 Review
Review

Affiliation:
1Discovery, Apellis Pharmaceuticals, Waltham, MA 02451, USA
Email: martin.kolev@apellis.com
ORCID: https://orcid.org/0000-0002-7208-3875
Affiliation:
1Discovery, Apellis Pharmaceuticals, Waltham, MA 02451, USA
Email: nageswara.kollu@apellis.com
Affiliation:
2Medical Affairs, Apellis Pharmaceuticals, Waltham, MA 02451, USA
Affiliation:
3Global Business Intelligence and Commercial Opperations, Apellis Pharmaceuticals, Waltham, MA 02451, USA
Affiliation:
1Discovery, Apellis Pharmaceuticals, Waltham, MA 02451, USA
Explor Immunol. 2024;4:577–615 DOI: https://doi.org/10.37349/ei.2024.00161
Received: May 03, 2024 Accepted: September 05, 2024 Published: October 18, 2024
Academic Editor: Uday Kishore, Brunel University London, United Kingdom
The article belongs to the special issue The Complement System in Health and Disease
Complement is both evolutionary and scientifically old. It predates the adaptive immunity by some 600 million years and was first described in 1905 by Jules Bordet and Paul Ehrlich. For the most of its, the existence complement system has been ignored by most scientists and clinicians due to the perception of it being complicated and its relevance for the pathogenesis of human disease being unclear. With the recent US Food and Drug Administration (FDA) approvals of pegcetacoplan for both paroxysmal nocturnal haemoglobinuria (PNH) and geographic atrophy (GA), avacincaptad pegol for GA and iptacopan and danicopan for PNH, we are at a crucial juncture for complement-targeting therapies. A number of companies and academic institutions are developing next-generation complement therapies, which is resulting in an increasingly competitive landscape. If one looks at the serum complement cascade, all 3 pathways now have biotechnology or pharmaceutical industry players with 1 or multiple clinical-stage inhibitors that are expected to be FDA approved within the next few years. Furthermore, with the limited number of clinically validated targets in complement-mediated disease, the competition in this space is set to further intensify in the coming years. In this review, we will discuss the timeline of the academic discoveries that led to the development of the current crop of FDA-approved complement therapeutics. We follow with a discussion of an increasingly crowded complement therapy space and of the scientific advances that have emerged in recent two decades underpinning future innovation, including advances in our understanding of complement biology, such as local and intracellular complement, emerging complement targets, combinational approaches of complement and non-complement therapeutics to unlock new disease indications and new technologies such as gene therapy. We will also give a comprehensive overview of the gene therapy landscape and how it can be utilized to target complement dysregulation.
Several discoveries over the last century provided the knowledge backbone that led to successful clinical development of complement targeted inhibitors (Figure 1). The discovery and characterization of the proteins that formed our understanding of the complement system as 3 distinct serum complement pathways were significant [1]. The period between the initial identification of complement as a heat-labile serum system and the late 1990s was marked by the discovery and characterization of multiple new complement proteins and their functions by numerous scientists, including Professors Hans Muller-Eberhard, Douglas Fearon, Mohamed Daha, KBM Reid, Teizo Fujita, Peter Gal, Peter Zipfel, Robert Sim, Jens Jensenius, Paul Morgan, Marina Botto, John Lambris, Sir Peter Lachman and many other distinguished complementologists. It is through the efforts of these researchers that a critical mass of knowledge of the way in which complement functions was accumulated, and this knowledge shaped both the way in which we view complement as a serum effector arm of innate immunity protecting the host from infections and current therapeutics that aim to block it.
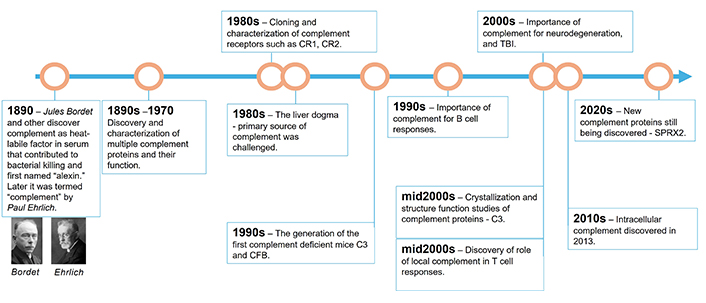
Timeline of key discoveries in the field of complement research. CR1: complement receptor 1; CR2: complement receptor 2; SPRX2: sushi repeat-containing protein 2; CFB: complement factor B
Briefly, complement is comprised of more than 60 cell membrane-bound, intracellular, and plasma proteins [2, 3]. Most of these circulate as inactive precursors in the blood and interstitial fluid and function to activate one another following an initial trigger in a cascade-like manner similar to the coagulation system [4]. The activation of serum complement occurs via the following 3 pathways: the classical pathway (CP), lectin pathway (LP), and alternative pathway (AP) [3, 5]. Complement can be activated by numerous danger signals, such as immune complexes containing IgG or IgM antibodies (CP), pathogen-derived molecules such as lipopolysaccharide (AP), acetylated surfaces or mannan (LP), and apoptotic cells (CP). A low level of spontaneous activation of the third complement component, termed C3, occurs continuously in a process called “tickover” activation of AP. All 3 serum complement pathways converge first at the level of C3 and share a common terminal pathway, initiated with the cleavage of C5 [6]. With the activation of each pathway an enzymatic cleavage event occurs [i.e., C1s or mannan-binding protein-associated serine protease 2 (MASP-2) cleave C4 and C2 to form C4bC2b, C3 convertase of CP, or LP] [7], while factor D cleaves factor B to Bb and Ba, with Bb forming a complex with either C3(H2O) or existing C3b. The C3(H2O)Bb complex is termed proconvertase, while the C3bBb complex is the C3 convertase of AP [6] (Figure 2). C3 convertases and multiple proteases such as cathepsin L, thrombin, or factor Xa can cleave C3 to biologically active fragments C3b (large fragment) and C3a (small fragment) [8–10]. Additionally, C3b generated by any pathway can, in turn, lead to the activation of AP via the amplification loop. It has been estimated in vitro that up to 80% of complement activity of LP and CP comes from AP amplification [11–13].
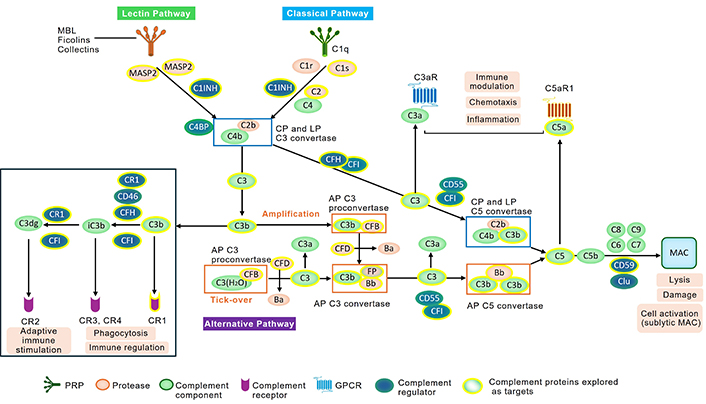
Schematic of serum complement activation cascades. The three activation pathways—alternative pathway (AP), classical pathway (CP), and lectin pathway (LP) are shown. Their activation leads to C3 activation and generation of C3 activation products C3b, iC3b, and C3dg via the action of complement regulators such as complement factor H (CFH), cluster of differentiation 46 (CD46), and CR1 which act as cofactors for the protease complement factor I (CFI). These C3 fragments can be recognized by a number of receptors on immune and non-immune cells and lead to phagocytosis of opsonized particles via complement receptor 3 (CR3), complement receptor 4 (CR4) or adaptive immune activation via CR2, C3a receptor (C3aR) or C5a receptor 1 (C5aR1). Furthermore, C3a and C5a act as anaphylatoxins and recruit immune cells such as monocytes, neutrophils, or T cells to sites of complement activation. C3b association with C3 convertases of AP (C3bBb) or CP/LP (C4bC2b) leads to generation of C5 convertases that cleave C5 and activate the terminal pathway of complement leading to generation of C5b-9 or membrane attack complex (MAC). MAC can lyse erythrocytes or Gram-negative bacteria or in case of sublytic MAC activate some cells. Complement proteins which have been targeted in the clinical studies are highlighted in yellow. GPCR: G-protein coupled receptors; Clu: clusterin; PAR1/4: protease-activated receptor 1 or 4; MASP: mannan-binding lectin serine protease; C4BP: C4 binding protein; C1INH: C1 inhibitor; CR1: complement receptor 1; CR2: complement receptor 2
C5 convertases are formed on surfaces covered with large amounts of deposited C3b and serve to cleave C5 to a large cleavage fragment termed C5b and a low-molecular-weight anaphylatoxin called C5a. These powerful effector molecules are generated during complement activation—C3a and C5a exert their function through cognate G protein-coupled receptors (GPCRs), C3aR, C5aR1, and C5a receptor 2 (C5aR2, formerly known as C5L2)—and drive a diverse set of processes such as inflammation, immune cell modulation and chemotaxis, cell survival, tissue repair and regeneration, vasodilation, and smooth muscle contraction [4–6, 14]. C5b generation gives rise to MAC, a 1,500 kDa complex that forms a pore in the membrane of target cell or a microorganism. It consists of C5b, C6, C7, C8, and multiple copies of C9 [15]. The MAC-induced lysis is important in the defence against invading Gram-negative bacteria, enveloped viruses, and parasites [15]. There are many excellent reviews that cover the intricacies of extra- and intracellular complement, so we will focus on the therapeutic implications in this review [1, 4, 6, 14, 16, 17].
Since there are many important contributions that are beyond the scope of this review, we will focus on a few key points from its discovery until today (Figure 1) [1]. Some of the most important discoveries were made possible by an advancement in cloning technology that enabled the cloning of complement receptors such as complement receptor 1 (CR1), complement receptor 2 (CR2), and CR4, as well as the characterization of their specific patterns of cellular expression [18–20]. The discovery of the membrane complement regulators CD46, CD55, and CD59 happened in the mid to late 1980s [21–23]. The “liver dogma” that ruled the complement world, and stipulating that the liver is the primary organ of complement protein production, was also challenged in the 1980s [24, 25], thus establishing the prerequisite paradigm shift in thinking for the discovery of local complement in late the 2000s [26, 27] and then of intracellular complement in early the 2010s [9].
