 Review
Review

Affiliation:
Diagnostic & Research Institute of Pathology, Medical University Graz, 8036 Graz, Austria
Email: helmut.popper@medunigraz.at
ORCID: https://orcid.org/0000-0003-4970-1265
Explor Immunol. 2024;4:285–308 DOI: https://doi.org/10.37349/ei.2024.00141
Received: August 30, 2023 Accepted: February 28, 2024 Published: April 30, 2024
Academic Editor: Jinming Han, Capital Medical University, China
Dendritic cells (DCs) are part of the specific and innate immune system and present antigens for lymphocytes but also regulate the actions of cells of the innate immune system such as eosinophilic and neutrophilic granulocytes. There are several types of DCs, which might have opposite functions: some enhance an immune reaction, and others activate regulatory T (Treg) lymphocytes and thus can induce tolerance towards an antigen. Normally DCs migrate to regional lymph nodes and there they present modified antigens to cells of the immune system, however, in disease this might not function, resulting in the accumulation of DCs. The role of DCs in lung disease has not gotten much attention in the past, as investigations were predominantly focused on lymphocytes, macrophages, and granulocytes. Only in the last decades, DCs been more recognized. Several investigations are focusing on their role in immunotherapy in lung cancer, another focus is on inflammatory disorders including infections and allergies. In this review, non-tumor and non-infectious lung diseases with a focus on smoking-induced, autoimmune, and allergic diseases are discussed.
Dendritic cells (DCs) are part of the immune system, presenting antigens to immune cells. Within the lung different types of DCs are present, and all of them exert different functions. The cells are known as conventional or classical DC (cDC), monocytoid DC (mDC), interdigitating DC (iDC), follicular DC (fDC), plasmocytoid DC (pDC), and Langerhans cell (LHC). The great majority of them express common markers, such as S100 protein and HLA-DR, but can also express more specific markers, which helps in sorting them into disease states (Figure 1A and B) [1–3]. Conventional ones can be stained in tissues by cluster of differentiation 1c (CD1c), CD11c, and CD33, monocytoid by CD163 and CD64, interdigitating by CD14 and CD83, follicular by CD21 and CD35, plasmocytoid by CD123 [interleukin-3 receptor α (IL-3Rα)], CD303, CD304, and Fas death factor ligand (FasL), and LHC by CD1a and langerin [4–7]. Also, immature DCs (imDCs) are known, thought to be responsible for signaling tolerance during development, but these imDCs can also be found as resting cells in all organs [8]. imDCs can promote regulatory T (Treg) cells and induce T cell anergy. They will stain for common DC markers, but may also express CD1c and CD303. imDC expresses CD40 and CD83, but when exposed to antigens they undergo maturation upregulating major histocompatibility complex (MHC) class I and II molecules (for markers of DC subtypes see Table 1). Most DC will stain for common DC markers, such as S100 protein, and HLA-DR; staining for CD14 and CD15 can also be seen in a majority, but there are exceptions. CD68 and CD163 are usually expressed on macrophages but may stain some DCs. There are many more markers for subsets of DCs outside the CD classification, which are not included here. In addition, during migration to lymph nodes, they also will express CD54 and CD86 [9, 10]. Some authors separate histiocytes such as LHCs from DCs, which in my opinion does not make sense, as the primary function of these cells is similar to DCs. Macrophages will be excluded, although they can function as antigen presenting cells, because their predominant function is phagocytosis and maintaining homeostasis within the peripheral lung. Despite existing markers for subsets of DCs, it is well known, that they can change their phenotype. For example, imDC when exposed to antigens or drugs, can change into either cDC or pDC depending on factors especially different cytokines within the stroma [11, 12].
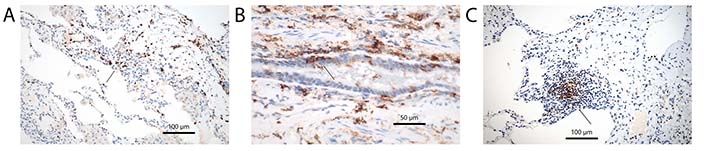
General and specific markers characterizing DCs. (A) S100 protein antibodies, arrow point to DC clusters. Immunohistochemistry, bar 100 µm; (B) antibodies for HLA-DR, arrow point to intraepithelial DCs. Immunohistochemistry, bar 50 µm; (C) antibodies for CD35 characterizing fDCs, the DCs are closely associated with unstained lymphocytes, arrow point to a DC cluster. Immunohistochemistry, bar 100 µm
DC subsets and their surface markers
| Subtype of DC | Surface markers |
|---|---|
| cDC | CD1c, CD11c, CD33 |
| mDC | CD163, CD64, CD14, CD11b |
| iDC | CD14, CD83 |
| fDC | CD21, CD35 |
| pDC | CD123, CD303, CD304, FasL |
| LHC | CD1a, langerin (CD207) |
| imDC | CD1c, CD303 |
In this review, a focus will be on the occurrence of DCs in asthma, chronic obstructive lung disease, LHC histiocytosis (LHCH), other smoking-related interstitial lung diseases, autoimmune diseases, and hypersensitivity pneumonia (HP). The role of DCs in infectious pneumonia will not be discussed in detail, because such discussion would require comparing different subpopulations of DC acting in viral, bacterial, and parasitic diseases, and the role of different signal cascades, selected by different Toll-like receptors (TLRs). This too much would expand this review.
Mouse models will not be discussed for several reasons: Models of hypersensitivity are predominantly based on ovalbumin sensitization, which does not recapitulate HP in humans; models for asthma also focus on specific features of the disease, such as effects of nerve receptors or muscle contractibility; cigarette smoke exposure in laboratory animals focused predominantly on the induction of tumors. Due to anatomical differences, these models cannot reproduce the spectrum of human smoking-associated lung diseases. In rodents, bronchial branching is dichotomous in contrast to asymmetrical branching in humans, which results in deposition of the toxins and carcinogens in the peripheral lung, in contrast to humans with additional impaction at bifurcations. Another problem is that in humans the CD classification of antigens is not always used in experimental mouse research, limiting comparison. Finally, there are many differences in the immune system between mice and humans, which also makes a comparison difficult.
PubMed was searched for reports dealing with DCs in lung diseases, to this, personal experience derived from diagnosing patient cases presenting with one of the below-discussed diseases has been added.
