 Review
Review

Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
†These authors contributed equally to this work.
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
†These authors contributed equally to this work.
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
†These authors contributed equally to this work.
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
†These authors contributed equally to this work.
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
†These authors contributed equally to this work.
Affiliation:
Laboratory of Inflammation Research, School of Life Science, Handong Global University, Pohang 37554, Republic of Korea
Email: jbaek@handong.edu
ORCID: https://orcid.org/0000-0003-4439-5545
Explor Immunol. 2024;4:358–375 DOI: https://doi.org/10.37349/ei.2024.00146
Received: November 07, 2023 Accepted: May 20, 2024 Published: June 20, 2024
Academic Editor: Margherita Sisto, University of Bari Medical School, Italy
The article belongs to the special issue Chronic Inflammation and Autoimmunity
Rheumatoid arthritis (RA) is a complex autoimmune disorder characterized by a spectrum of hypersensitivity reactions, encompassing Type II, Type III, and Type IV responses. Firstly, RA is marked by Type II hypersensitivity reactions driven by autoantibodies, such as rheumatoid factor (RF) and anti-(cyclic) citrullinated protein antibodies (ACPAs). These autoantibodies serve not only as serological markers for RA but also actively participate in inflammation, bone erosion, and clinical outcomes, with concurrent activation of the complement system involving C1q, C3, and C5 components specifically linked to RA progression and bone damage. Secondly, RA exhibits traits of Type III hypersensitivity, marked by the formation of immune complexes inciting inflammatory reactions. Immunoglobulin G (IgG) autoantibodies like RF and ACPA play pivotal roles in immune complex formation and the ensuing inflammatory responses. RA also demonstrates Type IV hypersensitivity propelled by CD4+ T cells, encompassing T helper 1 (Th1) and Th17 subsets. Th1 cells release interferon (IFN)-γ, promoting proinflammatory cytokines, while Th17 cells secrete IL-17, IL-22, and granulocyte-macrophage colony-stimulating factor (GM-CSF), contributing to synovial inflammation, bone and cartilage damage, and angiogenesis. RA concurrently exhibits features of Type II, Type III, and Type IV hypersensitivity. It is crucial to comprehend the presence and complex interplay of hypersensitivity responses and specific immune cell subsets in RA to create precise and efficient therapeutic approaches for the management of this incapacitating autoimmune condition. Thus, in this review, we aim to provide a comprehensive overview of the hypersensitivity features of RA.
Rheumatoid arthritis (RA) is a complex autoimmune disease characterized by chronic joint inflammation caused by the immune system aberrantly targeting synovial cells in the joints. The hallmark of RA is joint-related symptoms, including joint swelling and stiffness. Of note, RA is not limited to joint issues alone; it can lead to systemic inflammation affecting various parts of the body. This systemic inflammation can increase the risk of other conditions such as stroke, heart disease, diabetes, and osteoporosis [1, 2].
While the exact cause of RA remains unclear, it is believed to involve a combination of genetic and environmental factors. RA displays a higher prevalence in women compared to men, as is often observed in other autoimmune diseases. This suggests that hormonal factors [3] and gender-specific genetic variables [4] likely contribute to the disease onset. Furthermore, RA tends to be more common among older individuals, underscoring the role of aging in its development [5, 6].
Genetic factors are thought to play a crucial role in the development of RA. For instance, the human leukocyte antigen (HLA)-DRB1 gene has been associated with the production of rheumatoid factor (RF), which is an antibody produced as part of an autoimmune response [7]. Additionally, abnormal activity of peptidyl arginine deiminase (PAD) enzymes can trigger a process known as citrullination, leading to the formation of cyclic citrullinated peptides (CCP). These CCPs can act as autoantigens, provoking an immune response and the production of anti-(cyclic) citrullinated protein antibodies (ACPAs) (Figure 1) [8].
