 Review
Review

Affiliation:
1Haplomic Technologies Pty Ltd, Melbourne 5087, Australia
2Department of Medicine, University of Melbourne, Victoria 3010, Australia
Email: btait@haplomictechnologies.com; briandt@unimelb.edu.au
Explor Immunol. 2024;4:568–576 DOI: https://doi.org/10.37349/ei.2024.00160
Received: April 04, 2024 Accepted: August 14, 2024 Published: September 25, 2024
Academic Editor: Esma Isenovic, University of Belgrade, Serbia
Type I diabetes susceptibility is caused by both environmental and genetic factors, the latter comprising approximately half of the total risk as evidenced by the fact that identical twins have approximately 50% concordance, suggesting 50% of the disease risk is environmental. The human leukocyte antigen (HLA) genes account for approximately half of the genetic risk, as demonstrated by the concordance between HLA identical siblings. Because environmental and genetic differences vary between racial groups, the incidence of type 1 diabetes (TID) differs across the world, being highest in Caucasians. Recent GWAS (genome-wide association studies) studies have suggested there may be up to 50 genomic regions contributing to the non-major histocompatibility complex (MHC) genetic risk contribution. This review presents and discusses the latest research on the MHC and non-MHC genes. Only the non-MHC regions, which have been confirmed in multiple studies and which are considered definite regions of genetic susceptibility, are included in the review.
Type 1 diabetes (TID) is an autoimmune disease where cytotoxic T cells (CD8) with specificity for the β cells of the pancreas (islet cells), are totally destroyed over time, resulting in failure of insulin (INS) production and a state of hyperglycaemia in the affected individual. The pancreas is able to make sufficient INS with the majority of islet cells destroyed, thereby becoming one of the few diseases where diagnosis of disease is made at the virtual endpoint of the immune process. The distinction between disease onset and date of diagnosis must be made as the difference can be measured in weeks to years depending on the genetic makeup of the individual. Pre-clinical individuals can be identified by the presence of autoantibodies to INS, glutamic acid decarboxylase 65 (GAD65), and islet cells. There is clearly heterogeneity within the clinical diagnosis of TID [1]. Autoimmune destruction of beta cells occur rapidly in most children. Auto immunity can also occur in adults, with the slow destruction of beta cells being referred to as latent autoimmune diabetes (LADA). Although often INS-dependent genes that confer susceptibility to LADA are similar, but not identical, to childhood-onset TID. This review focuses on the genetics of childhood autoimmune diabetes, referred to as TID.
The MHC region, which contains the HLA genes accounts for approximately half of the genetic risk [2]. This review reports the latest findings on the genetic susceptibility to TID dealing with both MHC and non-MHC genes [3].
There are approximately 270 genes and transcripts described within the MHC, which is located on the short arm of human chromosome 6. The HLA genes are located within the MHC and consist of three sub-regions termed class I, II, and III which collectively cover approximately 3 megabases of DNA. The class I region contains the classical class I genes HLA-A, HLA-B, and HLA-C, while the class II region contains the DRA, DRB, DQA, and DQB genes. The class III region contains genes with a variety of immune functions, such as the fourth component of complement (C4) and the TNF genes. One of the confounding issues when analysing HLA associations with disease is the phenomena called linkage disequilibrium (LD). LD occurs when alleles of two neighbouring genes occur together more or less commonly than would be expected based on their individual frequencies. A classic example is the 8.1 haplotype which occurs commonly in Caucasians and consists of the alleles A1-B8-DR3-DQ2. This haplotype exhibits the strongest LD observed and is associated with many autoimmune diseases, including TID. It follows, that historically, there has been little recombination between alleles on this haplotype.
There are two points concerning the HLA associations with TID, which need to be made, namely that the associations should be seen as dynamic and not stationary, reflecting changes in the environmental milieu. This was demonstrated elegantly by Fourlanos et al. [4], who showed in juvenile patients (less than 18 years at diagnosis) that different HLA associations were observed, based on the decade of diagnosis.