In human DCs originate within the hematopoietic system, mainly bone marrow, and enter the circulation most often in an immature state. They populate not only lymph nodes but also non-lymphoid organs, such as the lungs. These DCs are constantly produced and released from the bone marrow [13]. They can be found along the bronchial tree but also in the peripheral lung. Maturation is induced again by antigen exposure, including haptens, but also by cytokines released from other cells [14]: examples are tumor necrosis factor-α (TNF-α), IL-1, or lipopolysaccharide (LPS), which induce maturation of CD14-positive DCs [15]. A very important interaction of molecules for the maturation of DCs is the CD40-CD40L system. CD40 is expressed on B lymphocytes, whereas CD40L is expressed on activated CD4+ T lymphocytes [16, 17]. Exposure of imDC to CD40-CD40L complexes induces maturation followed by a downregulated antigen uptake and an upregulation of MHC complexes. This will force DCs to migrate with their antigen load to regional lymph nodes, where they present processed antigens to lymphocytes (under normal conditions); in the lung under pathological conditions’ antigen presentation can also happen in tertiary lymph follicles (Figure 1C). Formation of tertiary lymph follicles is usually a sign of non-resolving inflammation, and in the setting of autoimmune diseases might be a response to self-antigen amplification [18]. The C-X-C motif chemokine ligand 13 (CXCL13) and C-C motif chemokine ligand 21 (CCL21) are involved in this type of reaction and cDCs play a prominent role in the early phase [19, 20]. Besides cDCs also fDCs are important. These fDCs provide a long lasting ‘depot’ of antigens and interact with B cells helping the maturation of B cell receptors against the antigens [21]. When stimulated DCs upregulate C-C chemokine receptor 7 (CCR7), which regulates migration to lymph vessels also within secondary lymphoid organs [22]. Migration of DCs is facilitated by the rearrangement of cytoskeletal proteins, adhesion molecules, and cytokine receptors [23]. If a normal antigen presentation can occur only in lymph nodes, and if DCs entering tertiary lymphoid structures will not induce a normal immune reaction is unknown and needs further investigation [24].
Induced pluripotent stem cells (iPSC) have been used to stimulate the differentiation of various immune cells. T cells, natural killer (NK) cells, macrophages, and DCs can be grown in culture [25]. However, in contrast to precursor DC derived from bone marrow iPSC derived DC cannot be induced to stimulate a Toll-receptor-mediated inflammatory cytokine response and do not activate T cells. These cells first need to be transdifferentiated into hematopoietic stem cell precursors, before being stimulated to become DCs [26].
A number of DC subpopulations have been identified in the lung, including myeloid or conventional DCs that initiate T cell immunity and antibody production and plasmacytoid DCs that have an important role in antiviral immunity and immune tolerance [27]. And there are still reports coming up subdividing myeloid DCs into type 1 [blood DC antigen 1 (BDCA1+)/HLA-DR+], myeloid DC type 2 (BDCA3+/HLA-DR+), and plasmacytoid DC (BDCA2+/CD123+) [3].
DCs are located at the basolateral site of the lungs and continuously scan the environment to detect the presence of pathogens and subsequently initiate an immune response. They are a heterogeneous population of antigen presenting cells that exert specific functions. Compelling evidence is now provided that DCs are both sufficient and necessary to induce allergic responses against several inhaled harmless allergens. How various DC subsets exactly contribute to the induction of allergic asthma is currently a subject of intense investigation [28].
cDC are important, as they act in concert with T lymphocytes in protecting the organism from infections, but also act against cancer development. Recently glutamine and folliculin (FLCN) have been identified as relevant for the interaction of cDC and CD8+ cytotoxic lymphocytes. FLCN and FLCN-interacting protein 2 (FNIP2) form a complex, which mediates glutamine uptake by cDC and furthermore stimulates antigen presentation to CD8 lymphocytes [29]. The detection of this signaling system might open a new avenue for stimulating an immune reaction.
mDC are potent inducers of T helper type 1 (Th1) and Th2 cytokines in naive T lymphocytes. The type of Th response is determined by the level of IL-12. Bacteria, bacterial compounds, and also interferon-γ (IFN-γ) derived from memory T cells or NK cells, will shift the immune reaction towards a Th1 response. Conversely, Th2 responses will be induced by an inhibition of IL-12 release, and also by IL-10 and prostaglandin E2 [30–32].
mDC and pDC uptake inhaled antigens and present it to T lymphocytes. However, pDC has an additional role, as they suppress effector T cell generation thus contributing to an immune balance. When pDCs are experimentally depleted, inhaled inert antigens cause immunoglobulin E (IGE) sensitization, airway eosinophilia, goblet cell hyperplasia, and Th2 cell cytokine production, all features of asthma [33]. pDC are also activated by CD40L, but in contrast to mDC induce Th2 differentiation [34]. pDC in contrast to mDC produces low amounts of IL-12 and usually dies by apoptosis if IL-3 is not present in the stroma. Interestingly, if pDC is exposed to the virus, it will produce high levels of IFN-I and also can promote Th1 polarization, and in addition, stimulate CD8+ T lymphocytes. This means that pDCs can act differently depending on the antigen and the composition of the stroma [34, 35].
The role of TLR is well known in immune reactions. Accordingly, they play a role in DC differentiation. mDC can express all TLRs, except TLR7 and TLR9, which however are selectively expressed by pDCs. pDCs in contrast, do not express TLR3 [36–38].
In viral infections primarily imDC are seen, which rapidly differentiate into cDC. They rapidly migrate from the lung to draining lymph nodes and present the antigens to T cells. Maturation and migration are stimulated by CCL5 and CCR5 [22]. pDC is involved too in early viral detection. They synthesize IFN-II, which inhibits viral replication and spread [39]. They also can induce tolerance for antigens and therefore might inhibit the development of allergies. mDC similar to cDC plays a role in inflammatory lung diseases. Here another cytokine, CCR2, induces accumulation of mDC in the perivascular region of the lung. To induce migration into the bronchiolar region the formylpeptide receptor 2 (Fpr2) is required. In an experimental model CCR2, its ligand CCL2 and Fpr2 could be detected in bronchoalveolar lavage (BAL) [40]. cDCs express integrin a(E)b7 and CD11c and reside in the lung mucosa and the vascular wall. They produce IL-12. In addition, they express langerin, and the tight junction proteins Claudin-1, Claudin-7, and zonula occludens 2 (ZO-2) [41]. They are involved in antigen uptake and transport across the airways towards the regional lymph nodes for antigen presentation. Besides their role in antigen-induced lung diseases, DCs also play a role in immune responses induced by environmental pollutants. In an experimental cell culture system exposure to diesel-exhaust particles caused a maturation of imDC to iDC (positivity for CD83). Granulocyte-macrophage colony stimulating factor (GM-CSF) seems to be the major cytokine responsible for this maturation [42].
On regular hematoxylin and eosin (HE)-stained sections DCs are hard to recognize, as the cytoplasm does not show distinct cell borders, and the nuclei do not present with a specific morphology. Cells can be either spindly or epitheloid (Figure 2A and B). By immunohistochemistry this changes especially when langerin antibodies are applied (Figure 2C). The long slender cell processes present with a spider-like appearance, which separates them easily from other cells expressing the S100 protein (Figure 2D). When CD68 antibody is applied, some DCs will stain but much more faintly compared to the intense staining of alveolar macrophages. Although macrophages also can uptake, process, and present antigens to lymphoid cells, DCs are far more specific in this task [43]. DCs with a more epitheloid appearance are even harder to separate from monocytes and macrophages. This is more complicated when analyzing cells within the alveolar lumen. A distinction from macrophages is impossible. pDC can sometimes resemble plasma cells with oval or indented nuclei and a perinuclear pale zone [44]. Here the expression of DC markers is the only way to separate them (Figure 3). However, not all DCs function in the same manner, as we have explained above and will further discuss under the different diseases.