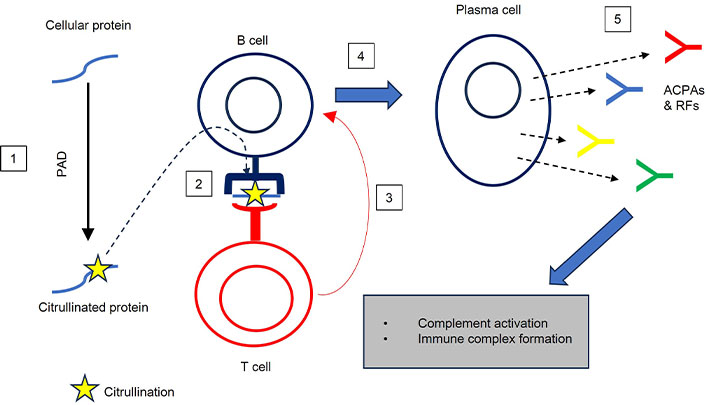
B cell-dependent hypersensitivity. 1: Citrullination of cellular proteins; 2: processing and presentation of citrullinated protein; 3: activation of B cells by T cells; 4: differentiation of naive B cells into plasma cells; 5: production of ACPAs and RFs. ACPAs: anti-(cyclic) citrullinated protein antibodies; PAD: peptidyl arginine deiminase; RFs: rheumatoid factors
In synovium, the infiltration of leukocytes leads to inflammation of the synovial membrane, a condition known as rheumatoid synovitis [9]. This infiltration involves both innate and adaptive immune cells [10], fostering an environment, in which fibroblast-like synoviocytes (FLSs) exhibit invasive and inflammatory behaviors, ultimately resulting in synovial lining hyperplasia [11]. FLSs release enzymes like collagenase and matrix metalloproteinases (MMPs), which contribute to cartilage destruction and the formation of pannus, which can obliterate the joint [12, 13]. The degradation of bone is facilitated by the maturation of pre-osteoclasts into osteoclasts, a CXCR4-dependent process favored by leukocyte infiltration and the release of pro-inflammatory cytokines [14]. Synovial hyperplasia, combined with hypoxia and the cytokines and growth factors generated by infiltrating cells, promotes angiogenesis [15], establishing a feedback loop that sustains ongoing leukocyte infiltration and chronic inflammation.
The distinctive feature of RA-associated inflammation is the recruitment of various immune cells, including monocytes, macrophages, neutrophils, B cells, and CD4+ and CD8+ T cells, to the synovial compartment [16]. CD4+ and CD8+ T cells within the synovium produce pro-inflammatory cytokines and chemokines that significantly contribute to the pathogenesis of RA [17–19]. It is widely recognized that aberrant inflammatory CD4+ T cell activity fundamentally influences the development and progression of RA [20]. Among the CD4+ T cell subsets involved in RA, T helper 1 (Th1) and Th17 cells have been identified as prominent contributors [21, 22]. The inflamed synovium is also noted for its enrichment in memory CD4+ T cells, which support B cell activity and immunoglobulin (Ig) synthesis [23].
Significantly, RA displays characteristics of both Type II, Type III, and Type IV hypersensitivity reactions simultaneously. It is crucial to comprehend the presence and complex interplay of hypersensitivity responses and specific immune cell subsets in RA to create precise and efficient therapeutic approaches for the management of this incapacitating autoimmune condition. Thus, in this review, we aim to provide a comprehensive overview of the hypersensitivity features of RA.
IgG and IgM antibodies are key to humoral immunity. They bind to microbes, such as bacteria, and facilitate their direct elimination through various antibody-dependent mechanisms. Unfortunately, the humoral response can turn against the host when these antibodies recognize self-antigens, potentially causing serious harm. Type II hypersensitivity reactions can be categorized into two subtypes: Type IIa and Type IIb hypersensitivity. Firstly, Type IIa hypersensitivity represents antibody-mediated cytotoxic reactions, where antigen-antibody complexes trigger the complement pathway and generate the membrane attack complex (MAC), leading to the lysis of target cells. Both IgG and IgM antibodies can also bind to Fc receptors (FcRs) on B lymphocytes, macrophages, and dendritic cells, thereby activating antibody-dependent cellular cytotoxicity (ADCC). Secondly, Type IIb hypersensitivity involves antibody-mediated cell-stimulating reactions driven by autoantibodies. For instance, in Graves’ disease, antibodies bind to thyroid cells, stimulating the excessive production of thyroid hormone.
Type II hypersensitivity has been associated with RA. Studies have consistently shown elevated levels of complement components, such as C2, C3, C3a, C5a, and the terminal complement complexes C5b-9, in synovial fluid and synovial tissue [24]. This suggests that complement activation plays a pivotal role in the early stages of RA and contributes to the amplification of inflammation as the disease progresses [25].
