class switching and by what degree and conformation.
The potential treatment strategies borrow from different aspects of IgG4-RD and cancer therapy: Glucocorticoids are typically the initial medication choice, but they may not lead to complete remission in all patients [96, 97]. Rituximab offers a promising alternative, especially for those with recurrent or refractory disease [97, 98]. However, treatment success can be influenced by the extent of fibrosis present in affected organs [97]. In mRNA vaccinated individuals with elevated IgG4 levels, further investigation is warranted due to the potential pro-fibrotic effects of the spike protein due to its potential mimicry of Gal-3 which is known to be pro-fibrotic [99]. With regard to glutathione, different therapeutic approaches with either direct or indirect targeting in cancers have been proposed [100]. Oxidized vitamin C has previously been shown to deplete glutathione in cancer cells, leading to the death of cancer cells due to oxidative stress injury [101]. However, this effect was only observed against specific genetic backgrounds [101]. In terms of M2-like macrophage reduction, various strategies have also been proposed in conjunction with the use of immune checkpoint inhibitors, including clodronate used to reduce the occurrence of bone metastasis [102].
In conclusion, healthcare authoritative bodies should commence monitoring the effect of IgG4 antibody levels in the populations to start assessing the true impact of this unsuspected long-term side effect, including the impact of subsequent boosting and natural infections. As alluded to earlier, based on prior literature, lymphoma cancer rates in such groups might possibly indicate such undesirable non-specific effects. Finally, it is also proposed that the scientific and medical communities expand the investigation of how to potentially treat this phenomenon reminiscent of IgG4-RDs now observed in certain mRNA vaccinated individuals. In turn, vaccine developers might consider further investigation into optimal genetic content quantity and delivery to minimize the potential risk of inducing IgG4 class switching in vaccine recipients.
ADCP: antibody-dependent cellular phagocytosis
COVID-19: coronavirus disease 2019
CSPG4: chondroitin sulfate proteoglycan 4
EGFR: epidermal growth factor receptor
Fab: antigen-binding fragment
Fc: fragment crystallizable
FcγRIIB: fragment crystallizable gamma receptor IIb
Gal-3: galectin-3
IgG4: immunoglobulin G4
IL-10: interleukin-10
mRNA: messenger RNA
NK: natural killer
NTD: N-terminal domain
PBMCs: peripheral blood mononuclear cells
RD: related disease
S1-NTD: N-terminal domain of the S1 subunit
SARS-CoV-2: severe acute respiratory syndrome coronavirus 2
TAMs: tumor-associated macrophages
TNBC: triple negative breast cancer
MR: Conceptualization, Validation, Writing—original draft, Writing—review & editing. DC: Conceptualization, Validation, Writing—review & editing. EMR: Validation, Formal analysis, Data curation, Writing—review & editing. VNU: Validation, Formal analysis, Data curation, Writing—original draft, Writing—review & editing, Supervision. ARC: Conceptualization, Validation, Formal analysis, Data curation, Writing—original draft, Writing—review & editing, Visualization, Supervision. All authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Marc H.V. Van Regenmortel
Christine Jacomet
Vladimir N. Uversky
Brent Brown ... Ingo Fricke
Chittaranjan Baruah ... Bhabesh Deka
Brent Brown ... Enrique Chacon-Cruz
Om Saswat Sahoo ... Subhradip Karmakar
Ankit Kumar ... Vijay Mishra
Yulia Desheva ... Irina Isakova-Sivak