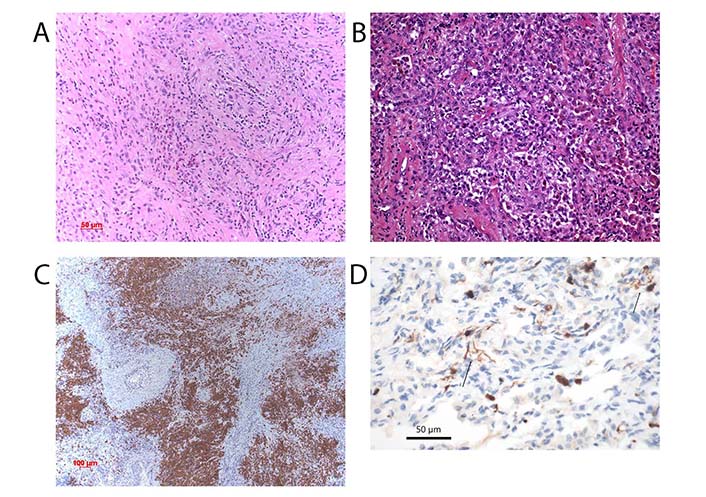
DC in LHCH. (A) Proliferation of LHCs in a case of LHCH. The morphology of the cells is either spindly or epitheloid (arrows). The infiltration of eosinophils and small lymphocytes. HE, bar 50 µm; (B) macrophages and DCs in a case of respiratory bronchiolitis-interstitial lung disease (RB-ILD). HE, bar 50 µm; (C) expression of langerin in a case of LHCH. The dense nodular accumulation of LHC. Immunohistochemical stain for langerin, bar 100 µm; (D) DCs highlighted by an antibody for S100 protein. The spider-like processes of the cells are characteristic, two arrows point to these cells. Immunohistochemical stain for S100 protein, bar 50 µm
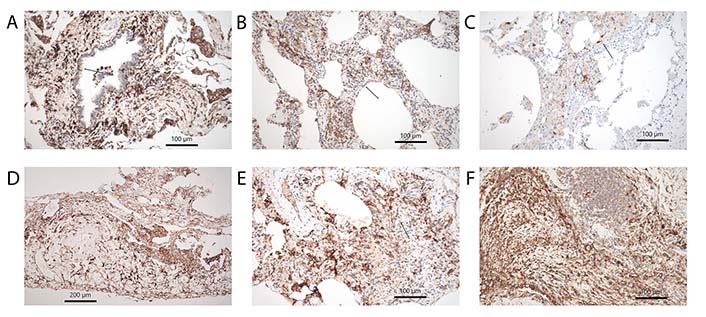
Subtypes of DCs. (A) CD11c showing cDC in the bronchial mucosa and surrounding stroma, note also positively stained intraalveolar cells, morphologically looking like alveolar macrophages, arrow points to an intraepithelial group. Immunohistochemistry, bar 100 µm; (B) CD14 another marker staining cDC in the alveolar walls with fibrosis and inflammation, arrow points to a group. Immunohistochemistry, bar 100 µm; (C) LHC hyperplasia in RB-ILD, here stained by langerin antibodies, arrow point to such cells. Immunohistochemistry, bar 100 µm; (D) CD15+ DCs within pleura in a tissue sample from a rheumatoid arthritis patient. Immunohistochemistry, bar 200 µm; (E) imDC stained by CD303, arrow points to such group of cells. Immunohistochemistry, bar 100 µm; (F) pDC highlighted by CD123 antibody in fibrosing lung disease. Few pDC are also infiltrating the tertiary lymph follicle. Immunohistochemistry, bar 100 µm
Chronic obstructive pulmonary disease (COPD) is morphologically characterized by emphysema and chronic bronchitis/bronchiolitis. Some variations can be seen, such as chronic bronchitis involving predominantly the large airways, whereas in other cases the smaller airways are affected. Clinically the patients present with recurrent airway infections, chronic cough, and purulent expectorations. Recurrence is often seasonal with peaks in spring and autumn. Almost all patients are active or former cigarette smokers. Clinically disease onset is most often in the fourth decade of life. Over time centrolobular type emphysema develops. Tobacco smoke is composed of a large number of carcinogens, toxins, and also incompletely combusted plant proteins can be identified, which possess an antigen property [45, 46]. Many of them induce the formation of oxygen radicals, and this mixture of substances results in chronic bronchitis and emphysema [47–49]. Cigarette smoke is well known to induce an activation of cyclo-oxygenase 2 followed by the secretion of prostaglandin E2, which in turn induces DC maturation. This mechanism indicates oxidative stress [50]. In addition, CXCL8/IL-8 released by neutrophils increased the infiltration of immature and CD40L-positive cDCs. There are ill-defined patient-intrinsic mechanisms, mainly focused on the selection of TLR, which direct the selection of DCs and ultimately predict the morphological picture [51, 52]. The maturation of mDC and iDC is reduced as seen in reduced expression of CD83 and CCR5. As CCR5 is important for the uptake and procession of microbial antigens; this could be one reason, why COPD patients are prone to bacterial colonization and increased sensibly to bacterial infections [51]. In a study of COPD patients by flow cytometry a significant decrease of maturation markers CD40 and CD83 in iDC and mDC was seen [53]. This decrease was more pronounced in current smokers compared to former smokers, pointing to tobacco smoke as the most relevant factor [53]. The above-mentioned effects of smoking are further supported by another study, showing that mDC in smokers presented with reduced CCR7, which is a lymph node homing receptor [54]. This not only explains, why bacterial antigens are not efficiently presented to immune cells but also fits to recurrent bronchitis and airway obstruction as a result of impaired immune defense. Besides functional impairment of mDC also LHC decreased in number, pointing to an impaired antigen uptake. On the contrary, in long standing and severe COPD, an increase of LHC was found at the basis of the bronchial epithelium (interface of epithelial cells and stroma) [55]. In this study, it was shown, that there was also an increase of physical contact between LHC and T cells. The severity of parenchymal remodeling (emphysema severity) correlated positively with DC and lymphocytic contacts as well as the numbers of cells found at the alveolar surface interface. DCs seem to stay longer in tissues probably induced by heme-oxygenase 1 and the antiapoptotic protein B cell lymphoma-extra-large (Bcl-XL) [56]. The appearance of LHC in long standing COPD suggests, that there seems to be an increase of antigens trapped at the alveolar lining epithelium. In this setting, an aggravated inflammation and the appearance of tertiary lymph follicles can be seen [57]. In these lymph follicles, CD4+ T cells as well as Treg cells are found. Unfortunately, no information about pDCs was provided, which seems to be likely. In an experimental study by Givi et al. [58] the role of different DCs was evaluated. Emphysema was induced by cigarette smoking. Different populations of DCs were either expanded or depleted. If mDC and pDC populations were expanded by fms-related receptor tyrosine kinase3-ligand (Flt3L) treatment the numbers of macrophages declined, and the levels of fibroblast growth factor (FGF) and vascular endothelial growth factor (VEGF) increased in BAL fluid, contributing to emphysema severity. If the pDC population was depleted, the severity of emphysema was reduced, as evaluated by a decrease in the mean linear intercept (mLI, it is a marker for the severity of emphysema; a line is drawn between the two nearest bronchioles and the number of alveolar septa is counted; a normal number is 7, in emphysema this can be reduced to 2-3 septa). So, it seems that both mDC and pDC can contribute to and inhibit emphysema development, probably by either activating the immune reaction or inducing tolerance.