Following the differentiation of plasma cells, autoreactive B lymphocytes, driven by Toll-like receptors (TLR), primarily secrete autoantibodies [26]. The expansion of autoreactive B cells in RA may result from interactions between specific proteins with post-translational modifications and external antigens (Figure 1). Key autoantibodies in RA include RFs, ACPAs, and anti-PAD-4 antibodies [27]. The presence of RF is one of the diagnostic criteria for RA, but it is not definitive on its own and is used in conjunction with other clinical and laboratory findings for diagnosis and disease monitoring, as RF is not present in every RA patient. Among autoantibodies, ACPA holds significant prognostic value for the onset of RA in at-risk patients. ACPAs have emerged as a more specific serological biomarker than RF, which has higher sensitivity but lower specificity. This has led to the development of diagnostic tests based on ACPA detection. Approximately 60 percent to 70 percent of RA patients exhibit ACPAs, which target epitopes modified through citrullination by PAD [28]. Overexpression of PAD in the RA synovium leads to the production of ACPAs against citrullinated antigens. ACPAs are closely associated with inflammatory markers and often appear in serum years before the onset of RA symptoms [29]. They can activate immune cells and upregulate the production of pro-inflammatory cytokines. Studies have observed the involvement of ACPAs in bone erosion, arthralgia (joint pain), and arthritis induction, all common symptoms of RA. Bone erosion is linked to early joint inflammation and correlates with high ACPA levels. However, debates continue regarding whether ACPAs are the primary antibodies responsible for these effects, or if other antibodies contribute. Some studies suggest that ACPAs have an osteoclast-activating effect, enhancing inflammation. Arthralgia, an early symptom of RA, has been examined in models like collagen antibody-induced arthritis (CAIA), highlighting the relationship between antibodies and pain associated with immune complex formation [30]. Notably, ACPA-positive RA patients tend to experience worse clinical outcomes than ACPA-negative individuals, emphasizing the importance of early diagnosis and therapy guided by ACPA markers [31].
The complement system, a component of the innate immune system, is responsible for recognizing foreign antigens and clearing dead cells. Complement proteins, primarily synthesized in the liver, can rapidly activate when danger is detected, acting as opsonins, proinflammatory mediators, and lytic agents. The complement system can be activated through three major pathways: classical, lectin, and alternative. Although these pathways differ in their triggering mechanisms, they all lead to C3 cleavage, the release of anaphylatoxins, C3a and C5a, and the formation of the MAC. Studies have indicated a strong connection between the complement system and RA pathogenesis, particularly the pathogenic role of C1q in RA and the involvement of C3 and C5 in disease progression [32–35]. The interplay between the complement system and RA is evident through the induction of osteoporosis due to ACPA detection, complement activation, and subsequent bone damage. The role of complement cascades in RA has been confirmed through observations of C3-deficient and C5-deficient mice, which were found to be resistant to animal models of collagen-induced arthritis (CIA) [36, 37]. As synovial inflammation progresses, both the complement system and TNF-α secretion are activated. Anti-TNF-α treatment results in reduced levels of C3 and C4 complement components and shows potential in reducing cardiovascular disease associated with RA [38]. These findings support the use of anti-TNF-α therapies in the treatment of RA. Using complement receptor 1-related gene/protein y (Crry) targeted by means of CR 2 (CR2-Crry), a complement inhibitor that targets C3 breakdown products deposited in RA-affected joints, has shown promise in reducing inflammation, cartilage degradation, and bone damage [39]. It also significantly reduced the production of anti-collagen IgG and inflammatory cytokines TNF-α and IL-1β [39]. This approach may offer a therapeutic avenue for RA, although clinical trials of such inhibitors in RA patients have remained without success so far.
The formation of immune complexes is a natural part of the humoral response to an antigen. In the body, antigen-antibody complexes are typically removed via phagocytosis, with CRs and FcRs recognizing these complexes. These immune complexes often contain denatured proteins, antigens from gut bacteria, or dead cells and are cleared by macrophages. However, when the immune complex concentration exceeds a certain threshold, inflammatory reactions occur, leading to what is known as Type III hypersensitivity. The pathogenic effects of these immune complexes have been studied to understand their inflammatory consequences.
Type III hypersensitivity, which leads to the formation of immune complexes, is associated with the development of RA [40]. Studies have consistently shown that these immune complexes play a crucial role in the pathogenesis of autoimmune diseases [41]. Autoantibodies, such as RF and ACPAs, are found in RA patients and contribute to inflammation in the joints. These autoantibodies, specifically IgG antibodies, are known to be involved in further inflammatory responses, making them widely present in RA synovial fluids (Figure 1) [42].