Secondly, studies in pre-clinical siblings have shown that based on HLA type, there are two stages of TID development [5]. The first is the auto-immune stage as evidenced by the fact that the HLA class II profiles of siblings who are deemed pre-diabetic, by the presence of TID autoantibodies, are almost identical to the TID patients. The pre-clinical individuals who go on to develop TID are selected on the basis of HLA class I specificities, susceptibility being associated with A24, A30, and B18 while protection seemed to be associated with A1, A28, B14, and B56. This observation is consistent with current knowledge concerning the immunopathology of TID. The damage to the islet cells is caused by CD8+ T cells which recognize auto-antigen in the context of HLA class I molecules, hence the association with HLA class I of those who develop TID. In short, the data indicates an association of autoimmunity with HLA class II alleles, while the progression to TID is associated with HLA class I alleles.
The original genetic association with TID was shown to be the HLA class I specificities B8 and B15, both coded for by the HLA-B gene [6, 7]. This was demonstrated to reflect LD with the serologically defined HLA class II specificities DR3 and DR4 respectively [8] and later the two haplotypes A1-B8-DR3 and A2-B15(62)-DR4 were shown to confer susceptibility to TID in family studies [9]. By contrast, the HLA class II specificity DR2 appeared to be highly protective for the development of TID as was DR5. DR1 was positively associated but less so than DR3 and DR4 [10].
The introduction of DNA sequence genotyping permitted the further definition of these haplotypes and this became a major focus of the Type I Diabetes Genetic Consortium (TIDGC) [10], which was established in 2004. Results from family studies reported in 2008 by Erlich et al. [11] demonstrated that the following DR/DQ haplotypes showed a positive correlation with the development of TID. The most susceptible haplotypes were the DR3 and DR4/DQ8 (*0302) haplotypes. The full allelic specificities were: DRB1*0301-DQA1*0501-DQB1*0201, DRB1*0405-DQA1*0301-DQB1*0302, DRB1*0401-DQA1*0301-DQB*0302, DRB1*0402-DQA1*0301-DQB1*0302. Two DR4 haplotypes showed a lesser degree of susceptibility: DRB1*0404-DQA1*0301-DQB1*0302, DRB1*0801-DQB1*0401-DQB1*0402. The most protective haplotypes were: DRB1*1501-DQA1*0102-DQB1*0602, DRB1*1401-DQA1*0101-DQB1*0503, DRB1*0701-DQA1*0201-DQB1*0303. This data indicates that specific combinations of DRB1 and DQAI/DQBI alleles are associated with susceptibility to TID.
DQAI and DQBI gene products can form trans heterodimers, as first shown by Nepom et al. [12]. DR3/DR4 haplotypes have been demonstrated in many datasets to confer the highest risk, and this has been interpreted as indicating that the highest risk is conferred by the DQAI*0501/DQB1*O302 heterodimer.
The important point to highlight in this dataset is the fact that in Caucasians DRB1*0401 is in LD with DQB1*0301 and the DRBI*0401-DQB1*0302 combination is not a common haplotype. This brings into question the issue of using LD to predict disease risk in a variety of ethnic groups [13].
Horn et al. [14] reported an association of amino acid position 57 in the DQBI molecule with TID. Aspartate at this position seemed to confer protection to the development of TID while a non-negatively charged amino acid was associated with susceptibility. Morel et al. [15] further demonstrated in 27 families, compared with 123 normal non-diabetic controls, there was an impressive association of non-aspartate/non-aspartate homozygotes among the diabetic patients (96% vs. 19.5%), giving a relative risk of 107 [15].
The view at the time was that the highly negatively charged amino acid at position 57 formed a salt bridge with arginine at the non-polymorphic position 79 in the DQA1 molecule. This salt bridge was hypothesized to limit the size of the “diabetogenic peptide” able to bind to the dimeric DQ molecule [16]. The position 57 finding in the DQB1 molecule was confirmed in the Norwegian population a year later, albeit not as strong as in the Horn paper [14], by Ronningen et al. [17] in Erik Thorsby’s laboratory in Oslo.