One of the major problems in evaluating subsets of DCs in COPD is the dynamic evolution of the disease, which is not only reflected by the subtype of DC but also by the subsequent activation of different subpopulations of lymphocytes.
COPD patients constantly expose themselves to tobacco smoke, and these toxins and antigens cause an inflammatory reaction. Early on cDC and mDC dominate the reaction, whereas later on pDCs are more numerous. In addition, during the course of COPD, there is bacterial colonization of bronchiectatic airways, which elicits a response by granulocytes, lymphocytes, macrophages, and DCs. Infection can follow this initial colonization, either with purulent bronchitis or in the worst case with bronchopneumonia. Besides bacteria, also viral infections do occur, especially during cold weather periods. And this all affects the selection of DC, lymphocytes, and granulocyte subpopulations. There is still a lot of work to better characterize the disease course of COPD.
The production of autoantibodies directed towards elastin was induced in mice by cigarette exposure. An elastin challenge caused a specific T cell response and release of metalloproteinase 12, emphysema could be induced. Antibodies for elastin were also detected in COPD patients [59, 60]. These findings were further expanded by two independent reports from Mehraban et al. [61] and Fagiola et al. [62] using intratracheal elastase instillation combined with LPS to induce an inflammatory reaction in hamsters. In this investigation fragments of elastin had a proinflammatory effect.
A major problem for a longitudinal study of COPD is the access to tissues. The severity of COPD is classified according to the Global Initiative for Chronic Obstructive Lung Disease (GOLD) system [63]. In most studies, patients are evaluated using pulmonary function tests. Biopsies are usually taken at late stages when patients already present with severe symptoms, or in cases of lung cancer and associated COPD. Therefore, changes of infiltrating cells of the innate and specific immune systems have not been investigated along the time course of the disease.
LHCH presents as either a single organ-based or a multiorgan disease. Multiorgan-based LHCH is most often seen in children, whereas the lung-only-based one most often affects young patients, almost all of them heavy cigarette smokers (the multiorgan type of LCH will not be discussed in this review). In a few instances, they can present with acute lung insufficiency at the clinic. On computed tomography (CT) scans an unspecific nodular pattern is seen but in long standing disease a more specific so called starry-like picture is represented by scars surrounded by translucent emphysematous lung tissue (traction emphysema). Morphologically the nodules are composed of LHCs admixed with eosinophilic granulocytes and macrophages, less lymphocytes. Eosinophils are usually seen in early and florid diseases (Figure 2A and C). LHCs, as well as the less numerous T cells, secrete eosinophilo-tactic interleukins 4 and 5, which facilitate the tissue influx of eosinophils as well as their release from the bone marrow [64]. Smoking-induced LHCH is thought to be induced by an overwhelming exposure of plant antigens, due to incomplete combustion of tobacco plants. LHC accumulates along the bronchial tree down to the bronchioles. Due to the release of cytotoxic proteins from eosinophils, necrosis of the bronchial mucosa occurs, resulting in decreased airflow and hypoxia. It should however be noted, that a hyperplasia of LHC can occur in other settings, including COPD and desquamative interstitial pneumonia (DIP).
With respect to the current understanding, LHC is derived most often from mDC [65]. Activating mutations were detected in BRAF and mitogen-activated protein kinase (MAPK) genes [65, 66]. This results in the activation of the MAPK-extracellular regulated protein kinase (ERK) pathway and finally drives the nodular proliferation of LHC. Mutated MAPK confers a higher risk [67] for disease progression and multiorgan involvement. A confirmation of these findings is pending. Another open question is the impact of these gene mutations: Smoking-induced single-organ LHCH can present with but also without these mutations. A study comparing DC subpopulations, mutations, and the correlation with clinical presentation is still missing. Another probable progression factor lies in the origin of LHC: if derived from mDC the disease progresses more slowly, whereas if derived from immature hematopoietic precursor cells progression occurs much faster [66]. Finally, the Notch pathway is activated in LHC. The cells express the Notch ligand Jagged 2 and by that activate the Notch pathway. Downstream this induces the expression of matrix-metalloproteinases 1 and 12, which in concert with acidic proteins released from eosinophils might induce well-known tissue destruction [68–70]. LHC remains for a long time in the lung tissue. On CT scan controls of patients who quit smoking, the nodules very slowly disappear over months. An abnormal retention of LHC could be explained by an aberrant expression of chemokine receptors or dysregulation of chemokine production. Another explanation was raised by Chu and Jaffe [1]. It was discussed that LHC in histiocytosis might be defective in their capability of antigen presentation, and also do not migrate to lymph nodes, which might explain the retention at the bronchial mucosa [1]. Together with LHC also imDCs were found. imDC expressed CCR6 in LHCH lesions together with the ligand CCL20/macrophage inflammatory protein (MIP)-3α, which causes inhibition of migration [71]. LHC expressed CCL5/RANTES and CXCL11, which on one hand stimulates migration, but also induces maturation. Due to the presence of imDC, it seems that LHCH nodules remain in an immature state [71].
RB-ILD and DIP are two other diseases related to cigarette smoking. In both an accumulation of macrophages is seen and forms the diagnostic clue (Figure 2B); in the former macrophage accumulation is confined to respiratory bronchioles and extends into the centrolobular portion of the peripheral lung, in the latter the accumulation is diffuse within the alveolar periphery but do not extend into bronchioles. In contrast to bronchiolitis obliterans, where studies were published in the background of either respiratory syncytial virus infection or lung transplant rejection, not many reports have evaluated these diseases. Looking up LHC in different smoking-induced lung diseases, Masunaga et al. [72] found an increase in RB and smoking-related interstitial fibrosis. However, other DCs were not evaluated. A similar finding was seen in cases of DIP. For a better understanding, a more detailed investigation of these smoking-induced lung diseases is necessary. Acute onset but also periods of improvement and stable disease point to the possibility of changes in the composition of DCs (Figure 4A–C). This might be used to improve therapeutic intervention. Currently, corticosteroid therapy and smoking cessation are the only options.