Type III hypersensitivity arises from the deposition of immune complexes due to overproduced IgG and IgM reacting against foreign or self-antigens. When an excessive number of immune complexes are formed and cannot be efficiently cleared from tissues, it also triggers the activation of the complement system (Type II hypersensitivity) and leads to the recruitment of inflammatory mediators, ultimately activating immune cells like basophils and neutrophils in the tissue. These recruitments can persist for hours, weeks, or even years. Immune complexes are a normal occurrence but become problematic when their reaction becomes hypersensitive, resulting in Type III hypersensitivity. This condition results from an overproduction of antigen-antibody immune complexes, a process that often requires complement system activation for phagocyte recruitment to the site of the immune complex. This can occur both via complement-dependent factors, such as C1q and C5a, and complement-independent factors, including P-selectin, Mac-1, substance P, FcRs, and phagocytes [40].
The formation and accumulation of immune complexes, specifically those involving the interaction of IgG and IgM, play a crucial role in autoimmune responses. These immune complexes may cause inflammation in the joints, contributing to autoimmune antibody responses [43].
IgG is recognized through FcγRs present on various immune cells in the body, which are key components of both the humoral and cellular immune systems. The activation and downregulation of the immune system are mediated through FcγRs. These receptors are diverse, each having different specificities for isotypes [40].
Activation signals are associated with a common FcR γ chain, containing an immunoreceptor tyrosine-based activation motif (ITAM). Activation signals induce phosphorylation of tyrosine residues in the ITAM and initiate a cascade of responses, including the release of cytokines by macrophages and ADCC mediated by natural killer (NK) cells. The binding of IgG to FcγRIIB, which harbors an intracellular immunoreceptor tyrosine-based inhibitory motif (ITIM), regulates immune cell activity. The co-expression of B-cell receptors (BCRs) and FcγRIIB on B cells enables them to bind to IgG and antigens forming immune complexes. Dysregulation of FcγRs is associated with RA, making these receptors crucial in the protection against IgG-complexed antigens and the development of RA [44].
The immune complexes are also formed when an FcγR binds with IgG and complexes with antigens, as evidenced by the development of animal models for arthritis [45]. In autoimmune arthritis, FcγRs play a critical role, with some receptors inducing arthritis (e.g., FcγRI and FcγRIII) and others suppressing it (e.g., FcγRIIB). The expression level of the FcγRIIA gene has been observed to be abnormal in RA patients, and it can affect RA activity, whereas the increase in expression of FcγRIIB can affect stabilizing FcγR function in patients [46]. This receptor also influences the uptake of immune complexes and may be used in the therapeutic progress of RA. Therefore, molecules targeting FcRs have become a potential approach in RA treatment [47].
Immune complexes are internalized through the action of FcγRs, and immune cells, particularly macrophages, are crucial for the uptake of antigen-antibody complexes. The binding of immune complexes by FcγRs leads to the activation of phagocytes and the release of inflammatory cytokines. These antigen-antibody complexes can promote cellular immune responses, often having a stronger effect than responses initiated by antigens alone. The involvement of FcγRs extends to influencing bone erosion in RA, a common consequence of the disease [48]. Different types of FcγRs are associated with various aspects of RA. The absence of FcγRs in models has been used to study their role, with the FcR γ chain being associated with disease development [45].
Type IV hypersensitivity, also known as delayed-type hypersensitivity, manifests slowly, typically taking a day or more to develop [49]. Unlike other (I-III) hypersensitivity types, it is a cell-mediated, not an antibody-mediated reaction. Key players in this response include T lymphocytes, monocytes, and macrophages. This reaction is initiated when antigens are presented on class II MHC by antigen-presenting cells, leading CD4+ T cells to recognize the complex and trigger a hypersensitivity response. Macrophages release the cytokine IL-12, promoting the expansion of additional CD4+ T cells [50]. In cases of intracellular pathogens, activated macrophages produce hydrolytic enzymes and transform into multinucleated giant cells [51]. However, an exaggerated response by helper T cells, along with excessive cytokine production, can damage tissues, leading to inflammation, and cell death. Genetic (e.g., HLA haplotype) and environmental factors may cause enzyme modification, failure of tolerance, and upregulated lymphocyte activation, inducing Th1 and Th17 responses to self-antigens. This contributes to the influx of T lymphocytes into the joints [52].