The generation of the first complement deficient C3 and factor B mice allowed for a rapidly increased understanding of the physiological roles of complement in healthy and disease states [28]. This led to the generation of complement disease mouse models, such as membranoproliferative glomerulonephritis [29] and other rare AP-driven diseases that are a result of mutations in key complement activators and regulators [30]. Another key step in increasing our understanding of complement system dysregulation and contribution to disease came from human genetic studies, unveiling the gain or loss of function mutations in key complement genes and complement deficiencies [31], which was complemented by work done in mouse complement deficient models allowing for better understanding of molecular underpinnings of human disease [32]. Classic examples of this are the deficiencies in CP complement proteins C1, C2, and C4, which are often associated with an increased risk of developing systemic lupus erythematosus (SLE) [31]. MASP-2 deficiency is known to increase susceptibility to recurrent infections [33]. Deficiencies in complement regulators have also been described. For example, complement factor I deficiency leads to an increased risk of atypical haemolytic uremic syndrome (aHUS) and age-related macular degeneration (AMD) [34]. Co-factors for the enzyme factor I are key complement regulators such as CD46, CR1, and complement factor H (CFH) [6]. Mutations in factor H, a key complement regulator in the fluid phase, lead to complement activation and C3 consumption which can lead to aHUS [35]. Deficiencies in key complement proteins such as C3 and C5 have been associated with an increased risk of infections [36, 37]. Interestingly, this increased infection risk did not translate to the clinical use of C3 and C5 inhibitors, most likely due to the requirements for vaccination prior to treatment and most patients being adults with fully developed immune system [38, 39].
Another key advancement in our understanding of complement was the crystallization of key complement proteins such as factor B in 2000 [40], C1s and C1q in 2003 [41, 42], and C3 in 2005 [43]. Important advances in our understanding of the molecular mechanisms of complement activation were also made shortly thereafter, particularly on key complement-activating enzymes such as the convertases of the AP [44–48]. C5 and the terminal pathway deserve their own mention. While structural information on C5a has been available since at least the late 1980s [49], it took until 2008 for the C5 crystal structure to be available [50]. Since 2016, the MAC structure has also been available [51]. The availability of crystal structures helps with developing better therapeutics targeting those proteins.
Along with those preclinical advances, an increased understanding of the aetiology of new, potentially complement-driven diseases has been achieved in recent decades. For example, Dr. Stevens and colleagues [52] led a mini revolution uncovering the role of complement in normal brain development and during neurodegeneration. In 2007, Stevens et al. [52] showed that mice deficient in complement protein C1q or C3 have defects in central nervous system synapse elimination. Furthermore, the authors show that this complement-mediated synapse elimination mechanism can become hijacked in neurodegenerative disease. C1q, C3, and CR3 expressed on microglia were implicated as key players in tagging and removing synapses during Alzheimer’s disease (AD), based on data from mouse AD models [53]. Thus, it is not surprising that a recent study by Veteleanu and colleagues [54] showed that C1q levels are significantly increased in the plasma of patients with AD. Furthermore, single nucleotide polymorphisms (SNP) in other complement genes such as CR1, C1S, and CFH were found to influence their plasma levels, which suggests a deeper involvement of complement in the pathogenesis of AD. The MAC inhibitor clusterin and CR1 were the first complement genes to be associated with AD in the initial large-scale, genome-wide association studies [55]. Currently, there are no complement inhibitors being tested in AD, but this is probably going to change in the future due to the large and growing body of preclinical evidence in this disease.
Genetic studies have also been very useful in deciphering the contribution of the complement system to disease. For example, SNPs in C3, complement factor B (CFB), and CFH genes were long known to be associated with an increased risk of developing AMD [56]. The genetic data along with animal model data [57] and the discovery of C3 and MAC deposits in the retinal tissue of patients with geographic atrophy (GA) [58], have served as a platform for multiple clinical trials for drug candidates such as for pegcetacoplan (a C3/C3b inhibitor), GT005 [a recombinant adeno-associated virus 2 (rAAV2) based gene therapy delivering FI in the eye], iptacopan (a factor B inhibitor) and avacincaptad pegol (a C5 inhibitor) [59].
A great example of the successful clinical translation of mechanistic complement knowledge is that in paroxysmal nocturnal hemoglobinuria (PNH). The sensitivity of PNH patients’ erythrocytes to complement lysis has been postulated since the 1960s [60, 61]; however, it was not until the mid-1980s that researchers started to attribute this sensitivity to a lack of complement regulatory proteins on the erythrocyte surface [62, 63]. The mounting knowledge resulted in the recognition of PNH as a complement-mediated disease in late the 1980s [64]. The next decade marked further the understanding of the molecular causes of PNH as a somatic deficiency of the GPI-anchored proteins CD55 and CD59, which inhibit complement activity on the surface of red blood cells [65]. The add-back of CD59 to CD59-deficient erythrocytes resulted in protection from complement-mediated lysis [66, 67]. The accumulation of preclinical evidence eventually led to clinical trials testing the anti-C5 monoclonal antibody (mAb) eculizumab in PNH [68]. Eculizumab was shown to be both safe and efficacious in the treatment of PNH and was approved by the Food and Drug Administration (FDA) in 2007, marking the first approval of a complement inhibitor in the clinic [69]. Since then, anti-C5 therapeutics have established themselves as a dominant class of complement inhibitors in multiple diseases, with C3 inhibitors more recently emerging as a class of differentiated complement therapeutics [3].
Complement has long been suspected as a potential driver or contributor in a number of human diseases. While the exact number of diseases with complement involvement is not known, we speculated recently that it might be over 400 [3]. As discussed in the previous section, significant advances in our understanding of complement biology have been made over the last 35 years. The merit of the therapeutic targeting of the complement system has long been proposed; however, the approval of eculizumab was when complement therapeutics entered the clinical stage and established the complement system as a target in human disease [70].
The development of anti-C5 antibodies started in the 1990s. The first publication from Alexion Pharmaceuticals (now part of AstraZeneca Rare Disease) was released in 1995 when Evans and colleagues [71] reported the reformat of an anti-C5 mAb into a single-chain variable fragment (scFv). This fragment was shown to bind to C5 and inhibit complement in vitro and ex vivo, similar to the parental clone. The scFv was based on the N19-8 clone, which was a murine anti-human C5 antibody first described by and Würzner and colleagues [72] in 1991 and later by Rinder and colleagues [73] in 1995. By the following year, Alexion reported the successful humanization of the antibody (now termed h5G1.1), while retaining the complement-inhibiting properties of the original clone and continuing to work on the scFv version (Figure 3A) [74]. The monoclonal version of the antibody (i.e., eculizumab) was targeted for multiple indications, such as membranous nephritis, lupus nephritis (LN), rheumatoid arthritis (RA), and dermatomyositis, while the scFV version (i.e., pexelizumab) was to be used for cardiovascular indications [75]. Indeed, Alexion started phase 1 clinical development in dermatomyositis and RA a few years later (Figure 3A) [75, 76]. It was until 2007, however, that Alexion had its first clinical approval in PNH after successful phase 2 and 3 clinical trials [68, 77]. Multiple clinical trials followed, and eculizumab was eventually FDA approved in aHUS in 2011 [78], generalized myasthenia gravis (gMG) in 2017 [79], and neuromyelitis optica spectrum disorder (NSMOD) in 2019 (Figure 3B) [80]. To date, there are 127 studies listed on ClinicalTrials.gov that have eculizumab as part of the intervention or treatment agent (ClinicalTrials.gov, accessed March 21, 2024). Apart from marketing authorized indications, eculizumab has been used off-label in several other diseases with varying success, including C3 glomerulopathy (C3G), antibody-mediated rejection (AMR) of kidney transplantation, and hematopoietic stem cell transplantation-associated thrombotic microangiopathy (HSCT-TMA) [81]. This shows the immense success of the first FDA-approved complement treatment. Eculizumab, while very effective, requires a relatively high weekly dose of 600 mg for PNH and 900 mg for aHUS/gMG in the first month [81]. The weekly or bi-weekly administration is not very characteristic for most therapeutic monoclonal antibodies, so Alexion eventually addressed this by the introduction of a long-acting version of eculizumab (i.e., ALXN1201 or ravulizumab) [82]. Apart from mutations in the Fc domain (M428L, N434S) that enable more efficient neonatal Fc receptor (FcRn) recycling, ravulizumab also has histidine insertions to reduce binding to C5 at pH 6.0 and target mediated drug disposition, both of which result in improved half-life in mice [82] and humans [83]. Ravulizumab eventually gained FDA approvals in all diseases in which eculizumab was previously shown to be efficacious i.e., PNH, aHUS, gMG, and NSMOD (Figure 3B) and [84–86].
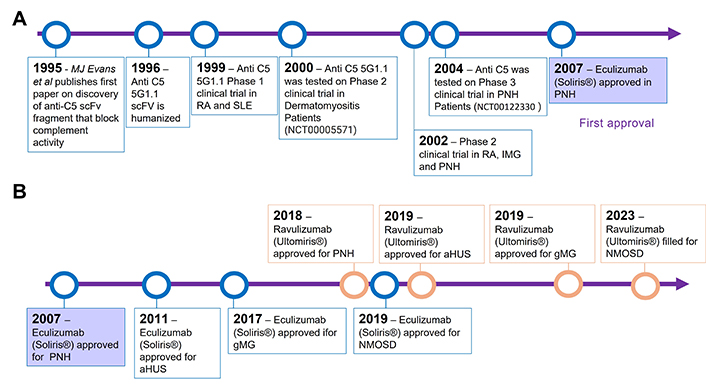
Timeline of key events in C5 antibody inhibitors development. A. Preclinical and early clinical milestones in eculizumab development; B. timeline of FDA approvals of eculizumab and ravulizumab
The clear clinical and commercial success of targeting C5 spurred a lot of interest in both preclinical and clinical development in the C5 inhibitor space. Currently, C5, C5a, and C5aR are the most clinically targeted complement proteins (Table 1). While detailed descriptions of each of these therapeutics are beyond the scope of this review, recent reviews are available [1, 70, 87]. Notable mentions in this category are avacopan, a C5a receptor antagonist that was approved in 2021 for the treatment of anti-neutrophile cytoplasmic autoantibody (ANCA)-associated vasculitis (AAV) [88] and avacincaptad pegol (also known as Zimura), which was approved in 2023 for the treatment of GA, an advanced form of AMD [89]. Vilobelimab (also known as IFX-1) is an anti-C5a mAb developed by InflaRx that was granted an Emergency Use Authorization by the FDA in 2023 for use in patients with severe coronavirus disease 2019 (COVID-19) [90]. This antibody is also in phase 3 clinical testing for pyoderma gangrenosum with data expected to be released in 2025.