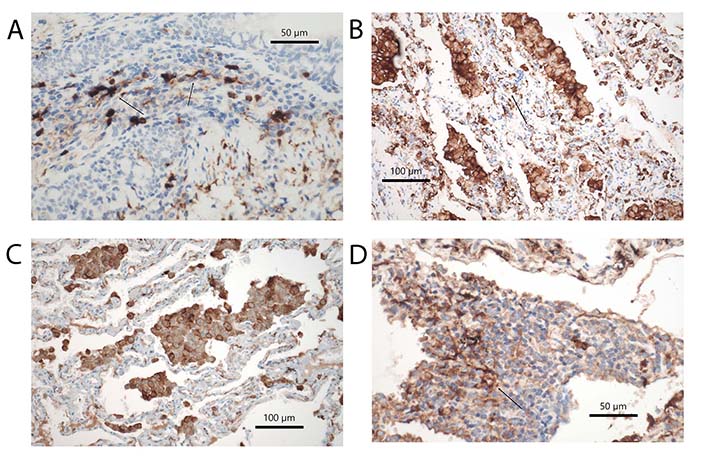
Subtypes of DCs in different diseases. (A) DCs expressing S100 protein in rheumatoid arthritis, arrow points to a DC with cytoplasmic extension. Immunohistochemical stains, bar 50 µm; (B) HLA-DR+ DCs in RB-ILD, arrow points to a group of DC. Immunohistochemical stains, bar 100 µm; (C) CD123+ pDCs in DIP; among macrophages also some intraalveolar pDCs are seen, but also others within the alveolar walls. Immunohistochemical stains, bar 100 µm; (D) CD35+ fDCs in a tertiary follicle in RhA. The arrow points to a spider-like net of DCs in close contact with lymphocytes. Immunohistochemical stains, bars 50 µm
Todate et al. [2] reported infiltration of LHC, cDC, and mDC in diffuse panbronchiolitis. This is a bronchiolar disease found in children with a polymorphism in HLA genes, which causes an impaired bacterial clearance, making these children susceptible to infections with the same rare bacteria. HLA-B54 and HLA-A11 were significantly associated with the disease [73]. Increases in the number of CD1a+, CD1c+, and CD83+ iDCs and cDCs were found in both the bronchiolar epithelium and submucosal tissues. In addition, the authors found an expression of GM-CSF in bronchiolar epithelial cells, which is responsible for DC maturation. Although not a smoking-induced lung disease, it might serve as an excellent model to compare DC subpopulations with those in RB-ILD and DIP.
Bronchial asthma is a chronic inflammatory disease of the conducting airways, in which the epithelium, and cells of the innate and adaptive immune system are involved. Asthma affects approximately 300 million people worldwide, and its incidence is increasing especially in developed countries. The leading symptom is hyperreactivity of the airway smooth muscle cells. Clinically it is characterized by shortness of breath, wheezing, and chest tightness. Traditionally asthma was separated into allergic (intrinsic) and non-allergic (extrinsic) asthma, but in recent years within non-allergic asthma, several so called endotypes have been identified. So asthma is no longer regarded as a single disease but rather a syndrome [74]. These endotypes differ with respect to genetic susceptibility, environmental risk factors, age of onset, clinical presentation, prognosis, and response to treatment [75].
Several factors have been identified as possible causes of the increasing incidence of allergic diseases, among them asthma: maternal weight or obesity, use of drugs, and maternal stress. External factors are infections, exposure to mold and fungi, and outdoor pollution [76]. Exposure to polyaromatic hydrocarbons present in the environment has been identified as an additional factor [77]. Different environmental allergens such as house dust mites stimulate oxidative DNA damage in airway cells. During repair secretion of Th2-type cytokines is found, which might induce an allergic inflammation [78].
A huge amount of literature has accumulated on immune mechanisms, which is impossible to discuss here, therefore, the reader is directed to several relevant reviews on the subject [74, 75, 79–86]. For understanding, the most relevant aspects will be summarized.
Allergic asthma is a Th2 driven disease, however, in patients Th2high and Th2low clusters have been identified [87], characterized by high IL-4, IL-5, and IL-13 and eosinophilia in blood and tissues in the former variant. In Th2high clusters there is also high IGE, which characterizes these asthma patients as driven by IL-4-induced class switching of immunoglobulins synthesized by B cells—these patients will often present with a history of atopy [88]. These patients are sensitized against a wide array of allergens, such as house dust mites, tree pollen, animal dander, and fungal spores [89]. Children in a family with atopy have a higher propensity to develop asthma. A good marker for this asthma endotype is a high serum level of IL-25 and periostin, which also correlates well with tissue eosinophilia [90]. In addition to a specific allergen oriented immune reaction by primed lymphocytes also cells of the innate immune system are involved: In asthma innate lymphoid cell 2 (ILC2) plays a major role. ILC2 do not have antigen specific receptors, but they similarly produce IL-13, IL-5, and IL-9 as Th2 cells when stimulated by epithelial derived IL-25, IL-33, and thymic stromal lymphopoietin (TSLP) [75, 91, 92]. ILC2 are activated early on after allergen exposure. They produce IL-5 and elicit an eosinophil infiltration. Much research has also been done evaluating the role of Treg. Treg are decreased and/or functionally impaired in asthma; experimentally it has been shown that they can suppress asthma by secretion of IL-10 and suppress IL-17-induced bronchial hyperreactivity. However, their role is not entirely clear, probably because there are different populations of Treg acting such as inducible costimulatory 1+ (ICOS1+) Treg, which are probably capable of counteracting asthma [83, 93].
In bronchial mucosa of asthma patients 3 types of DCs have been identified: CD11b+CD172+(SIRP1a) expressing cDC sufficient to induce allergic sensitization, and CD11b–CD103+XCR1+imDC, which will need additional IFN regulatory factor 8 (IRF8) and Basic Leucine Zipper Transcriptional Factor ATF-like 3 (BATF3) stimulation for sensitization; in contrast to the former two types pDCs counteract the immune reaction by inducing tolerance via attraction of Forkhead box protein P3 (FOXP3+) Treg [28, 83, 84]. This is another example of pDC orchestrating or regulating tolerance. There is some evidence that a primary defect in pDC function may underlie the development of asthma [39].
How do antigens reach DCs within the mucosa? First DCs always interact with epithelial cells within the mucosa. Second, many allergens possess a protease activity, which can open the epithelial barrier. For example, papain cleaves the tight junction proteins and stimulates innate cytokine response. Aspergillus fumigatus spore protease leads to fibrinogen cleavage, and metabolites activate TLR4 on epithelia; airway cells in response secrete IL-33, TSLP, and GM-CSF, which in turn activates CD11b+cDC and also ILC2. This again induces a Th2 polarization orchestrating the allergic response [82, 83, 94–96]. Some allergens contain an endotoxin fraction. In this case, an additional Th1 reaction is mounted. This links to so called neutrophil asthma, characterized by an influx of neutrophils into the bronchial mucosa. An example is a high exposure to diesel exhaust. This type of asthma is associated with Th17 cells. The simultaneous activation of Th2 and Th17 profiles induces CD4IL-4+IL-17+ T cells and has been termed overlap syndrome [75, 97–99].