RA was identified as a Th1-driven disease, as it was found that CD4+ T cells inclined to release interferon (IFN)-γ are present in the synovial fluids of RA patients [51]. This subset of CD4+ T cells expresses CXCR3, which binds the chemokines CXCL9 and CXCL10 induced by IFN-γ. In RA, synovial fluids exhibit higher concentrations of CXCL9 and CXCL10, and CD4+ T cells in these fluids express CXCR3 [53]. Notably, most CD4+ T cells in synovial joints are of the memory phenotype (CD45RO+), but their antigen specificity remains uncertain. Th1 cells typically stimulate macrophage activation, characterized by an increased capacity to release pro-inflammatory cytokines such as TNF-α, especially in the synovial joint [54]. Investigating whether Th1 cells can alter the characteristics of local macrophages is crucial, as it might contribute to disease maintenance (Figure 2) [55].
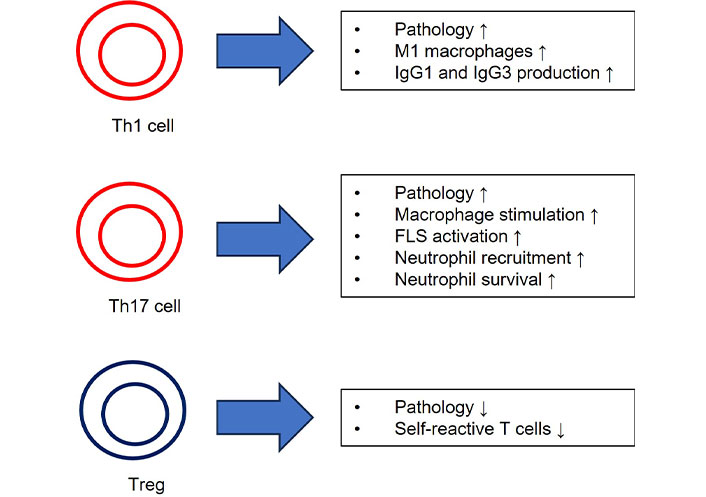
Effector functions of RA-specific T cell subsets. ↑: promoting effect; ↓: inhibitory effect. FLS: fibroblast-like synoviocyte; IgG1: immunoglobulin G1; Th1: T helper 1; Treg: regulatory T cell
Additionally, Th1 cells may influence class switching toward IgG1 and IgG3 antibodies, potentially impacting the antibody profile in RA [56]. Polyclonal antibodies to type II collagen are primarily of the IgG1 and IgG3 subclasses in RA while autoantibodies to citrullinated fibrin are primarily of the IgG1 subclass, indicating a prior interaction with IFN-γ-producing cells [57]. However, this process is influenced by a variety of factors throughout inflammation, not solely by a specific CD4+ T-cell subset. While CD8+ T cells have been identified in synovial joints, their interaction with CD4+ T cells and their activation mechanisms are not yet fully understood [58].
Th17 cells, a distinct subset of T cells, secrete IL-17 independently of cytokines from Th1 or Th2 cells [59]. IL-17 bridges innate and adaptive immunity, exerting both positive and negative effects on immune responses. In addition to IL-17, Th17 cells produce TNF-α, IL-21, IL-22, and granulocyte-macrophage colony-stimulating factor (GM-CSF), making them significant contributors to immune responses [60, 61]. Th17 cells are implicated in the maintenance of immunological homeostasis, such as at mucosal membranes in the intestine, and are increasingly linked to persistent inflammation in RA and other autoimmune diseases [62, 63]. Notably, Th17 cells have been found to play a critical role in the early stages of RA due to immune dysfunction [64]. IL-17 is substantially expressed in the synovial fluid of RA patients, and there is a strong correlation between the proportion of Th17 cells in the synovial fluid and the severity of arthritis inflammation [65, 66]. Furthermore, plasma levels of IL-21 and IL-23, cytokines associated with Th17 cells, strongly correlate with RA severity [67]. Th17 cells release various cytokines, including IL-17A, IL-17F, and IL-22, which stimulate synovial fibroblasts and macrophages to produce proinflammatory substances such as IL-1, IL-6, TNF-α, and prostaglandin E2 (PGE2), exacerbating synovial inflammation [68]. Th17 cells also induce the release of GM-CSF, promoting joint inflammation [69]. IL-17 directly stimulates osteoclastogenesis, leading to bone degradation and cartilage damage [70]. Therefore, Th17 cells contribute to RA pathogenesis by inducing synovial inflammation, pannus development, cartilage and bone degradation, and angiogenesis [71–75]. Additional research into the interactions between Th17 cells and other T-cell subsets can provide insights into the immunological processes underlying RA.