Non-exhaustive list of disclosed C5 therapeutics
| Drug name | Company | Modality | Highest clinical stage achieved | Target indications |
|---|---|---|---|---|
| Eculizumab | Alexion/AZ | Humanized mAb | Marketed | aHUS (market), gMG (market), PNH (market), NMOSD (market), gMG pediatric (filed), NMOSD pediatric: phase 3 |
| Ravulizumab | Alexion/AZ | Humanized long acting mAb | Marketed | PNH (market), aHUS (market), PNH pediatrics (market), aHUS pediatrics (market), gMG (market), NMOSD (filed), HSCT-TMA (phase 3), CSA-AKI (phase 3), Renal Basket (phase 2), Dermatomyositis (phase 2) |
| Avacopan | ChemoCentryx/Amgen | C5aR antagonist small molecule | Marketed | AAV: approved (United States, Japan, and European Union)C3G: phase 2HS: phase 2 |
| Zimura (Avacincaptad pegol) | Iveric Bio | Pegylated RNA aptamer | Marketed | GA: marketed (in the United States)Stargardt disease: phase 2 |
| Zilucoplan | UCB | 15-amino acid macrocyclic peptide | Marketed | gMG (market) |
| Vilobelimab | InflaRx | Anti-C5a mAb | Marketed | COVID-19: emergency authorization in the United States, Pyoderma Gangrenosum: phase 3 |
| Pozelimab | Regeneron | mAb | Approved (CHAPLE) | PNH: phase 3gMG: phase 3GA: phase 3 (planned) |
| Eculizumab biosimilars | Multiple | mAb | Multiple phases including marketed | PNH (marketed) |
| Nomacopan (Coversin, AK576) | Akari | Small protein | Phase 3 | HSCT-TMA: phase 3GA: preclinical |
| Cemdisiran | Alnylam | GalNAc conjugated siRNA | Phase 3 | PNH: phase 3gMG: Phase 3GA: phase 3 (planned) |
| Crovalimab | Chigai/Roche | Humanized long acting mAb | Phase 3 | PNH: filed (Japan, China, United States, and European Union)aHUS: phase 3LN: phase 1GBS: phase 3Scleroderma: phase 2 |
| CAN106 | CanBridge | Humanized long acting mAb | Phase1/2 | PNH: phase 1b/2 (China) |
| RLYB-114, RLYB, 116 | Rallybio | Antibody mimetic fusion protein + HSA binder, pegylated mAb | Phase 1 | Rare disease, ophthalmology |
| KP-104 | Kira Bio | Bi-functional mAb (include FH1-5 tail) | Phase 1 | IgAN, C3G, SLE-TMA, PNH: all indications phase 2 |
| CB-301, CB-401 | Cascade Biotech | Not disclosed | Preclinical | Not disclosed |
| PBP1603 | Prestige | Eculizumab biosimilar | Preclinical | Not disclosed |
| HMI-104 | Homology Medicine | rAAV gene therapy | Preclinical | Not disclosed |
| STP145G | Sirnaomics | siRNA | Preclinical | Not disclosed |
| IAB-101 | ImmunAbs | mAb | Preclinical | Not disclosed |
GalNAc: N-acetylgalactosamine; aHUS: atypical haemolytic uremic syndrome; AAV: antineutrophilic cytoplasmic antibody-associated vasculitis; C3G: C3 glomerulopathy; CSA-AKI: cardiac surgery-associated acute kidney injury; COVID-19: coronavirus disease 2019; FH: factor H; gMG: generalized myasthenia gravis; GA: geographic atrophy; GBS: Guillain-Barre syndrome; HSCT-TMA: hematopoietic stem cell transplantation-associated thrombotic microangiopathy; HS: hidradenitis suppurativa; HAS: human serum albumin; IgAN: IgA nephropathy; mAb: monoclonal antibody; NMO: Neuromyelitis Optica; PNH: paroxysmal nocturnal haemoglobinuria; rAAV: recombinant adeno-associated virus; siRNA: silencing ribonucleic acid; SLE-TMA: systemic lupus erythematosus-associated thrombotic microangiopathy; LN: lupus nephritis
The recent FDA approvals of pegcetacoplan for PNH and GA further galvanized interest in the field of C3 complement therapeutics. Pegcetacoplan contains 2 C3-, C3b-, and C3c-inhibiting cyclic peptides that belong to the compstatin family. Those peptides are linked by a 40 kDa polyethylene gycol (PEG) for improved serum half-life [3]. The first compstatin, a 13-mer synthetic C3 inhibitor, was first published in mid-1990s by Sahu and colleagues [91]. It was discovered after screening a phage display library for C3 binders [91]. Since 1996, the compstatin family of C3 inhibitors has grown with at least 7 new members [92]. The newer members of the family are characterized by better stability in serum, increased half-life, and higher potency in inhibiting complement activation [93]. The development of the compstatin family of inhibitors split in 2007 when Potentia Pharmaceuticals licensed the rights to compstatin derivative under the name POT-4. POT-4 was tested in a phase 1 study in patients with AMD [94]. From that initial trial in AMD, it took until 2015 for the next clinical study in PNH to be initiated (NCT02264639) [95]. Like eculizumab, pegcetacoplan was shown to be safe and efficacious in patients with PNH, which led to its first FDA approval in 2021 (Figure 4) [96]. In 2023, pegcetacoplan intravitreal injection was also approved as the first ever treatment for patients with GA secondary to AMD based on positive data from 2 phase 3 studies [97, 98]. It was joined a few months later by avacincaptad pegol, a pegylated RNA aptamer targeting complement at the level of C5 as the second approved treatment for GA [89]. Its proposed mechanism of action and comparison with C5 inhibition has been covered extensively elsewhere [3].
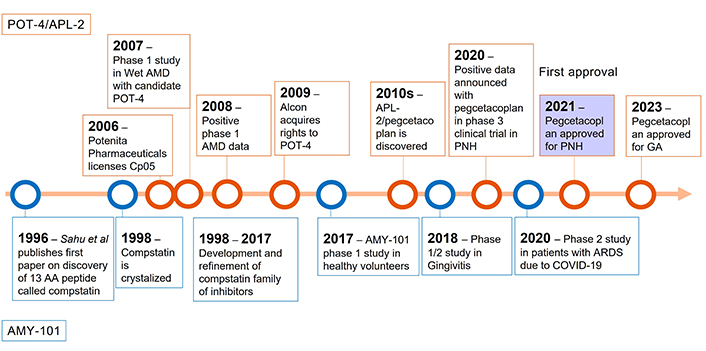
Timeline of key events in development of C3 inhibitors from compstatin family. Top: preclinical and clinical milestones in pegcetacoplan development; bottom: timeline of milestones of AMY-101 development. AMD: age-related macular degeneration; PNH: paroxysmal nocturnal haemoglobinuria; GA: geographic atrophy; AA: amino acid; ARDS: acute respiratory distress syndrome; COVID-19: coronavirus disease 2019
Amyndas Pharmaceuticals took a different approach and instead started clinical development of their lead asset AMY-101, a cp40-based, fifth generation compstatin, in periodontitis (Figure 4) [99] and NCT03694444 after completing a successful phase 1 safety study in healthy volunteers (NCT03316521). AMY-101 was also tested in patients with acute respiratory distress syndrome (ARDS) due to COVID-19 infection in 2021 (NCT04395456). The preliminary results published in 2022 showed a trend towards a reduction in the use of supplemental oxygen at day 14 and a reduction of C-reactive protein (CRP), ferritin, and sustained C3 inhibition [100]. Phases 3 and 2 studies on AMY-101 are planned in periodontitis and in C3G, PNH, and kidney transplantation, respectively (Table 2), www.amyndas.com.
Non-exhaustive list of disclosed C3 targeting therapeutics
| Drug name | Company | Modality | Highest clinical stage achieved | Target indications |
|---|---|---|---|---|
| Pegcetacoplan (APL-2, POT-4) | Apellis | PEGylated cyclic peptide | Marketed | GA: marketed, PNH: marketed, C3G: phase 3 |
| AMY-101 | Amyndas | Cyclic peptide | Phase 2 | Periodontitis: phase 2, COVID-19: phase 2 ongoing, C3G: phase 2 planned, PNH: phase 2 planned, kidney transplantation: phase 2 planned |
| ARO-C3 | Arrowhead | siRNA | Phase 1 | G3G: phase 1/2, IgAN: phase 1/2, development stopped? |
| ALXN2030 | Alexion/AstraZeneca/Novo Nordisk | siRNA | Phase 1 | Chronic active AMR: phase 1 |
| APL-3007 | Apellis | GalNAc conjugated siRNA | Phase 1 | Healthy volunteers: phase 1 |
| CB2782 | Catalyst Biosciences | PEGylated C3-protease | Preclinical | GA: planned, development stopped? |
| ALN-CC3 | Alnylam | GalNAc conjugated siRNA | Preclinical | Development stopped? |
| SLN-501 | Silence/Mallinckrodt | GalNAc conjugated siRNA | Preclinical | Not disclosed |
| CB-101/501/601/801 | Cascade Biotech | Not disclosed | Preclinical | Not disclosed |
| STP146G | Sirnaomics | GalNAc conjugated siRNA | Preclinical | Not disclosed |
| KNP-301 | Kanaph/Samsung Biologics | Bi-specific C3b and VEGF mAb | Preclinical | Retinal disease |
| KNP-302 | Kanaph | Bi-specific C3b and CD59 mAb | Preclinical | Not disclosed |
GalNAc: N-acetylgalactosamine; aHUS: atypical haemolytic uremic syndrome; AMR: antibody-mediated rejection; C3G: C3 glomerulopathy; COVID-19: coronavirus disease 2019; IgAN: IgA nephropathy; mAb: monoclonal antibody; PNH: paroxysmal nocturnal haemoglobinuria; siRNA: silencing ribonucleic acid; GA: geographic atrophy; VEGF: vascular endothelial growth factor
The clinical and regulatory success of pegcetacoplan and, the wealth of preclinical data highlighting the importance of C3 as a complement target have sparked an increased interest in C3 by a number of companies (Table 2). The publicly disclosed C3-targeting pipeline seems to be dominated by silencing ribonucleic acid (siRNA) as a modality compared with C5 pipeline (Table 1). This could be reflective of the specific mechanisms related to the target since C5 knockdown alone might not be sufficient to reduce AP to therapeutically meaningful levels. An example of this is the inability of cemdisiran (C5 targeting with GalNAc-siRNA) alone to control lactate dehydrogenase (LDH) levels in patients with PNH [101]. An alternative explanation could be that this is due to the fact that therapeutic pipelines that target C3 are newer compared with C5 for the historic reasons outlined above.