Tait et al. [18] originally demonstrated that the DQB1*302 allele which was Asp57 negative was replaced by DQB1*0301 in DR1/4 TID individuals. Erlich et al. [19] subsequently cast doubt on the universality of the DQB1 Asp57 story by publishing findings that suggested the association did not apply to Caucasian DRB1/4 diabetic individuals. In these individuals, the association seemed with the DRB1*04 gene (DW4) and the association with the DQB1 Asp57 molecule did not hold for Chinese patients. The conclusion drawn from this data was that although there was a general association with DQB1 Asp57, this association did not confer complete protection to the development of TID, and further that specific combinations of DRB1/DQB1 alleles appeared to confer susceptibility to the development of TID.
In recent years the focus has shifted to HLA haplotypes (combinations of genes) as the MHC contribution to TID susceptibility, in the absence of definitive proof that the susceptibility can be explained, at a functional level by a single gene or allele.
Clark et al. [20] recently published data demonstrating that there are 89 unique miRNA transcripts located within the MHC. These short-length miRNA molecules of approximately 25 mer are intimately involved in controlling the expression of MHC genes. Approximately half of these miRNA molecules are found in LD blocks containing many disease-associated SNPs (single nucleotide polymorphisms), suggesting the RNA molecules may play a part in the etiology of numerous diseases demonstrating an HLA association. One miRNA, miR-6891-5p was found to be located in a conserved intronic region of the HLA-B gene and it modifies the expression of several immune-related transcripts, such as the heavy chain of IgA. The authors point out that miRNA molecules that reside in polymorphic regions of the HLA-B and other MHC genes may also play a role in regulating the expression of genes involved in other biological processes, which may be pertinent to the etiology of TID.
Mengkai Shieh, one of the co-authors of this paper published a second paper in 2018 [21], in which the point is made that most complex diseases like TID involve numerous other genes apart from the MHC, which should always be borne in mind when analyzing HLA data, concerning disease associations. We are entering a new era of thinking about complex auto-immune diseases, such as TID. The last 45 years have been spent cataloguing HLA associations with numerous diseases. The number where a proposed mechanism has turned out to be correct, such as coeliac disease, is very small. It is time for a new approach. The concept of level of expression and the role of miRNA molecules is an algorithm worthy of consideration and study.
When all genome-wide association studies are considered some 60 genetic regions have been shown in individual studies to be associated with susceptibility to TID. A nomenclature has been introduced that labels the susceptibility genomic regions according to their level of influence. The MHC region is designated IDDM1 (INS-dependent diabetes mellitus), the INS gene region IDDM2, and so on up to IDDM18 [22]. However, this review will be restricted to those non-MHC genes that have been shown repeatedly to be associated with TID susceptibility and are now universally recognized as such and are based on the studies of Howson et al. [23]. These 4 genes are the INS gene, CTLA-4, protein tyrosine phosphatase non-receptor type 22 (PTPN22), and interfero induced helicase domain containing protein 1 (IFIH-1).
Bell et al. [24, 25] demonstrated in 1984 before the utilization of genome-wide association scans (GWAS) to study the genetics of TID, that there were three classes of length polymorphism in the 5’ region of the INS gene on chromosome 11, which were termed class I, II, and III characterized by differences in the number of repeat sequences of the consensus sequence ACAGGGGTGTGGGG. Class I contains approximately 40 repeat sequences, class II, approximately 95, and class III, approximately 170 repeat copies of the sequence. Caucasian TID patients were shown to contain an excess of the class I repeats. The class I polymorphisms were associated with a low level of mRNA compared with the class III polymorphism, which supports the contention that INS gene expression is affected in TID. These VNTRs (variable number of tandem repeats) are extremely useful in assessing risk, but the question remains, since they are in a non-coding part of the gene, are they a marker for a polymorphism in the coding part of the gene?
There are no polymorphisms identified in the coding part of the gene which is associated with susceptibility; however, it was subsequently demonstrated that three SNPs in the non-coding part of the gene are in LD with the VNTR region. They are rs842748, rs3842753, and rs689 (-23Hph1), the latter consisting of an A to T mutation. The T allele has been reported as being associated with lower levels of RNA in the thymus [26], leading to the hypothesis that the lower level in the thymus results in a failure to eradicate auto-reactive clones of T cells, in turn leading to clinical autoimmune TID.