Regulatory T cells (Tregs), a subset of CD4+CD25+ lymphocytes expressing FOXP3, play a vital role in maintaining immunological tolerance [76]. Tregs release TGF-β and IL-10 to suppress the immune system, and they express markers like CTLA-4, GITR, and CD39, with changing expressions based on circumstances [77]. Tregs inhibit self-reactive T cells through cell-to-cell contact mediated by substances like CTLA-4, which suppress T-cell activation [78]. In peripheral blood, Tregs can be categorized into naive and activated forms based on CD45RA expression [79]. While FOXP3 is expressed in human effector T cells, its transient upregulation does not restrict it to Tregs. Therefore, assessing Tregs for their ability to inhibit immune responses and demethylation of the FOXP3 Treg-specific demethylated region (TSDR) is essential for their characterization [80]. FOXP3+CD25+CD4+ T cells accumulate in the synovial fluid and tissue of RA patients and exhibit suppressive capacity in vitro, indicating their functionality [81]. However, effector T-cell proliferation and proinflammatory cytokines can reduce Treg function in vitro, a phenomenon expected to occur in situ. It has been observed that introducing TNF-α during co-culture experiments inhibits Treg functions. However, it is not clear whether this is due to direct interactions with Tregs or effector T cells [82, 83]. There have been contradictory reports about the frequency of Tregs in RA patients. More stringent characterization of Tregs has revealed that RA patients have decreased numbers of Treg cells in peripheral blood but increased numbers in synovial fluid, raising the question about the functionality of Tregs in RA [84]. The origin of FOXP3+ Tregs in synovial tissues is not well understood; they may develop in situ from conventional T cells (induced Tregs) or originate from the thymus (thymic Tregs) [85]. There is evidence that Tregs with a distinct T cell receptor V gene segment are present in juvenile arthritis patients, indicating that Tregs develop from a separate lineage from conventional T cells [86]. The synovial fluid of RA patients contains cytokines like IL-2 and TGF-β, essential in facilitating the development of Tregs. Treatments that reduce inflammation may improve Treg function, as they have been shown to increase the frequency of Tregs in RA patients. Targeting molecules like IL-2R or IL-6R can shift the balance between Treg and Th17 cells in favor of Tregs [87, 88]. Recent treatment approaches involve administering modest doses of IL-2 to stimulate Treg growth in autoimmune patients, as low IL-2R signaling is crucial for Treg proliferation and development [89, 90]. Clinical trials exploring these treatments for RA are currently underway [91].
The initial phase of differentiation from recently migrated monocytes to mature macrophages takes place within the RA synovial membrane. These subgroups colonize the superficial and deep layers of the lining and the synovial sub-lining and lining layer, respectively. The expression of various activation markers and adhesion molecules highlights the potential functional variations in these regions, which may contribute differentially to the progression of the disease [92].
The synovial joint is a complex structure that connects skeletal elements, facilitating movement. Articular bones are separated by synovial fluid, with articular cartilage covering the corresponding bones. The joint is externally supported by a joint capsule, while the inner synovial membrane encloses the synovial cavity, separating its fluid from exterior joint structures. The synovial membrane consists of a lining layer enriched with macrophages (type A synoviocytes) and synovial fibroblasts (type B synoviocytes). In addition to these, the sub-lining interstitial synovial tissue houses mast cells, adipocytes, blood vessels, lymphocytes, and diverse populations of interstitial macrophages, and fibroblasts [93]. Cytokines produced by macrophages and T cells stimulate synovial fibroblasts, creating feedback loops that polarize and activate macrophages. Synovial fibroblasts respond to TNF-α and IL-1 by producing TNF ligand superfamily member 11 (RANKL) and GM-CSF, crucial for the development of osteoclasts, the bone-resident macrophages [94]. Synovial fibroblasts produce GM-CSF, which possibly enhances the survival and activity of neutrophils and macrophages, promoting arthritis.
Macrophages are among the most abundant cell types in sites of inflammation in RA. Human and mouse synovial tissue macrophages express macrophage markers such as CD64, CD11b, CD68 (human), or F4/80 (mouse). Based on the cell membrane expression of MerTK, a tyrosine-protein kinase receptor involved in efferocytosis and inflammation resolution, and CD206, a C-type lectin mannose receptor functioning as a pattern recognition receptor for pathogens, two primary macrophage populations within the human synovium can be distinguished [16]. These populations include tissue-resident MerTK+CD206+ macrophages and tissue-infiltrating MerTK-CD206- macrophages. In response to arthritis, MerTK+CD206+ synovial tissue macrophages express CD163. Single-cell RNA sequencing (scRNA-seq) and multiparameter flow cytometry studies reveal numerous transcriptionally unique phenotypic clusters within these two synovial tissue macrophage populations, each with varying functions and distribution across different stages of arthritis progression [95].