C3 and C5 have emerged as the most clinically attractive complement targets over the past 2 decades; however, many other complement-targeting therapeutics are being tested in clinical studies or have recently gained regulatory approval. A recent example is the December 2023 FDA approval of the small molecule factor B inhibitor iptacopan (also known as LNP023) for the treatment of PNH in adult patients. This compound, which was developed by Novartis Pharmaceuticals, is the first small molecule therapeutic targeting the complement system to be approved. Currently, there are 13 active clinical studies with iptacopan on ClinicalTrials.gov in multiple complement mediated diseases such as PNH, C3G, aHUS, immune complex membranoproliferative glomerulonephritis (IC-MPGN), IgA nephropathy (IgAN), GA, and LN. Another small molecule, factor D inhibitor, danicopan (also known as ALXN2040) developed by AstraZeneca was recommended for marketing authorization in Europe for the treatment of patients with PNH who were previously treated with C5 inhibitors but still present with significant extravascular hemolysis. It was FDA-approved on the basis of positive phase 3 clinical trial data [102]. In addition to PNH, danicopan is currently being tested in a phase 2 study in patients with GA. Sutimlimab (also known as SAR445088 and developed by Sanofi, is a mAb inhibiting C1s and thus the CP [103]. It was approved in 2022 for the treatment of cold agglutinin disease (CAD), making it the first FDA-approved CP inhibitor [104]. Sanofi is also testing sutimlimab in other disease indications, such as AMR and chronic inflammatory demyelinating polyneuropathy (CIDP). Sanofi is also developing an anti-factor Bb mAb for the treatment of rare renal disease. For further information on the discussed complement therapies, readers are referred to a recent review by Bortolotti et al. [105].
The extracellular complement system is comprised of more than 60 soluble and membrane-bound proteins [3, 6]. It has been growing steadily in numbers in recent decades as additional new complement proteins such as the neuronal complement inhibitor SPRX2 are discovered [106, 107]. Despite this, there is a limited number of non-function redundant complement targets. The 3 complement activation pathways converge first on C3 and then on C5 (Figure 2), thus targeting those initially provided good control of complement activity no matter which pathway was initially activated. Recently, companies have been moving upstream in the complement cascade targeting the enzymatic complexes or upstream activators of a specific pathway. Examples of this are the recently approved or prospective treatments such as factor B (iptacopan, LNP023), factor D (danicopan), C1q (ANX005, Annexon Biosciences), C1s (sutimlimab, SAR443809), C2 (ARGX-117, empasiprubart, Argenx) [108], and MASP-2 (OMS721, narsoplimab, Omeros Corporation) (Figure 2) [109]. Another growing area is the utilization of soluble complement inhibitors such as factor I, factor H (see Gene therapy section for more details), and C1 inhibitors (for acute attacks of hereditary angioedema) to control complement dysregulation.
With these new complement inhibitors being developed, the “obvious” targets in the complement system have already, more or less been covered, including both pathway-specific and broad-range inhibitors. This presents a challenge for future development in the complement therapeutics space since the remaining complement proteins have not been validated clinically and their function and contribution to human disease in many cases are much less clear.
The complement system, as discussed above, is an ancient and flexible part of innate immunity. As such, it has been postulated to play a role in the pathogenesis of a significant number of human diseases (Figure 5A) [3, 13, 14, 70, 87]. However, the list of diseases with clinically proven complement involvement is much shorter (Figure 5B). This presents another challenge for the commercial development of future therapies due to the increased competition in those limited number mostly very rare diseases. Thus, in the future, there might be a lack of patients available for a clinical trial since they are well served by approved therapies. Furthermore, the current set of complement-validated diseases is unlikely to be able to support a full commercial launch for new therapies. PNH is a good example of this. With approximately 500 new patients being diagnosed with PNH in the United States and approximately 220 in the United Kingdom each year [110], it is classified as a rare disease. There are at least 5 marketed therapeutic treatments (not counting the Soliris biosimilars) with multiple therapeutics in phases 2 and 3 trials (Figure 6). While the case of PNH is special due to it having a very clear complement-driven pathogenesis [3] and, consequently, being a “model” disease for which many companies are testing the efficacy of their inhibitors, the situation in other diseases with the clinical validation of complement inhibition is fast becoming more and more competitive.
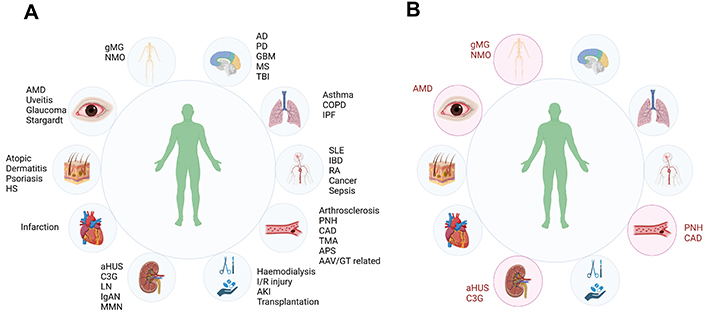
Diseases with suspected and clinically validated complement involvement. A. Schematic representation of disease for which there is pre-clinical evidence to be complement-mediated; B. schematic representation of disease for which there is marketing authorization of complement-targeting therapeutic. * Vilobelimab was granted emergency authorization by the FDA but has not been approved. AAV: antineutrophilic cytoplasmic antibody-associated vasculitis; AD: Alzheimer’s disease; aHUS: atypical hemolytic uremic syndrome; AMD: age-related macular degeneration; C3G: C3 glomerulopathy; CAD: cold agglutinin disease; COPD: chronic obstructive pulmonary disease; GBM: glioblastoma multiforme; gMG: generalized myasthenia gravis; GT: gene therapy; IBD: inflammatory bowel disease; IgAN: IgA nephropathy; I/R: ischemia/reperfusion; IPF: idiopathic pulmonary fibrosis; LN: lupus nephritis; MN: membranous nephropathy; MS: multiple sclerosis: NMO: neuromyelitis optica; PD: Parkinson’s disease; PNH: paroxysmal nocturnal hemoglobinuria; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; TBI: traumatic brain injury; TMA: thrombotic microangiopathy. Figure 5A is created in BioRender. Kolev, M. (2023) BioRender.com/u59k787; Figure 5B is created in BioRender. Kolev, M. (2023) BioRender.com/v03r343
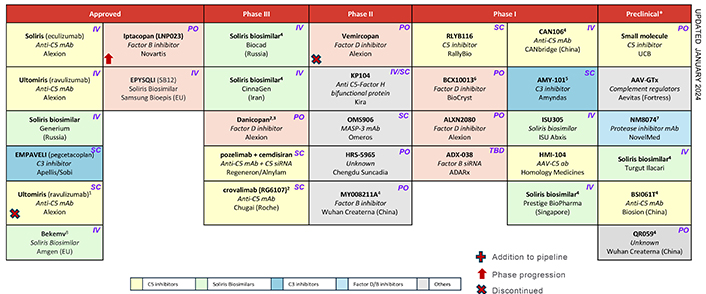
List of therapeutics in development for the treatment of PNH. 1: Approved in the US, EU approval was expected in 1H 2023. 2: Filed in combination with C5 inhibitors. 3: Current development in specified regions only. 4: First phase II will be in periodontal inflammation and gingivitis. Phase II in PNH planned since 2019. 5: Lead indication IgAN. 6: Phase II to start in 1H 2024. 7: Filed in US. 8: Filed in US. * Preclinical list not exhaustive. Alexion has preclinical complement assets: peptide therapies with Zealand Pharma, GalXC RNAi with Dicerna. Alnylam and Silence/Mallinckrodt have C3 siRNAs in development. IgAN: IgA nephropathy; PNH: paroxysmal nocturnal haemoglobinuria; siRNA: silencing ribonucleic acid
Furthermore, the number of companies that have developed or are developing complement targeting therapies has been growing steadily over the past 2 decades. Both larger pharmaceutical companies such as AstraZeneca, Sanofi, and Novartis and smaller biotechnology companies such as Annexon, Kira, Q32Bio, and Omeros have publicly disclosed therapeutics. It seems feasible to expect that the interest from the industry will continue to grow; however, the biggest obstacles that might limit the potential future growth is the limited set of clinically validated targets in mostly rare diseases.
As discussed in the previous section, the current trajectory of the development of complement therapeutics suggests that, in near future, competition in this space might significantly increase, which will benefit patients but might also delay investments from pharmaceutical or biotechnology companies due to very competitive markets and unclear paths for new players. In this section, we will share our thoughts on where the future innovation in the complement therapeutics might come. In our opinion, there are a few key factors that will drive innovation in the coming decades, including, validation of complement inhibition in new diseases, which can be achieved by using combinational therapies with other non-complement therapeutics or leveraging new discoveries in the complement field to design novel complement therapeutics. Finally, the emerging new therapeutic modalities such as CRISPR/CRISPR-associated protein 9 (Cas9) editing, base editing, and messenger ribonucleic acid (mRNA) editing, might allow for more targeted and specific complement inhibitors with new therapeutic properties.