Initially, mast cells were regarded as important in asthma, mainly attributed to Th2high, IGEhigh, and atopy associated asthma. But more likely basophils within epithelia play a more prominent role. Both cells interact with eosinophils in promoting the release of cytotoxic eosinophilic granules [100]. Thus, they are responsible for some of the morphologic changes seen in asthma biopsies.
Biopsies or autopsy lung specimens of patients with bronchial asthma will show infiltration by eosinophils within the bronchial mucosa, spissated mucus with masses of eosinophils, Curschmann spirals, and different amounts of lymphocytes depending on disease activity [101]. Mast cells and more important basophils highlighted by immunostains for tryptase and chymotryptase are seen in IGEhigh atopic asthma. Goblet cells within the mucosa and the bronchial glands are increased, and smooth muscle cells are hyperplastic in early stages but may be replaced by fibrosis in long standing asthma. Epithelial disruption is another characteristic feature of asthma bronchitis: the epithelial layer shows shedding of columnar cells, and only basal cells remain firmly attached to the basal membrane. The basal lamina is typically thickened and on electron microscopy will show several layers, each newly formed after repair of an acute asthma attack and induced by transforming growth factor-β (TGF-β), which is also called airway remodeling.
The major differential diagnosis is chronic bronchitis in COPD. There is no single feature that allows a certain distinction of both diseases, however, a combination of features can most likely be of help: Eosinophilia, epithelial shedding, hyperplasia of smooth muscle cells, and extreme thickening of the basal membrane are features favoring asthma bronchitis [101].
Several autoimmune diseases are discussed here: sarcoidosis, systemic vasculitis confined to the respiratory system, and collagen vascular diseases [systemic sclerosis (SSc), rheumatoid arthritis, systemic lupus erythematosus (SLE), dermatomyositis]. Investigations analyzing DCs in these diseases are scarce. Most reports primarily focus on lymphocytes, T cell receptor analysis, and the role of Treg and NK cells. Since in most diseases, the primarily acting auto-antigen is unknown research concentrated more on the effector site.
Sarcoidosis is a systemic granulomatous disease, involving predominantly the lungs among several other organs. The etiology and pathogenesis are incompletely understood. The research focus has always been on the role of T lymphocytes, macrophages, epitheloid cells, and giant cells. However, DCs were recognized a long time ago by their positive staining for S100 protein and lysozyme [102]. Decades later DC subtypes were investigated in sarcoidosis. Lommatzsch et al. [103] studied DCs in BAL and found an increase of mDC with increased CD80 and decreased CD86 costimulatory molecules. Comparing blood derived DCs and DCs in tissue sections, Ota et al. [104] identified mature DCs within the granulomas but decreased mDC and pDC in the blood. Their interaction with T lymphocytes was demonstrated. The study of Ten Berge et al. [105] confirmed this finding, showing an increase of mDC in BAL. These mDCs induce T cell proliferation and release TNF-α when cocultured with CD4+ lymphocytes [105]. In the present understanding, macrophages undergo differentiation into epitheloid and giant cells. As morphologically DCs and macrophages are not easily separated, epitheloid cells should be investigated, if these are derived from DC. Functionally they can present antigens, and secrete cytokines very similar to DCs [106].
Anti-neutrophil cytoplasmic antibodies (ANCA)-associated vasculitis is the predominant type of systemic vasculitis involving the lung. Although these are diseases associated with an immune reaction directed against myeloperoxidase or protein 3, and thus should also involve DCs, only one report has investigated these antigen presenting cells. mDC and imDC were identified, where imDC formed clusters with T lymphocytes [107]. Again, based on the disease course with activity, relapse, and indolent periods, the dynamics of DCs would be worth investigating.
SSc is characterized by lymphocytic interstitial pneumonia in the acute phase and later by fibrosing pneumonia, either usual interstitial pneumonia (UIP) or non-specific interstitial pneumonia (NSIP), respectively. DCs have been identified especially in the early phase. pDCs were responsible for the IFN-α production; increased monocyte chemoattractant protein-1 (MCP-1) or IFN-α serum levels were associated with lung fibrosis [108]. pDC might be the most important DC population. They produce IFN-I and also CXCL4 [109]. The release of CXCL4 is under the control of phosphatidylinositol 3-kinase δ and is due to the aberrant upregulation of TLR8. If pDC are experimentally depleted in a mouse model this prevents disease and reverts fibrosis. TLR8 seems to be the key molecule sensing for inflammation and fibrosis [36]. A reduction of inositol requiring enzyme 1-Xbox binding protein 1 (IRE1α-XBP1, a member of the unfolded protein response system) was seen in pDCs. IRE1α-XBP1 expression correlated inversely with IFN-I stimulation and CXCL4 release. IRE1α-XBP1 activation induces phosphoglycerate dehydrogenase (PHGDH) expression and reduces pyruvate access to the tricarboxylic acid cycle, which however is essential for pDC IFN-I response. PHGDH expression was reduced in pDCs from patients with SSc and SLE. Therefore, modulation of the IRE1α-XBP1-PHGDH axis may represent a therapeutic strategy to inhibit the deleterious function of pDC [110]. Fibrosis of the pulmonary (and skin) blood vessels is a common phenomenon in SSc. van Bon et al. [111] showed that CXCL4 induced markers of endothelial-cell activation and forced an influx of inflammatory cells into vessel walls. Another factor, RUNX family transcription factor 3 (RUNX3) secreted by DCs was identified to inhibit fibrosis and disease susceptibility. The polymorphism rs6672420 found in patients with SSc induced lower RUNX3 expression. Experimentally deletion of RUNX3 in DC leads to spontaneous induction of skin fibrosis and aggravated bleomycin-induced skin fibrosis [112].
Not much data exists comparing the early stages of SSc versus the late stages. Choreño-Parra et al. [11] in their work analyzed T cell populations in patients with limited cutaneous sclerosis and found Th2, Th17, and dual Th2-Th17 phenotypes, and increased production of Th17-related cytokines IL-1β, IL-17F, IL-21 and IL-22. mDCs were identified as producing IL-33. These changes were pronounced in the early stage and less in the late stage [11]. Two micro-RNAs (miRs) were identified in pDCs of patients with SSc again in the early preclinical stage. miR-126 and miR-139-5p were upregulated and correlated with the expression of IFN-I genes. Ubiquitin specific peptidase 24 (USP24) was identified as a target of miR-139-5p. TLR9 stimulated USP24 expression in pDC and contributed to their activation [113]. Still, further work is necessary to elucidate the role of different DCs in early and late SSc, probably opening new treatment options.