Several significant clinical observations have provided initial evidence for the pivotal contribution of macrophages to RA pathogenesis. These include the positive correlation between the abundance of synovial tissue macrophages and the extent of joint damage and the association between a high disease activity score in local joints and the abundance of macrophages in the synovial sub-lining layer [96, 97]. Macrophages play an active role in disease, not only by presenting antigens but also by producing reactive oxygen species (ROS), nitric oxide intermediates, and matrix-degrading enzymes. Macrophages contribute to the maintenance of the inflammatory response by attracting innate immune cells like neutrophils and monocytes and promoting T-cell differentiation [98, 99]. In contrast, RA sustains chronic osteoclast activity, resulting in widespread bone erosion. Macrophages isolated from synovial tissue in RA secrete monocyte chemoattractant protein 1 (MCP-1, also known as CCL2) and CXCL8 (also known as IL-8), essential for recruiting monocytes and neutrophils [100]. They also secrete IL-23 stimulating the growth and activation of Th17 cells [101]. Immunohistology and the analysis of dissociated synovium have indicated that macrophages are the primary producers of TNF-α in inflamed RA joints. The therapeutic efficacy of TNF-α inhibitors in treating RA underlines the significance of macrophages in this condition. The precise mechanisms leading to the overproduction of TNF-α by synovial macrophages remain unclear but may involve various factors such as cytokines, T cells, immune complexes, TLRs, or neutrophil extracellular traps (NETs) [95].
Interestingly, in RA, anti-TNF-α medication has proven more effective than blocking IL-17 signaling, suggesting the involvement of other effector pathways. Clinically effective treatments often reduce the number of macrophages in the affected joints, and complete macrophage elimination has been advantageous in RA animal models. The data suggest that macrophages play a crucial role in RA due to their control over the cytokine environment, promoting inflammation and contributing to bone and cartilage deterioration [100]. Macrophage-produced TNF-α, IL-6, and IL-1 can further enhance osteoclast activities, either directly or indirectly. In healthy individuals, osteoclast activity is episodically stimulated, leading to localized bone erosion, typically at cartilage-pannus junctions [102].
Neutrophils are the most prominent cell type in the synovial fluid of RA patients. In RA joints with inflammation, extensive recruitment of neutrophils occurs. Various cell types, including Th17 cells, neutrophils, and epithelial cells, produce the chemokine CXCL8, which is crucial for neutrophil migration and recruitment. Particularly in RA patients, cytokines produced by Th17 cells significantly enhance neutrophil migration. In the synovial fluid of RA patients, both Th17 cells and neutrophils are abundant. Th17 cells activate neutrophils directly by releasing IL-8, and they also produce IL-17, IL-23, TNF-α, various chemokines, and leukotriene B4 (LTB4), all of which facilitate neutrophil migration [103].
Additionally, the cytokine IL-22, related to Th17, is highly expressed in the synovial tissue of mice and humans with RA induced by antigens. This emphasizes the significant role of Th17-induced production of IL-17, -22, and -23 in the migration and recruitment of neutrophils, thereby contributing to the pathogenesis of RA by intensifying joint inflammation. Recent research has revealed that serum complement activation induces Th17 cell-dependent autoimmune arthritis, primarily through the activation of C5a. This finding underscores the role of Th17 cells in the pathogenesis of RA [104].
Although less prevalent in synovial tissue, they are abundant in the cartilage-pannus junction, where synovial tissue invades cartilage. In areas of inflammation, neutrophils exhibit prolonged activity and can inflict substantial damage on RA joints. Neutrophils in RA synovial fluid secrete two critical cytokines and chemokines, RANKL and TNFSF13B (also known as BLyS or BAFF). These substances are associated with the activation of osteoclasts and B lymphocytes, respectively. Moreover, RA synovial fluid neutrophils exhibit immunological activity similar to that of macrophages and dendritic cells, as they can present antigens to T lymphocytes and express MHC class II molecules [105].