One obvious answer to increasing competition is the validation of existing complement therapeutics in diseases for which there are no established complement therapies. This presents a challenge, as complement involvement in the pathogenesis of most of those diseases is not always clear. Some of the diseases where there is preclinical and anecdotal clinical information are listed in Table 3 and Figure 5. We will not discuss those in detail, however, we will use kidney disease and transplantation as an example since there has been a significant accumulation of preclinical and clinical data. There is active clinical development in these diseases suggesting that the FDA approval of complement therapy is just a matter of time. Kidney diseases with suspected complement involvement include aHUS, IgAN, C3G, and IC-MPGN. Atypical HUS is classified as thrombotic microangiopathy (TMA) that is predominantly renal in nature, unlike thrombotic thrombocytopenic purpura (TTP) and is not caused by Shiga-like toxin-producing Escherichia coli HUS (STEC-HUS) [111]. Atypical HUS can arise due to mutations in complement regulatory genes such as factor H [112], CD46 [113], factor I [112], and others. Autoantibodies targeting factor H have also been reported [114]. Mechanistically, the mutations in complement regulatory proteins and anti-factor H antibodies reduce complement regulation and lead to uncontrolled systemic complement activation.
Selected diseases with suspected complement involvement for which no complement therapeutics have been FDA approved
| Disease or condition | Preclinical complement involvement | Clinical complement involvement | Other treatment options |
|---|---|---|---|
| AD | Shown in multiple mouse models [132].Genome wide associations of complement proteins (clusterin, CR1) with AD [133]. Complement proteins present in plaques [134] and are upregulated in CSF [133]. | No clinical data | Lecanemab [135] aducanumab [136] |
| MS | Complement C3d fragments deposited in lesions and contribute to myelin destruction [137, 138].C3, C1q, and CR1 are associated with visual acuity loss in patients with MS [139].Reducing complement activation is protective in multiple EAE models [140]. | No clinical data | Anti-CD20 antibodies (rituximab, ocrelizumab, ofatumumab, and ublituximab) [141], IFN-1α, IFN-1β, glatiramer acetate, natalizumab, fingolimod, and dimethyl fumarate [142]. |
| Select secondary TMAs | Some drug-induced TMAs are suggested to be complement related [143].In TMAs associated with autoimmune conditions (SLE, catastrophic APS, scleroderma)—complement activation is present in some patients. The CP and the AP are triggered by anti-phospholipid antibodies in vascular and obstetric APS [144]. There is strong evidence for complement activation in TMAs after organ transplant (see text). | Eculizumab showed promising data in patients with SLE/APS in the treatment of TMA and improved kidney function [145].In catastrophic APS and scleroderma, eculizumab treatment showed promising results [146, 147]. A more recent study showed the efficacy of eculizumab in patients with scleroderma renal crisis [148].Eculizumab is frequently used off label in TMA secondary to transplantation [149].Narsoplimab (OMS721) administration resulted in improvement of organ function in phase 2 trial in patients with HSCT-TMA [109]. | Multiple depending on disease, most often steroids and primary disease specific treatments. |
| NAFLD including MASH | Complement is postulated to be activated in NAFLD (C3 fragments, C4d, and MBL/C1q) [150].An increase in complement components C3, C5, CFB, and C3a was associated with an increased risk and severity of NAFLD [151]. The role of AP activation in MASH has been reported [152]. | No clinical data but complement has been proposed as a therapeutic target [150]. | NR1A2 agonist—resmetirom [153]. |
| Insulin resistance and obesity | The level of C3 is increased proportionately to the total amount of adipose tissue and blood glucose levels [154]. C3adesArg might trigger a cytokine response and induce inflammation leading to insulin resistance [155]. C5aR1 signaling in obesity has also been linked to insulin resistance in muscle cells [156].Complement activation might be occurring in T1D, and Genetic variants of C2, CFB, C4A, and C4B appear to play a role in T1D risk [157]. | No clinical data | Anti-GLP-1 antibodies are the leading drug class for the treatment of obesity and also improves blood glucose levels [158]. |
| Acute and long COVID-19 | Patients with long COVID-19 were shown to have increased MAC [159], iC3b, and Ba generation, suggesting AP activation [160].Complement system activation plays a major role in acute COVID-19 [70, 161–163]. | AMY-101 tested in patients with acute COVID-19 showed some efficacy [100].Ravulizumab and zilucoplan were also tested in patients with acute COVID-19 with limited efficacy [164].Vilobelimab showed a reduction in mortality at 28 days in patients on mechanical ventilation and has emergency authorization [90]. | Corticosteroids (particularly dexamethasone), interleukin-6 receptor antagonists (e.g., tocilizumab), and Janus kinase inhibitors (e.g., baricitinib), molnupiravir, nirmatrelvir/ritonavir, and remdesivir. |
| LN | Complement protein deficiencies can lead to SLE (C1q, C1r, C1s, C4, C2).Multiple animal models suggest that complement drives the pathogenesis of SLE [165].The LP might be activated in LN [166].C3d/creatinine levels can discriminate between active and inactive LN, while C3d alone correlates with SLE disease activity index [167]. | Iptacopan (NCT05268289)Narsoplimab (NCT02682407)Ravulizumab (NCT04564339) | Glucocorticoids, MMF, cyclophosphamide, cyclosporin A, rituximab [168], belimumab, and voclosporin [169]. |
| RA | Antibodies against citrullinated proteins activate both the CP and the AP in vitro [170, 171].Multiple animal models using knockout mice suggest a pathogenic role of complement [172–174].TT32 (CR1/CR2 fusion protein) and inhibitor of AP, CP, and LP showed efficacy in 2 animal models of arthritis reducing disease activity [175]. | Blocking C5aR1 using PMX53 did not decrease synovial inflammation and did not change biomarkers associated with clinical efficacy in patients with RA [176]. | TNF-α inhibitors, rituximab, tocilizumab, and nintedanib [177]. |
AD: Alzheimer’s disease; APS: antiphospholipid syndrome; GLP-1: glucagon-like peptide-1; LN: lupus nephritis; MASH: metabolic dysfunction-associated steatohepatitis; MMF: mycophenolate mofetil; MS: multiple sclerosis; NAFLD: non-alcoholic fatty liver disease; RA: rheumatoid arthritis; SLE: systemic lupus erythematosus; TMA: thrombotic microangiopathy; AP: alternative pathway; CFB: complement factor B; COVID-19: coronavirus disease 2019; CP: classical pathway; CR1: complement receptor 1; CR2: complement receptor 2; HSCT-TMA: hematopoietic stem cell transplantation-associated thrombotic microangiopathy; IFN-1α: interferon 1α; LP: lectin pathway; MAC: membrane attack complex; MBL: mannan-binding lectin; TNF-α: tumour necrosis alpha
Complement has been long implicated in IgAN as part of the pathology since most patients with IgAN have C3 deposition in the kidney [115] and potential systemic complement activation exemplified by the presence of circulating C3 opsonized IgA-containing immune complexes [116]. Both the LP and the AP have been implicated [117]. Despite the anecdotal mixed efficacy data for eculizumab in case reports [118–120] today, there are multiple clinical trials testing complement inhibition in patients with IgAN and it seems that we are on the verge of FDA approval in near future [121].
Organ transplantation is another area where there is ample evidence for complement involvement in the rejection. In transplantation, complement activation can occur in both the donor and the recipient [122]. On the donor side, shock and inflammation after death can cause complement activation in multiple organs including the kidney [8, 13]. For example, during reperfusion in human transplanted kidneys, C5b-9 serum levels were shown to be noticeably higher in deceased-donor kidney transplantation compared with living ones [123]. Ischemia has been shown to result in complement activation mostly via the AP, although the LP and the CP were also implicated [13, 124]. The data on ischemia comes predominantly from mouse models of ischemia reperfusion injury (IRI) whereby C3, C3aR, or CFB knock-out mice were shown to be protected from injury [125, 126]. Consistent with those data, factor B-blocking antibody was shown to prevent C3b deposition in mouse kidneys [127]. Probably not surprisingly, complement inhibition pre-transplantation results in the reduction of delayed graft function. A recent study by Danobeitia et al. [128] showed that the complement recombinant C1 inhibitor (blocks CP at C1 complex level) pre-treatment of donor kidney resulted in a reduction of complement CP activation, a reduction in proinflammatory cytokines [i.e., TNF-α and monocyte chemoattractant protein-1 (MCP-1)], and improvement in delayed graft function in a primate transplantation model. The authors found that complement inhibition was associated with superior renal function and a reduction in urinary neutrophil gelatinase-lipocalin (NGAL) [128]. In a pig model of autotransplantation, pre-reperfusion treatment with recombinant C1 inhibitor therapy resulted in a faster recovery of glomerular function and was associated with improved long-term kidney function and reduced fibrosis [129]. On the recipient side, it is known that recipients can develop donor-specific antibodies such as anti-ABO or anti-HLA. These antibodies bind to epithelial and other cells in the transplanted organ and trigger complement activation via the CP in some patients, again pointing to the role of complement in rejection [130]. While the preclinical data are positive so far, the clinical use of eculizumab and C1 inhibitors has been more or less disappointing in that it failed to prevent chronic AMR [131].
While there is mounting evidence for the role of the complement system in cancer and cancer immunotherapy this is beyond the scope of this review, and readers are directed to our recent review on the topic [2].