The primary role of CTLA-4 is to provide a negative feedback signal to put a brake on T-cell stimulation. The molecule is coded for by a gene on chromosome 2 (2q33). CTLA-4 blockade has proved useful in stimulating the immune response in human cancer [27].
Polymorphism and its association with TID has been the subject of intense investigation. The CTLA-4 gene has the designation IDDM12 and produces a feedback-negative signal by binding to CD80 (B7-1) and CD86 (B7-2) on APC (antigen-presenting cells). CTLA-4 is a member of the immunoglobulin superfamily and deletion of this gene in mice causes excessive lymphocyte proliferation and an auto-immune-like condition.
The original report linking a SNP with susceptibility to TID [28] came from an international group that examined CTLA-4 polymorphisms in 48 Italian families with at least 2 affected offspring. An exon 2 polymorphism at position 49 A/G which produces a threonine to alanine substitution was associated with autoimmunity and specifically TID. However, whether this polymorphism is responsible for susceptibility remains open to discussion. Qu et al. [29] demonstrated, using data derived from the TIDGC that there were multiple SNPs in LD across a 43kb region from the 5’ to the 3’ region of the CTLA-4 gene.
A comprehensive GWAS study of the TIDGC consisting of 2,298 sib pairs families, resulted in the testing of over 11,000 DNA samples [30]. Two sets of SNPs were tested: those that had previously been reported as being associated with TID and had been replicated in at least one independent study (set 1, confirmed) and those that had not been replicated (set 2, replication study). 394 SNPs were tested on both the Illumina and platforms as a quality control exercise. Only those SNPs detectable with both platforms were deemed to associate with TID susceptibility or resistance.
24 SNPs were studied in the CTLA-4 region. Two SNPs in strong LD were found to be associated with TID (rs1427676 and rs231727) at the 3’ untranslated end of the gene. If the results indicate a non-coding polymorphism is responsible for the association with TID, it supports the hypothesis that the level of expression may be a determining factor, bringing into focus the role of miRNA molecules.
The IFIH gene codes for a protein called MDA5 (melanoma differentiation-associated protein 5), also termed IDDM19, and is located on chromosome 2 (q24.2). IFIH-1 is part of the viral RNA sensing system. The protein MDA5 initiates the immune response via interferon-beta (IFNβ). IFNβ is responsible in turn for the expression of HLA class I and for the activation of natural killer (NK) cells.
Multiple SNPs have been described, associated with the IFIH-1 gene [31–36], many of which have been shown to be associated with TID and other auto-immune diseases. Zhang et al. [35] showed an association of 3 SNPs with systemic lupus erythematosus (SLE) in Chinese patients, demonstrating that the rs1990760 T allele was associated with IL-18 and granzyme B levels in SLE patients. Rice et al. [36] associated gain of function polymorphism in 74 individuals from 51 families. They identified 27 pathogenic polymorphic changes in this group. Domsgen et al. [34] correlated the rs1990760 SNP T allele with an increased response of IFN produced in response to infection by the coxsackie virus, which has been implicated in the development of TID.
In contrast, Downes et al. [33] reported that the rs1990760 T SNP, the allele that codes for alanine at position 946 in exon 15, which was believed to resist the development of TID through loss of function, was not the causal variant, as this mutation does not cause loss of function. Resistance to TID appeared to be associated with individuals who were heterozygous for 3 SNPs; a premature stop codon rs35744605 (Glu627X), and two predicted splice variants, rs35337543 (IVS8+1) and rs35732034 (IVS14+1).
In summary, the IFIH-1 gene is highly polymorphic, containing many SNPs in strong LD. Some of the haplotypes, which produce loss of function are associated with protection from TID and autoimmune diseases generally, while gain of function, as measured by increased mRNA levels, causes increased susceptibility. There are still remaining questions demanding resolution, such as the responsible haplotypes involved and how this gene interacts with other genes to produce susceptibility to TID.