Survival of neutrophils in the synovial joint is promoted by hypoxia and the presence of anti-apoptotic cytokines, such as TNF-α, GM-CSF, IL-6, IL-15, and CXCL8, which can extend their lifespan for several days [106]. Th17 cells, prevalent in RA, produce IL-17, doubling the neutrophil lifespan [107]. IL-1 further delays neutrophil apoptosis, intensifying inflammation [108, 109]. Activated neutrophils pose a threat to host tissues by ROS and degranulating, either into the synovial fluid or directly onto the joint surface. This process is known as frustrated phagocytosis, arising from immune complex deposition on the joint surface, creating an environment where neutrophils release high concentrations of oxidants and cytotoxic chemicals directly onto the articular cartilage surface [110].
RA is characterized by synovial inflammation and the formation of pannus tissue, largely attributed to the infiltration and activation of neutrophils. Neutrophils in the synovial fluid, when exposed to immune complexes like RF and ACPAs, release ROS and proteases, which degrade hyaluronic acid and activate cytokines. Additionally, neutrophils near cartilage sites encounter immune complex deposition, leading to frustrated phagocytosis, resulting in the release of harmful substances onto the cartilage. Cathepsin G, a serine protease in neutrophil granules, is involved in RA pathogenesis [111]. Notably, RA synovial fluid shows elevated cathepsin G levels and activity, linked to cartilage degradation [112]. Of note, stimulated neutrophils release PAD4. As previously described, PAD4 stimulates and activates adaptive immune cells, resulting in APCA formation which contributes to RA pathogenesis [113].
One remarkable feature of neutrophils in host defense is their ability to combat pathogens through a process called NETosis, which involves the formation of NETs [114]. In RA, NETs play a critical role in pathogenesis. The NET formation is triggered by danger signals and can involve citrullination [115]. Of note, ACPA-targeted citrullinated peptides are present in NETs [116, 117]. On the other hand, researchers discovered that NET formation is increased by ACPA depending on its concentration [116].
Neutrophils interact with various immune cells to induce inflammation. B cells, stimulated by citrullinated proteins in the presence of ACPAs, activate neutrophil migration by producing CXCL8 [118]. RA synovial fibroblasts internalize NET peptides and present them to T cells, promoting T-cell immune responses [119]. NET formation primes macrophage activation, leading to cytokine production that activates Th17 cells, increasing inflammation [120]. Interestingly, neutrophils were found to express MHC class II, promoting antigen presentation and CD4+ T cell growth, suggesting a role in regulating adaptive immune responses [105, 121].
Conventional therapies for RA include anti-TNF-α and anti-IL-6 receptor treatments [122]. These therapies reduce inflammation and inhibit neutrophil-related functions.
Targeting molecules derived from neutrophils is an emerging approach. Modulating CCR2 [123] and CXCR1/CXCR2 chemokine receptors [124, 125] can inhibit neutrophil migration and function. PAD4 inhibitors have shown some promise in preventing RA severity [126].
RA is a complex disease with a multifaceted etiology, manifesting features of Type II, Type III, and Type IV hypersensitivity reactions simultaneously, engaging both adaptive and innate immune cells. Although the precise cause remains elusive, a combination of genetic and environmental factors is suspected to be involved. Unfortunately, RA cannot be cured yet, but ongoing research strives to formulate treatments targeting the autoimmune aspects of the disease. Despite a myriad of proposed approaches to support individuals grappling with RA, these solutions may fall short of fully addressing the intricacies of the condition. The pursuit of innovative strategies, such as comprehensive combination therapies addressing various facets of RA, holds the potential to significantly improve the prospects of conquering this challenging ailment.
ACPAs: anti-(cyclic) citrullinated protein antibodies
CR: complement receptor
FcRs: Fc receptors
GM-CSF: granulocyte-macrophage colony-stimulating factor
IFN: interferon
Ig: immunoglobulin
NETs: neutrophil extracellular traps
PAD: peptidyl arginine deiminase
RA: rheumatoid arthritis
RF: rheumatoid factor
ROS: reactive oxygen species
Th1: T helper 1
Tregs: regulatory T cells
JH, SH, JRK, JK, and YL equally contributed to: Investigation, Writing—original draft. JHB: Conceptualization, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This work was supported by the National Research Foundation of Korea (NRF) through the Ministry of Education [2021R111A3059820] (to Jea-Hyun Baek). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Yuto Sasaki ... Tadashi Matsuda
Aleksandra Kozlova ... Maria Zakharova
Vasiliy Ivanovich Reshetnyak, Igor Veniaminovich Maev
Nawal Labiad ... Nardjess Ouikhlef
Omer Okuyan ... Hafize Uzun