Another way to improve the efficacy of complement targeted therapy in diseases with more complex aetiologies is to use combination therapy with a complement inhibitor plus standard-of-care medicine. This has long been an established strategy in oncology. For example, combinational therapy with both anti-PD-L1 and anti-vascular endothelial growth factor (VEGF) antibodies, has shown improved efficacy in patients with hepatocellular carcinoma [178]. In addition, the combination of danicopan plus eculizumab or ravulizumab is a complement-specific example of improved control of extravascular hemolysis in patients with PNH who do not respond well to anti-C5 therapy alone [102]. Based on the reduction of C5 in serum after treatment with Cemdisiran, (C5 targeting GalNAc siRNA) one can infer that this will allow for lower doses of eculizumab or ravulizumab to be used for the treatment of PNH or other indications where there is clinical validation of C5 inhibition. A clinical stage example marrying both complement inhibition and oncology is the ongoing phase 2 clinical trial of pegcetacoplan in combination with either pembrolizumab [anti-programmed cell death protein 1 (PD-1) antibody] or pembrolizumab and bevacizumab (anti-VEGF antibody) (NCT04919629). It is expected that complement inhibition at the level of C3 in these patients will reduce C5a generation in the tumor microenvironment and thus aid the efficacy of pembrolizumab. This notion is based on data published by Markiewski et al. [179] showing that C5a can enhance tumor growth via the recruitment of myeloid-derived suppressor cells (MDSC) into tumors and block the function of antitumor CD8+ T cells thus complement inhibition is expected to reverse these processes. In our opinion, combinational approaches that include complement inhibitors plus a non-complement medication might be the best way to approach oncology [180] and autoimmune indications where complement activation is probably just one of the facets of a complex disease network.
Another potential opportunity in expanding the scope of complement therapies is targeting the more recently discovered roles of local and intracellular complement [3, 87]. Currently, the external facing drug pipelines of pharmaceutical and biotechnology companies are, for the most part, focused on targeting systemic complement activation. This is starting to change with the advent of ocular-delivered therapeutics such as pegcetacoplan and avacincaptad pegol as well as the emergence in the clinic of C3 tissue targeting bi-specific molecules such as Kanaph Therapeutics’ KNP-301, a fusion protein of C3b inhibitor and anti-VEGF antibody [181] or Q32’s ADX-097 (C3d targeted FH domains 1–5). These approaches, which have been first established by multiple academic labs [182–184] have the potential to reduce not only the amount of drug required while also reducing systemic complement inhibition and thus improving safety. This also results in improved efficacy via targeting local complement interaction with immune and non-immune cells. Locally produced and activated complement proteins can interact with complement receptors via both paracrine and autocrine signaling pathways which modulates the functions of several immune cell types [185]. For example, Strainic et al. [27] have shown that the endogenous production of C3a and C5a by antigen presenting cells and CD4+ T cells boost T cell proliferation and survival through signaling via C3a and C5a receptors. Another study by the same group revealed a role for the C5a-C5aR1 signaling axis in T cell survival and expansion [26]. Intracellular complement is another recent discovery that is yet to be explored in the clinic. Briefly, Liszewski et al. [9] first identified the presence of intracellular C3 in the endosomal and lysosomal compartments of CD4+ T cells. In resting T cells, C3 is constantly intracellularly activated by the cysteine protease cathepsin L, akin to the tickover mechanism described for serum C3. The generated C3a then engages C3aR on lysosomes to sustain the mammalian target of rapamycin (mTOR) activation required for cell survival [9]. During T-cell activation, T-cell receptor signaling drives activated C3a and C3b fragments to translocate to the cell surface and signal via surface located C3aR and CD46, which, in turn, drives a differentiation into T helper 1 (TH1) phenotype response [186]. Importantly, dysregulated C3 activation and CD46 signaling leads to hyperactive TH1 responses in some autoimmune disease such as RA [9, 187]. In addition to C3, intracellular C5 signaling induces the expression of NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome and IL-1β, further driving TH1 differentiation of CD4+ T cells [188]. The autocrine activation of CD46 is required to induce metabolic rearrangements and mount a TH1 response and allow the production of interferon γ (IFN-γ) [186]. CD46 was also shown to be an important costimulatory molecule for CD8+ T-cell function [189]. The field of intracellular complement has been rapidly expanding with more evidence accumulating in recent years [17, 190–196]. While it has not even been proven that it has relevance in vivo yet, the potential for controlling both innate and adaptive immunity with a single complement therapeutic seems like an enticing proposition.
Finally, emerging therapeutic modalities such as CRISPR-Cas9 editing, base editing, prime editing, mRNA editing, and rAAV-based gene therapy might provide differentiation from existing marketed products which are mostly protein or small molecules in nature. These technologies, along with specific challenges, and opportunities associated with using gene therapy approaches for targeting the complement system, will be discussed at length in the next section.
Gene therapy is an innovative breakthrough in medical science that marks a new section in the treatment of genetic disorders and diseases [197]. It represents a significant shift by targeting the fundamental causes of some diseases, which are defective genes, providing hope where traditional treatments often fail [198]. This section will provide a basic understanding of gene therapy by exploring its key principles, applications, and transformative potential for targeting diseases mediated by complement dysregulation.
Unlike traditional drugs that usually act on proteins or cells, genetic drugs modify gene expression to produce medicinal effects [199]. The treatment involves introducing external nucleic acid sequences into cells to counteract faulty genes, offering highly specific, long-lasting, and potentially curative results in both inherited and acquired disorders [200]. For example, a gene therapy approach is being applied to monogenic diseases such as cystic fibrosis, hemophilia, and muscular dystrophy [201, 202]. However, as our understanding of the genetic foundations of diseases deepens, the range of conditions amenable to gene therapy is expected to expand to more complex polygenic diseases such as cancer, cardiovascular disorders, neurological diseases, and metabolic syndromes [203].
Gene therapy for targeting complement activation represents a novel and still largely untapped approach to treating complement-mediated disease [204]. Various gene therapy approaches exist today including gene augmentation, interference, and deoxyribonucleic acid (DNA)/RNA editing [205]. Each technology can be tailored to the specific mechanisms of the disease of interest, highlighting the versatility and adaptability of gene therapy as a therapeutic option.
Gene augmentation involves the delivery of genetic constructs encoding a healthy gene to replace a dysfunctional one [206]. In the context of complement dysregulation, complement regulators that can be overexpressed to restore complement homeostasis and reduce inflammation associated with complement activation are most often targeted. Gene augmentation typically uses a virus as a carrier to enable the delivery of the gene of interest to the appropriate cells. Different types of viruses, such as adenovirus, adeno-associated virus, herpes simplex virus, and lentivirus, can be used to deliver gene therapies [174, 207]. Among these, rAAV vectors are currently the most common, and they constitute approximately 70% of ongoing clinical trials for one-time gene therapies [197]. Recombinant AAV vectors are preferred because they can transduce dividing and non-dividing cells, ensuring sustained transgene expression through circular concatemers that persist in the nucleus [208]. Recent breakthroughs have enabled the engineering of proprietary rAAVs with improved transduction efficiency and tissue specificity, further strengthening their utility as gene therapy vectors [209, 210]. Despite the advantages of rAAV-based gene therapies, some challenges remain. The major challenges are innate and adaptive immune responses against rAAV, determining the optimal dosage to balance therapeutic efficacy and minimize toxicity, and limited cargo capacity [208, 211]. Thus, a lot of companies and researchers have focused on utilizing this technology for specific tissues such as the eyes to minimize systemic exposure and limit side effects [211].
Gene editing is a groundbreaking technology that enables precise modifications to the genome, including insertions, deletions, and base substitutions [212]. It holds the potential for controlling genetic diseases by addressing mutations in single genes that lead to changes in gene expression in vivo. Over the years, gene-editing technology has evolved through 3 primary generations of development.
The first generation featured zinc-finger nucleases (ZFNs), followed by transcription activator-like effector nucleases (TALENs) in the second generation [212, 213]. However, third generation technology [i.e., clustered regularly interspaced short palindromic repeats (CRISPR)] and Cas are the most widely adopted [214]. This is because it utilizes a Cas9 nuclease and guide RNA (gRNA), the latter of which can be specifically designed for target gene. Thus, this technology enables the very precise editing of any sequence of eukaryotic cells [215]. Using the control of a sequence-specific gRNA, the Cas9 nuclease cuts, replaces, or inserts DNA sequences to precisely achieve the intended purpose of genome editing [216]. The emergence of CRISPR significantly enhanced the efficiency of gene editing and broadened the scope of gene-editing applications because CRISPR offers a crucial advantage over previous techniques: it is much simpler, faster, and cheaper to use [217]. Previous technologies often required the creation of a gene-editing protein from scratch for each specific DNA modification [218]. However, with CRISPR, the same Cas9 molecule can be directed to any sequence by designing a complimentary gRNA molecule, which is considerably simpler to design and easier to synthesize [219]. The gRNA guides Cas9 to the desired site, where it induces a double-stranded break (DSB) repairable via non-homologous end joining (NHEJ) or homology-directed repair (HDR) [216, 220]. NHEJ repair often results in insertions or deletions (indels) of DNA sequences at the Cas9/gRNA cleavage sites, enabling genetic disruption and functional protein knockout [221]. This approach shows promise in treating certain monogenic disorders, as demonstrated in the first human in vivo gene therapy, where liver-specific transcription ablation reduced pathogenic protein accumulation in patients with hereditary transthyretin amyloidosis (ATTR) [222]. There is an FDA-approved therapy using first generation CRISPR technology developed by CRISPR Therapeutics in collaboration with Vertex Pharmaceuticals for the treatment of sickle-cell disease and β-thalassemia [223]. Challenges with first generation CRISPR inlcude, unintended off-target mutations may occur due to editing in additional DNA sequences with blunt ends potentially interfering with repair, leading to various deleterious effects such as genomic rearrangements, duplications, truncations, micronuclei formation, chromosome bridges, and chromothripsis [224].