LYP is a 110 kDa protein coded for by the PTPN22 gene which is found on chromosome 1 (p13.2) and belongs to a large family of PTPN genes. LYP consists of a terminal phosphatase domain and a long C terminus rich in proline residues.
It is expressed in lymphocytes where it associates with the SH3 domain of the Csk kinase, the complex being a strong negative regulator of T-cell activation through the T-cell receptor (TCR). It acts by dephosphorylating TCR-associated kinases. Bottini et al. [37] first showed that a SNP, rsC1858T (arginine to tryptophan at position 620 in exon 14) is associated with TID. The “wild type” allele C binds Csk, the rarer allele T, which is associated with TID, does not. Functionally, this mutation means that the T allele does not provide a negative signal to the TCR, increasing the autoimmune response. Several other SNPs have been described which have been shown to be benign or of uncertain significance [38].
In summary, the coding variation at amino acid position 620 appears to influence autoimmunity in general, and TID specifically.
Despite years of investigation the MHC gene responsible for conferring susceptibility to TID has not been identified. Recent research on polymorphic miRNA molecules, and their targets in the non-coding regions of HLA genes, has raised the possibility that the control of the expression of HLA haplotypes may play a role in conferring susceptibility. Only non-MHC gene polymorphisms that have been shown to provide protection or susceptibility to TID in multiple studies are included for discussion. These genes are INS, CTLA-4, IFIH-1, and PTPN22. All the above are now considered part of the network of non-MHC genes involved in the aetiology of TID. There are almost certainly other genes remain to be confirmed, such as the alpha chain of the IL-2 receptor complex (CD25), which evidence in confirmatory studies suggests contributes to susceptibility or protection. One of the missing elements is a gene network linking these genes together in a coherent story, from a normal state through autoimmunity to a diagnosis of TID. This is addressed in Figure 1. What is required is the identification of genes involved in susceptibility to TID, within the approximately 50 genetic regions implicated by GWAS studies, and the construction of a gene network similar to that shown in Figure 1. The inclusion of environmental agents, the role of miRNA molecules and other epigenetic factors (e.g., methylation of responsible genes) will be required to complete the picture [39]. The use of overarching omics data would be invaluable in determining which genes are involved in TID susceptibility.