Rheumatoid Arthritis is an autoimmune disease, that primarily affects joints, but also involves the lung and pleura. The disease can involve the lung as granulomatous pneumonia, chronic inflammation with exudation into the pleura, and finally lung fibrosis, most often with the pattern of UIP [114]. mDC and pDC seem to play a major role in rheumatoid arthritis. Reports dealing with DCs have almost exclusively focused on the synovialis. However, it can be speculated, that a similar distribution of DCs is found in cases with lung involvement (Figure 3D, 4A and D). DCs strongly express Janus kinase 3 (JAK3), signal transducer and activator of transcription 4 (STAT4), and STAT6 in synovial tissue. These cells express phenotypes consistent with mDC and pDC types. They produce IL-12 as well as IFN-α and IFN-γ [115]. These DCs might be derived from mDC. When monocytes leave the blood and infiltrate inflamed synovial tissues, the differentiation into various DCs is influenced by cytokines and local acidosis. Other molecular factors, such as citrullination, can enhance osteoclast differentiation from mDC, which finally induces bone destruction in rheumatoid arthritis [116]. Treatment resistance is also influenced by DCs. imDCs in the synovialis are strongly associated with treatment resistance and lowered IFN-I signaling. A cluster of synovial DCs showed both features of imDC and pro-inflammatory cDCs [117]. pDCs very likely signal antigen tolerance. mDC in the synovialis induces Th17 cell expansion and enhances proinflammatory cytokine production. If catabolism is induced experimentally tolerogenic pDCs can be induced, probably by reprogramming of mDC [118]. In recent times better sensitivity of CT scans resulted in increased patient numbers with lung involvement in rheumatoid arthritis. Whereas acute disease is now effectively inhibited by new drugs (biologicals), chronic disease presentations with lung and pleura involvement increased. In this setting, more research is necessary to elucidate the immune mechanisms underlying these reactions.
Systemic Lupus rarely involves the lungs. However, in acute lung involvement, several features are present: acute hemorrhage, vascular thrombosis, and deposition of complement-activating antigen-antibody complexes within the lung stroma. Chronic or resolving features can be organizing pneumonia and unspecific fibrosis. A diagnostic hallmark of systemic lupus is the intravascular emperipolesis of neutrophils, which can also be seen in lung tissue sections. A missense variant of the neutrophil cytosolic factor 1 (NCF1) is one of the most significant associated risk factors for lupus, leading to excessive activation of pDCs. NCF1 is a subunit of the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 2 complex, impairment leading to an acidified endosomal pH, a well-known factor for pDC activation [119]. Different DC subpopulations can either promote autoimmunity or tolerance and link innate and adaptive immunity. pDCs are the main source of IFN-I cytokines, and take the leading part in the development of systemic lupus [120]. pDCs of SLE patients also showed increased expression of IRF1 and IRF5. There were also significant changes in the expression of APRIL, methylated DNA-binding domain protein 2 (MBD2), and E2-2 in mDCs [121]. A very interesting report by King et al. [122] showed, that there are differences between skin reactions in localized lupus versus systemic lupus. In a mouse model of lupus, the authors induced autoantibodies to desmoglein-3. Inducible ablation of LHCs in adult mice resulted in increased autoantibodies against skin antigens and accelerated lupus dermatitis. From skin-draining lymph nodes of LHC-ablated mice significantly fewer CD4+ T cells were harvested, and less anti-inflammatory IL-10. This was interpreted that a skin-resident DC population regulates local tolerance in lupus, but no effects on other organs such as kidneys, lungs, and liver were seen [122]. In another study, β-arrestin2 was identified as a regulator of DC migration and immune regulation [123]. β-arrestin2 directly interacts with zinc finger and BTB domain containing 46 (Zbtb46) a DC-specific transcription factor. CD74, nuclear receptor 4A1 (NR4A1), and ZFP36 are target genes regulated by the interaction between β-arrestin2 and Zbtb46 which negatively influences the migration of DCs. A deficiency of β-arrestin2 not only aggravates lupus nephritis but induces also experimental autoimmune encephalomyelitis.
Dermatomyositis rarely involves the lung, or maybe was overlooked in the past. In the acute phase, there is usually lymphocytic interstitial pneumonia, sometimes combined with histiocytic granulomas. In contrast to HP, the lymphocyte subpopulations are dominated by CD4+ T cells, however, as in almost all autoimmune diseases there is always a mixture of CD8+ T cells and CD20+ B cells. A constant feature is lymphocytic pleuritis. In a few of these cases, there was also a follicular bronchiolitis. In the chronic phase, fibrosis is present. This can be an unspecific interstitial fibrosis, but also NSIP, UIP, and organizing pneumonia have been described [124].
Recently dermatomyositis with expression of anti-melanoma differentiation-associated gene 5 antibody (MDA5) has been described, which is characterized by IFN-I expressing histiocytes in the lung [125, 126]. Anti-MDA5 expression in dermatomyositis seems to be associated with lung involvement and progressive fibrosis [125–128]. Fibrosis is associated with the prognosis in patients, showing the worst outcome for UIP pattern, although not as bad as in idiopathic UIP/idiopathic pulmonary fibrosis (IPF) [129]. Although the pathogenesis of anti-MDA5 antibodies is largely unknown, some aspects have been elucidated: By RNA sequencing an IFN-induced helicase C domain1 was identified, which encodes MDA5. MDA5 when tested on embryonal lung fibroblasts caused downregulation of a-smooth muscle actin, connective tissue factor, and the deposition of collagen I and III via suppression of TGF-β [127]. Thus, circulating MDA5 antibodies might cause the opposite reaction leading to lung fibrosis. In another report, IFN-β together with eukaryotic translation initiation factor 2 alpha kinase 2 (EIF2AK2) was identified in patients with MDA5 antibody-associated dermatomyositis and lung fibrosis [130]. The finding of IFN-I expression in histiocytes already raises the suspicion that these are DCs, which are known to secrete these IFN. Chen et al. [131] identified CD11c positive cells in skin lesions, and by mRNA analysis identified mDCs as the source of IFN-β. Interestingly pDCs were not seen in dermatomyositis patients but in the controls. In the reports by Khanna and Reed [132] and de Padilla and Reed [133], this was disputed, as these authors found IFN-I positive pDCs in tissues and peripheral blood. These authors also reported on the deregulation of the MHC class one complex. If this is related to the study population (juvenile dermatomyositis) needs to be investigated in further studies [134]. The involvement of pDC was also shown using the markers CD123 and CD303 on DC in peripheral blood. CXCR3, CXCR4, and CCR7 very likely induce the recruitment of pDC to the tissues [135]. It seems that imDCs mature when reaching the muscles, as shown by the expression of fascin [136]. However, this all requires confirmation and new studies using well-defined study populations: In the study by Page et al. [137], it was shown that imDCs migrate into the muscles and mature there. However, this study defined DCs by CD1a, which is a marker for LHCs, and lysosomal associated membrane protein 3 (LAMP-3) for mature DCs [137]. Other chemokines investigated, such as CCL20 and CCR6 were interpreted as controlling homing to tissues, however, the chemokines responsible for migration such as CCR7 were not found.
Sjøgren syndrome usually is seen in salivary and lacrimal glands. In rare instances, it can involve the lung causing massive lymphocytic interstitial pneumonia and hyperplasia of the bronchus-associated lymphoid tissue. Due to the rarity, a single report looked for the involvement of DCs. Wang et al. [138] reported increased TLR7 in salivary glands from Sjøgren patients; expression of TLR7 was also positively correlated with TNF-α, CXCL13, and CXCR5, cytokines closely related to DC [138].