As a result of these shortcoming of first generation CRISPR technology, researchers have adapted CRISPR-Cas9 to enable the direct generation of precise point mutations in genomic DNA or cellular RNA, bypassing the need for creating DSB, utilizing a DNA donor template, or relying on cellular HDR [225]. DNA base-editors (BEs) consist of hybrids between a catalytically impaired Cas nuclease and a base-modification enzyme that functions on single-stranded DNA (ssDNA) rather than double-stranded DNA (dsDNA). When bound to its target site in DNA, the gRNA forms base pairs with the target DNA strand, causing displacement of a small section of ssDNA in an “R-loop”. Within this single-stranded DNA bubble, the deaminase enzyme modifies DNA bases [226]. Two classes of DNA BEs have been described thus far: cytosine BEs (CBEs) and adenine BEs (ABEs) [226]. Together, these editors enable the installation of all 4 transition mutations (C→T, T→C, A→G, and G→A) using the CRISPR-Cas technology [227]. The challenges associated with base editing are concerns regarding off-target editing and the imperative to uphold high precision [228]. An example of clinical stage base editing aiming to correct a genetic defect is the BEAM-101 clinical trial which aims to ex vivo activate fetal hemoglobin expression in hematopoietic stem cells in patients with sickle cell disease (NCT05456880). The Verve Therapeutics clinical trial represents a different approach in the clinical translation of the technology (NCT05398029). Targeting the proprotein convertase subtilisin/kexin type 9 (PCSK9) gene, a key regulator of low-density lipoprotein (LDL) cholesterol levels implicated in cardiovascular risk, this trial employs precise base editing techniques to reduce the gene expression within liver hepatocytes. By reducing PCSK9 levels in the blood, LDL cholesterol levels will also be reduced, thus the approach holds promise for mitigating the risk of cardiovascular events in individuals suffering from heterozygous familial hypercholesterolemia using once-and-done approach. This trial's significance extends beyond cardiovascular disease, signalling the growing application of base editing in tackling complex genetic disorders.
Prime editing represents another recent advancement in genome editing technology, offering precise and versatile editing capabilities without the need for DSB or donor DNA templates [229]. It enables all 12 possible base-to-base conversions, as well as insertions and deletions, significantly expanding the scope of editable genetic mutations [230]. With its broad editing spectrum, prime editing holds the potential to correct up to 89% of human genetic diseases, making it a promising tool for therapeutic interventions. The prime editing approach functions as a “search-and-replace” mechanism, facilitated by an engineered Cas9 protein fused to a reverse transcriptase, along with a prime editing gRNA (pegRNA) [231]. This editing complex binds to the target DNA sequence recognized by the pegRNA, where it nicks the DNA strand and releases a 3’ DNA end. This end then undergoes reverse transcription, guided by the pegRNA, leading to the incorporation of the desired edit into the genome [231]. Since its discovery in 2019, prime editing has shown promise across various cell types, including postmitotic neurons, mice, organoids, and plants [232]. Despite these preclinical advancements, prime editing technology is still considered in its early stages, facing obstacles that need to be addressed to realize its full potential as an effective and broadly applicable gene editing tool. These challenges are mostly the same as those for base editing and are related to delivery methods due to large size, off-target effects, and efficiency variability [232].
The DNA editing technologies have many advantages, with a key advantage being highly specific and permanent DNA changes that can enable the use of one-and-done therapies in multiple diseases [229]. As discussed above, there are also challenges associated with off-target editing and the delivery to target cells. Recognizing the potential risks of a permanent alteration of DNA sequences, researchers have turned to epigenetic and RNA editing as alternative approaches.
Epigenetic editing does not change the DNA sequence itself but rather modifies the way genes are read and expressed within the cell. This modification, known as epigenetic regulation, involves adding or removing chemical tags to the DNA strand, such as methylation, which can silence or activate genes [233]. Epigenetic editing can be quite unspecific, but recent developments regarding coupling it with CRISPR-based systems can overcome this limitation [234]. It also offers several advantages over traditional DNA editing methods. First, it allows for the fine-tuned control of gene expression, unlike DNA editing, which is more akin to a simple on/off switch. An important advantage of epigenetic editing is its potential for reversibility. Unlike irreversible changes to the DNA sequence, epigenetic modifications can be introduced and erased, offering a level of control and flexibility in gene regulation [235]. This reversibility reduces the risk associated with off-target effects, as any unintended modifications can be corrected.
RNA editing has emerged as a promising alternative to DNA editing for treating genetic diseases, offering several advantages and unique capabilities. Unlike DNA editing, which permanently alters the genetic code, messenger RNA editing allows for temporary modifications to the molecules responsible for protein production, which reduces the risk of off-target effects and provides a safer option for genetic intervention [236]. While DNA editing requires the delivery of complex molecular machinery [e.g., Cas9 and gRNA encapsulated in lipid nanoparticles (LNP) for delivery [230]], RNA editing drugs require just a template, an RNA, and rely on cell intrinsic enzymes termed adenosine deaminase acting on RNA (ADAR), which can convert A→I with cells reading it as G [237]. There are multiple ADARs [238], and researchers and companies alike have been working on exploring their therapeutic potential [239, 240]. Another advantage of RNA editing is that it may be delivered more easily without the need for LNP or viral vectors, unlike DNA editors [237]. However, RNA editing also has its limitations, such as RNA editing targets multiple RNA molecules and long persistence of RNA editors in the cell, both of which raise concerns about lasting off-target effects and immunogenicity [241].
The clinical translation of CRISPR-based gene therapy presents a significant hurdle in the safe and efficient delivery of CRISPR components to target cells or organs [242]. While rAAVs are the primary vectors for in vivo gene therapy due to their high transduction efficiency and relatively low immunogenicity, their small packaging size poses a challenge in delivering large genome-editing tools such as base editors and prime editors [243]. This limitation has led to the development of dual rAAV vector approaches, where the gene of interest is divided into 2 parts and is packaged into separate rAAV vectors. However, these dual rAAV systems face several challenges, including low reconstitution efficiency, the production of unnatural proteins, and safety concerns related to immune responses and adverse effects [243, 244].
Due to the limitations of viral vectors, significant attention is being directed toward the development of non-viral delivery technologies [222]. Non-viral vectors such as LNP offer promising advantages such as a larger packaging capacity, simplified manufacturing processes, and the potential for re-dosing due to lower immunogenicity compared with rAAV [245]. LNPs have gained popularity for in vivo CRISPR gene editing due to their ability to deliver Cas9 mRNA and gRNA to the liver, enabling the transient expression of gene editing proteins and minimizing off-target effects [246]. Moreover, LNPs degrade within a couple of weeks, supporting long-term safety considerations [247]. LNPs also offer the potential for split-dosing, allowing for repeat administration due to their lower immunogenicity compared with rAAV [245]. An example of a successful clinical use of LNPs is patisiran, an FDA-approved LNP-encapsulated siRNA for the treatment of hereditary transthyretin (TTR)-mediated amyloidosis, which demonstrates the feasibility of chronic administration without safety and efficacy concerns [248]. Another example of a successful clinical use of LNPs include the Pfizer/BioNTech COVID-19 vaccine (alternative name, BNT162b2) which has been widely used across the world [249]. While less immunogenic than rAAVs, LNPs still activate the innate immune system and even complement, and this can lead to complications in some patients, such as low transduction efficiency and difficulty with extrahepatic delivery [250, 251]. Safety concerns related to transient liver injury have underscored the need for thorough evaluation and optimization. In conclusion, LNPs have emerged as a clinically mature non-viral platform for the safe and effective delivery of genetic medicines. LNPs played a pivotal role in enabling the FDA approval of groundbreaking therapies such as BNT162b2 and patisiran [252, 253]. However, intravenously administered LNPs predominantly accumulate in the liver and are internalized by hepatocytes, constraining their therapeutic utility in other organs [246, 254]. Targeted delivery is a critical area of current LNP research, aiming to improve delivery outside of the liver, mitigate toxicity and off-target effects, and enhance efficacy in challenging-to-transfect targets.
In terms of using gene therapies to target complement dysregulation, the gene augmentation therapies utilizing rAAV in AMD are the most clinically advanced and the only disclosed approach so far. The basic idea is to block complement activation by using rAAV to increase complement regulator expression for the treatment of AMD. This is perhaps unsurprising since rAAV-based gene therapy is currently the most widely clinically tested form and is particularly well suited for local rather than systemic delivery, and the recent FDA approvals of pegcetacoplan and avacincaptad pegol provided the clinical validation of complement as target. Table 4 summarizes some of the known gene therapies targeting complement dysregulation.
Recombinant AAV-based complement therapies in GA
| Therapeutic | Payload | Company | Target disease | Mechanism of action | Stage |
|---|---|---|---|---|---|
| 4D-175 | Optimized CFH | Aevitas Therapeutics/Molecular Therapeutics | GA | Express CFH to restore complement regulation | Phase 1 expected Q2 2024 |
| GT005 | CFI | Gyroscope Therapeutics/Novartis | GA | Express CFI to restore complement regulation | Phase 2 completed Development stopped |
| HMR59/JNK-1887 | Soluble CD59 | Hemera Biosciences/Johnson & Johnson | GA, wet AMD | Express CD59 to restore complement regulation | Phase 1 completed [255] |
AMD: age-related macular degeneration; CFH: complement factor H; CFI: complement factor I; GA: geographic atrophy
Apart from those examples, there is not a lot of publicly available information on targeting the complement system using advanced forms of gene therapy. Here, we want to share our thoughts on how utilizing base, prime, or RNA editing might have specific advantages and disadvantages compared with peptides, antibodies, and small-molecule-based therapies which are currently the clinically established modalities for complement inhibition.
One key benefit of gene therapy such as base or prime editing is dosing convenience. Unlike conventional therapies that often require frequent and long-term intravenous or subcutaneous administration, gene therapy typically involves a single dose [256]. This eliminates the need for frequent clinic visits and reduces the burden of treatment on patients and caregivers. Most complement therapies, such as pegcetacoplan, eculizumab, and iptacopan require either twice daily or weekly administration to achieve complement inhibition due to an abundance of the targets in serum [81, 257, 258]. A notable exception of this is ravulizumab, which has been specifically engineered to have a longer half-life [82]. Thus, a gene therapy involving complement inhibition might have an advantage over current standard of care complement inhibitors by requiring a single dose for the long-lasting control of the complement system.