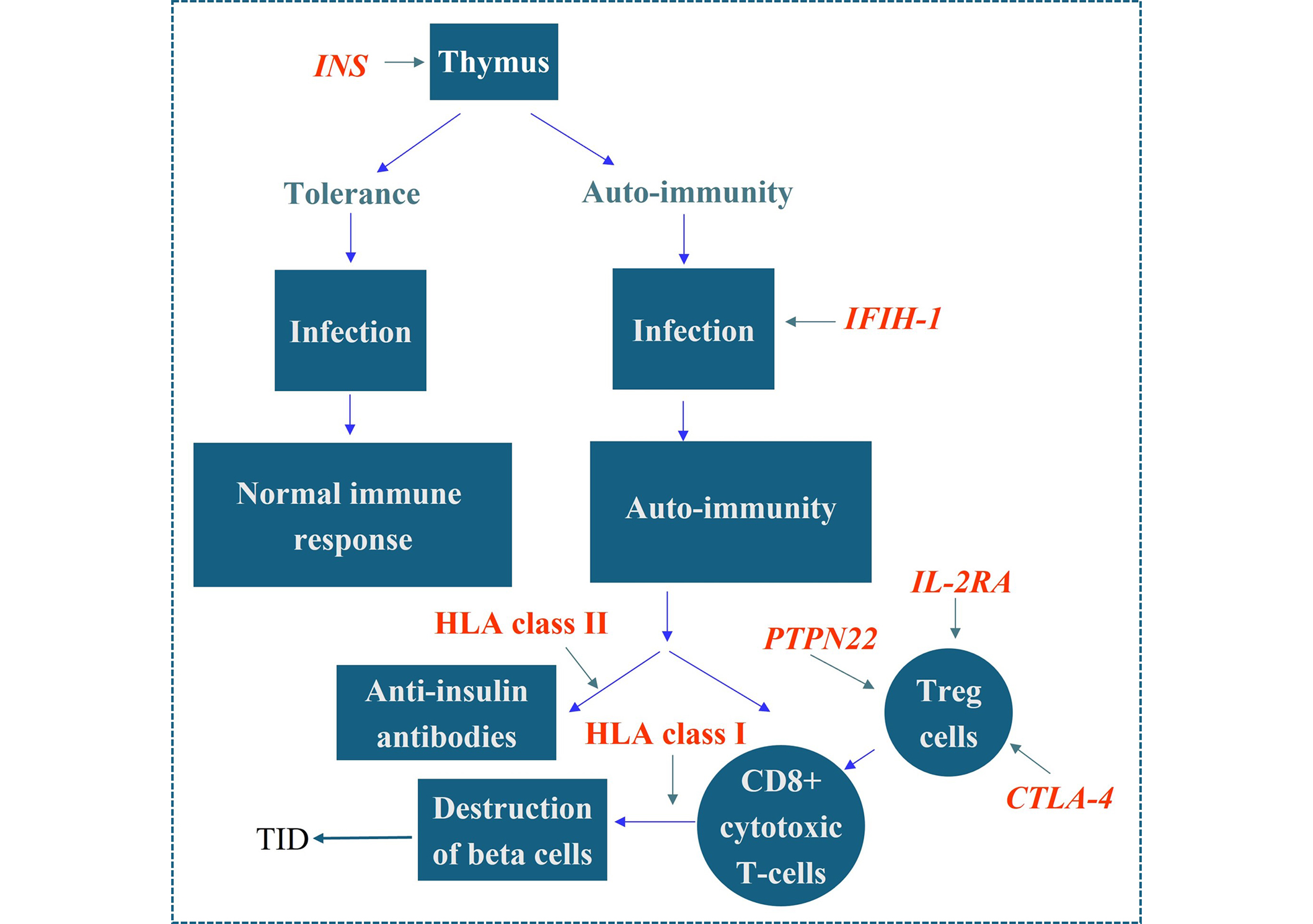
Proposed model for HLA and non-HLA gene effects in TID. 50% of the genetic risk associated with TID resides in the HLA system. The class II genes (DR, DQ) are associated with autoimmunity while the class I genes are associated with progression to TID. The mechanism underlying these associations has not been elucidated, although the general consensus is that it is related to their role in presenting exogenous peptide to CD4 T-cells, and presenting endogenous peptide to CD8 cytotoxic T-cells, respectively. Polymorphisms in the insulin gene produce reduced levels of insulin in TID. Additionally, a role has been demonstrated for the IFIH1gene, polymorphisms of which have been shown to be associated with TID and some appear protective. Polymorphisms in the gene IFIH1 may play a role in the infection stage of TID via IFIH1’s role as part of the viral DNA sensing mechanism. The gene is also responsible for activating the production of IFNβ which in turn upregulates HLA class I hence influencing the rate of β cell destruction. Polymorphisms in the CTLA-4, PTPN22, and IL-2A genes decrease the activity of T regulatory (Treg) cells in TID, thereby enhancing the degree of T-cell destruction of beta cells. HLA: human leukocyte antigen; TID: type I diabetes; INS: insulin; IFIH-1: interferon induced helicase domain containing protein 1; PTPN22: protein tyrosine phosphatase non-receptor type 22
GWAS: genome-wide association scans
GWAS: genome-wide association studies
HLA: human leukocyte antigen
IDDM: insulin-dependent diabetes mellitus
IFIH-1: interferon induced helicase domain containing protein 1
IFNβ: interferon-beta
LD: linkage disequilibrium
MDA5: melanoma differentiation-associated protein 5
MHC: major histocompatibility complex
PTPN22: protein tyrosine phosphatase non-receptor type 22
SNPs: single nucleotide polymorphisms
TCR: T-cell receptor
TID: type I diabetes
TIDGC: type I diabetes genetic consortium
VNTRs: variable number of tandem repeats
BDT: Conceptualization, Investigation, Writing—original draft, Writing—review & editing.
There are no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