HP is induced by inhalation of a wide variety of antigens, predominantly of fungal origin. Often these antigens are derived from thermolabile fungi. Patients present with acute onset of fever and fatigue. After cessation of exposure, the symptoms vanish. Morphologically HP is characterized by combined lymphocytic and granulomatous pneumonia, concentrated in the peripheral lung. The granulomas are composed of epitheloid and giant cells with a rim of lymphocytes, predominantly of CD8+ T cells. Which type of DC is involved in HP has not been studied extensively. In an experimental model, wild-type and CD103-knockout mice were exposed to Saccharopolyspora fungus antigens. CD103 expression on DCs was downregulated after exposure. CD103+ DC plays a regulatory role in Th17 cell response, driving airway inflammation [139]. However, it has to be mentioned, that CD103 is a molecule for activation of lymphocytes and not a molecule characterizing a subset of DCs. In HP patients CCL18 was elevated in lung tissues. DCs but also macrophages can secrete this chemokine. A correlation was found between the level of CCL18 and the number of lymphocytes in BAL [140]. However, the type of DC was not evaluated, but expression of CCL18 most likely points to the monocytoid type.
DC-associated vaccines have entered treatment options in immunological-based cancer therapy. Research is focusing on vaccines, where antigen was loaded on DCs and administered. For example, Vo et al. [141] loaded tumor antigens on DCs and used a mycobacterial vector for transfer. By this, they inhibited immune-suppressive cells and induced a polarization of T cells in favor of Th1 cells. Xiang et al. [142] in another investigation administered oral DNA vaccines against several tumor models and targeted different growth factors, survivin, and enzymes inducing suppression of tumor growth, and inhibiting angiogenesis. Here also activation of cytotoxic T cells, NK cells, and antigen presenting DC was achieved [142]. For an overview of vaccine-based strategies in immune-oncologic treatment trials, the reviews by Mogrão et al. [143], Gonzalez-Gugel et al. [144], and Benencia et al. [145] can be recommended. Also, antibodies directed against subsets of DCs are been used. As an example, Sung et al. [146] produced a bispecific antibody against lymphocyte activation gene 3 (LAG3) and programmed cell death ligand 1 (PDL1) and found not only an enhanced antitumor-activity of CD8+ T cells but also activation of antigen presenting DCs. Few investigations are focusing on non-tumor diseases. Experimentally tolerogenic DCs were generated and applied in a lupus erythematosus mouse model, which ameliorated disease activity [147]. In their review, Rosen et al. [148] discussed the possibility of using DC modulation to prevent rejection of transplanted organs.
Many open questions have been raised in this review, which could stimulate research. Are pDC subtypes impaired in RB-ILD and DIP, and can disease progression be modified by pDC activation? Are LHC dysfunctional in LHCH and can they be triggered to migrate and present antigens? How well functions antigen presentation in lymphoid aggregates within the lung, or is migration to regional lymph nodes required? Can the plasticity of DCs be demonstrated when analyzing different disease activities, and how can this be supplemented by cell cultures using three-dimensional (3D) models?
More research on the function of DC subtypes is required in autoimmune diseases. Here findings in experimental disease models need to be compared to human tissue samples using DC markers by immunohistochemistry. An analysis of TLRs expressed in different diseases together with DC subtype analysis should highlight inflammatory and anti-inflammatory downstream reactions.
When infectious diseases are studied, this should not focus on one agent, but compare subtype differentiation of DCs in parasitic, bacterial, and viral infections. There have been differences reported even between different viruses.
DCs have for quite a while been ignored. Several reasons are responsible, as morphology cannot easily identify them in human tissues, and CD markers for a while required staining in fresh frozen tissues. Improvement of immunohistochemical methods now enables one to study subtypes of DCs in formalin-fixed paraffin-embedded tissues. RNA sequencing might be included in these investigations, however, there is a need to discern genes specifically expressed in subtypes from secondary responses of the cells when stimulated, as there can be an overlap. For a while analysis of subtypes of lymphocytes predominated research and suppressed investigations for DCs. Input from experimental research and increased understanding of immune reactions, however, refreshed the interest in these cells. Efficient immuno-oncologic therapy in human cancers and subsequent resistance has accelerated a search for other cellular mechanisms to overcome resistance, the most important are DCs. Research focusing on immune mechanisms in viral infections, especially by severe acute respiratory syndrome coronavirus-2 (SARS-COV-2) in addition showed the importance of this family of antigen presenting cells. And finally, the possibility of therapeutically influencing their function and subtypes pushed research forward.
A major problem to be solved is the plasticity of DCs, which is still not fully understood. Research in non-tumor diseases is in its early phase, and as shown in this review much has to be elucidated. Disease models are being created for autoimmune diseases, and combined with human tissue evaluation will contribute to our understanding. Models for allergic diseases as well as those for smoking-associated diseases are still pending, or have to be refined.
BAL: bronchoalveolar lavage
BDCA1: blood dendritic cell antigen 1
CCL21: C-C motif chemokine ligand 21
CCR7: C-C chemokine receptor
CD1c: cluster of differentiation 1c
cDC: classical dendritic cell
COPD: chronic obstructive pulmonary disease
CT: computed tomography
CXCL13: C-X-C motif chemokine ligand 13
DCs: dendritic cells
DIP: desquamative interstitial pneumonia
fDC: follicular dendritic cell
FLCN: folliculin
GM-CSF: granulocyte-macrophage colony stimulating factor
HP: hypersensitivity pneumonia
iDC: interdigitating dendritic cell
IFN-γ: interferon-γ
IGE: immunoglobulin E
IL-1: interleukin-1
ILC2: innate lymphoid cell 2
imDCs: immature dendritic cells
IRE1α-XBP1: inositol requiring enzyme 1-Xbox binding protein 1
LHC: Langerhans cell
LHCH: Langerhans cell histiocytosis
MAPK: mitogen-activated protein kinase
MDA5: melanoma differentiation-associated gene 5 antibody
mDC: monocytoid dendritic cell
MHC: major histocompatibility complex
miRs: micro-RNAs
NK: natural killer
pDC: plasmocytoid dendritic cell
PHGDH: phosphoglycerate dehydrogenase
RB-ILD: respiratory bronchiolitis interstitial lung disease
RUNX3: RUNX family transcription factor 3
SLE: systemic lupus erythematosus
SSc: systemic sclerosis
TLRs: Toll-like receptors
TNF-α: tumor necrosis factor-α
Treg: regulatory T
UIP: usual interstitial pneumonia
HP: Writing—original draft, Writing—review & editing.
The author declares that he has no conflicts of interest.
According to the local Ethics Committee, all figures in this article are fully anonymized, so an ethical approval is not needed.
All figures in this article are fully anonymized, so consent to participate is not needed.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