A second potential benefit is with regard to improving safety. The frequent dosing of traditional therapies can increase the risk of adverse effects, including infusion reactions and complications associated with long-term drug exposure [259]. By contrast, a single dose of gene therapy minimizes the cumulative exposure to potentially harmful agents, reducing the risk of adverse events. Furthermore, one-and-done gene therapy has the potential to improve efficacy by providing sustained therapeutic benefits over an extended period. This has been shown by Musunuru et al. [260], who successfully used base editing to reduce PCSK9 and LDL in the blood of non-human primates for at least 8 months after a single treatment. This was an improvement over ALN-PCSsc, an RNA interference agent that reduced PCSK9 for up to 6 months after 2 administrations [261]. This continuous efficacy after a single dose, can slow disease progression, as it eliminates the disruptions in treatment adherence that ca be associated with conventional therapies [262]. The lessons learned from PCSK9 and ATTR inhibition can be applied in the future to complement inhibition.
Base and prime editing both present a highly promising avenue for tackling complement-mediated diseases, offering diverse strategies such as correcting point mutations that lead to protein dysfunction, silencing genes through splice disruption, creating stop codons, or directly disrupting proteins [263]. Diving deeper into the first point, multiple complement activators and regulators are known to have pathogenic variants associated with the disease of complement dysregulation, particularly of the AP [264]. Since AP lacks the specificity of activation and amplification, polymorphisms in complement regulators (CFH, CD46, CFI) or activators [C3, CFB, complement factor D (CFD)] can lead to an overactive AP [264]. In diseases such as AMD, where approximately 50% of the total genetic risk comes from a point mutation in CFH [265, 266], one can imagine that, by using gene therapy to correct the Y402H (rs1061170) variant, for example, long-lasting complement homeostasis can be achieved. Other examples include C3 polymorphisms such as C3102G, which is suspected to lead to an overactivation of the AP [266] and is associated with diseases such as AMD, IgAN, transplant dysfunction, and systemic vasculitis [267–269]. Editing C3102G (also known as C3 fast or C3F) to C3102R (also known as C3 slow or C3S) can improve regulation by factor H and reduce the overall activity of AP [266] thus reducing the risk of developing disease. Another advantage of this potential approach is that C3S is a naturally occurring variant and thus presents a low risk.
An alternative approach would be the inhibition of key components of the complement cascade that are responsible for its initiation and amplification, similar to the use of siRNAs to target C3 or C5 today but with the benefit of a single administration. This approach could be particularly beneficial in chronic systemic conditions where the complement system is constantly overactivated, such as in PNH or C3G. The main advantage here is that a single dose of gene therapy can achieve long-lasting control of the complement system. While infection risk with permanent complement inhibition will be one of the key issues to monitor for, to date, therapies such as C3 inhibitors have been well tolerated [38]. This emphasizes how important it is to choose the right target to avoid those safety concerns. The first probable application of gene therapy targeting complement inhibition could be as a maintenance therapy whereby 50% to 70% inhibition is sufficient to control disease progression in most patients, while minimizing bacterial infection concerns. Limitations such as the permanent nature of the DNA editing, off-target editing, and unknown long-term safety means that it will only be applicable to certain targets and mutations in a limited set of life-long and life-threatening disease until the wider clinical adoption of the technology.
The permanent nature of the base and prime editing might not be suited for all diseases and for the needs of all patients. This is where epigenetic and RNA editing might take center stage due to their unique advantages. Epigenetic editing technologies offer precise control over gene expression by altering epigenetic marks such as DNA methylation, histone modifications, and chromatin structure [235]. One strategy involves targeting specific epigenetic regulators or chromatin-modifying enzymes to increase the expression levels of complement regulators, similar to the rAAV approach discussed previously, with the view of restoring homeostasis. Since epigenetic editing is, in theory, reversible and could allow for the fine-tuning of protein expression rather than acting as an on/off switch it is expected to be safer than DNA editing [234, 235, 270]. One can imagine that reversible complement activator knock-down will be possible in the future using this technology. Chroma Medicine has already demonstrated the efficiency of epigenetic editing of its platform in reducing and restoring PCSK9 levels [271].
Silencing RNA targets specific mRNA molecules for degradation, reducing the expression levels of targeted proteins, and has been used to target C3, C5 and CFB protein expression in the clinic (Tables 1 and 2). RNA editing offers unique advantages, as it allows for precise modifications to be made to RNA molecules [241], thereby directly altering the protein-coding sequences without affecting the underlying genomic DNA [240]. This approach enables transient and reversible changes to gene expression, offering flexibility in modulating the cellular pathways involved in serum and intracellular complement activation and regulation for example. Furthermore, RNA editing can be used to modulate alternative splicing events or RNA stability, thereby influencing the expression levels of specific complement isoforms or regulatory factors similar to the strategy outlined above for DNA and epigenetic editing. The advantage of RNA editing over the other discussed gene therapy strategies is that it is easier to deploy since it does not require additional enzymes [223, 237].
While competition in the complement therapeutics space is set to increase in coming years, advancements in technology, such a gene therapies, can provide differentiation by unlocking new disease indications, and providing more convenient, longer lasting, and potentially safer treatments for patients. Today, the gene therapy toolbox has never been more diverse, with multiple options such as base editing, prime editing, epigenetic editing, and RNA editing. This variety both enriches and underpins the foundation of future precision medicine, providing scientists and clinicians with a multitude of potential strategies for combating complement-mediated diseases. There are still many challenges associated with gene therapy due to its relative novelty, such as unresolved delivery to target organs or cells outside of the liver, long-term safety that is not well understood, unclear regulatory path, and extremely high costs for patients. However, since 2013, when the initial use of CRISPR for genome editing was described, its use has been exponentially growing, and CRISPR-based first medicine was recently FDA approved [223, 272]. We believe that gene therapies have the potential to become one of the most dominant therapeutic classes alongside antibodies and small molecules. Despite their current shortcomings, gene therapies offer unique advantages of personalized treatments for complement-mediated disease, and this might be what drives future innovation in the complement therapeutics space.
AAV: anti-neutrophile cytoplasmic autoantibody-associated vasculitis
ABEs: adenine base-editors
AD: Alzheimer’s disease
ADAR: adenosine deaminase acting on RNA
aHUS: atypical haemolytic uremic syndrome
AMD: age-related macular degeneration
AMR: antibody-mediated rejection
AP: alternative pathway
APS: antiphospholipid syndrome
ATTR: transthyretin amyloidosis
BEs: base-editors
C3aR: C3a receptor
C3G: C3 glomerulopathy
C5aR1: C5a receptor 1
CAD: cold agglutinin disease
Cas9: clustered regularly interspaced short palindromic repeats-associated protein 9
CBEs: cytosine base-editors
CD46: cluster of differentiation 46
CFB: complement factor B
CFD: complement factor D
CFH: complement factor H
CFI: complement factor I
COPD: chronic obstructive pulmonary disease
COVID-19: coronavirus disease 2019
CP: classical pathway
CR1: complement receptor 1
CR2: complement receptor 2
CR3: complement receptor 3
CR4: complement receptor 4
CRISPR: clustered regularly interspaced short palindromic repeats
CSA-AKI: cardiac surgery-associated acute kidney injury
DNA: deoxyribonucleic acid
DSB: double stranded break
dsDNA: double-stranded deoxyribonucleic acid
FDA: Food and Drug Administration
GA: geographic atrophy
GalNAc: N-acetylgalactosamine
GBM: glioblastoma multiforme
GBS: Guillain-Barre syndrome
GLP-1: glucagon-like peptide-1
gMG: generalized myasthenia gravis
gRNA: guide RNA
HAS: human serum albumin
HDR: homology-directed repair
HS: hidradenitis suppurativa
HSCT-TMA: hematopoietic stem cell transplantation-associated thrombotic microangiopathy
I/R: ischemia/reperfusion
IC-MPGN: immune complex membranoproliferative glomerulonephritis
IFN-1α: interferon 1α
IgAN: IgA nephropathy
IPF: idiopathic pulmonary fibrosis
IRI: ischemia reperfusion injury
LDH: lactate dehydrogenase
LDL: low-density lipoprotein
LN: lupus nephritis
LNP: lipid nanoparticles
LP: lectin pathway
mAb: monoclonal antibody
MAC: membrane attack complex
MASH: metabolic dysfunction-associated steatohepatitis
MASP-2: mannan-binding protein-associated serine protease 2
MBL: mannan-binding lectin
MCP-1: monocyte chemoattractant protein-1
MDSC: myeloid-derived suppressor cells
MMF: mycophenolate mofetil
mRNA: messenger ribonucleic acid
MS: multiple sclerosis
NAFLD: non-alcoholic fatty liver disease
NGAL: neutrophil gelatinase-lipocalin
NHEJ: non-homologous end joining
NSMOD: neuromyelitis optica spectrum disorder
PAR1: protease-activated receptor 1
PCSK9: proprotein convertase subtilisin/kexin type 9
PD: Parkinson’s disease
PD-1: programmed cell death protein 1
pegRNA: prime editing guide RNA
PNH: paroxysmal nocturnal haemoglobinuria
RA: rheumatoid arthritis
rAAV2: recombinant adeno-associated virus 2
scFv: single-chain variable fragment
siRNA: silencing ribonucleic acid
SLE: systemic lupus erythematosus
SLE-TMA: systemic lupus erythematosus-associated thrombotic microangiopathy
SNP: single nucleotide polymorphisms
SPRX2: sushi repeat-containing protein 2
ssDNA: single-stranded DNA
STEC-HUS: Shiga-like toxin-producing Escherichia coli haemolytic uremic syndrome
TALEN: transcription activator-like effector nucleases
TBI: traumatic brain injury
TH1: T helper 1
TMA: thrombotic microangiopathy
TNF-α: tumour necrosis alpha
VEGF: vascular endothelial growth factor
ZFNs: zinc-finger nucleases
Authors want to thank Matt Ferenc for his suggestions in improving the manuscript.
MK: Conceptualization. MK and KNR: Writing—original draft. MY, AP, and PD: Writing—review & editing. All authors agree with the contents of the manuscript.
KNR, MY, AP, PD and MK are employees and shareholders at Apellis Pharmaceuticals Inc. Apellis researches, produces, and markets products that inhibit the complement system.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Célia Dos Santos ... Analía Sánchez-Luceros
Mariagiulia Spazzapan ... Roberta Bulla
Jeyaparthasarathy Narayanaperumal, Ganesh Gopal
