 Review
Review

Affiliation:
Department of Propaedeutics of Internal Diseases and Gastroenterology, Russian University of Medicine, 127473 Moscow, Russian Federation
Email: vasiliy.reshetnyak@yandex.ru
ORCID: https://orcid.org/0000-0003-3614-5052
Affiliation:
Department of Propaedeutics of Internal Diseases and Gastroenterology, Russian University of Medicine, 127473 Moscow, Russian Federation
ORCID: https://orcid.org/0000-0001-6114-564X
Explor Immunol. 2024;4:624–639 DOI: https://doi.org/10.37349/ei.2024.00163
Received: May 02, 2024 Accepted: September 24, 2024 Published: October 18, 2024
Academic Editor: Margherita Sisto, University of Bari Medical School, Italy
The article belongs to the special issue Chronic Inflammation and Autoimmunity
Primary biliary cholangitis (PBC) is a chronic cholestatic progressive liver disease associated with cholangiopathies. The detection of antimitochondrial autoantibodies (AMAs) plays an important role in the diagnosis of classical PBC. AMAs are formed against the antigenic component associated with the dihydrolipoyl transacetylase of pyruvate dehydrogenase complex (E2 PDC) localized on the inner membrane of mitochondria. The loss of immune tolerance of E2 PDC in PBC is thought to be the cause of the mechanism of AMA formation and immune-mediated destruction of biliary epithelial cells (BECs) of the small- and medium-sized intrahepatic bile ducts. E2 PDC is not only present in BECs, but is also abundant in the mitochondria of all nucleated cells. The question remains as to why E2 PDC of only small BECs is the target of autoimmune attack. There is no evidence that AMAs have a deleterious effect on BECs. New scientific data has emerged that explains the damage to BECs in PBC by the defect of the biliary bicarbonate (HCO3–) “umbrella” that protects BECs from the detergent action of bile acids under physiological conditions. Disruption of HCO3– production by BECs in PBC leads to changes in the pH of hepatic bile, accompanied by accumulation of bile acids in the small BECs. The detergent action of bile acids leads to damage of membrane structures of BECs and their apoptosis, development of ductulopenia, and intrahepatic cholestasis. For the first time, it has been suggested that under the influence of bile acids, the E2 PDC antigen may undergo conformational changes that alter its immunological properties. E2 PDC becomes a neoantigen that is recognized by the normal (“healthy”) immune system as a foreign antigen, leading to the production of AMAs. For the first time, the authors of this review provide an explanation for why only small BECs are damaged in PBC.
Primary biliary cholangitis (PBC) is a chronic cholestatic progressive liver disease with destruction, apoptosis, and necrosis of the epithelium of mainly small intralobular and septal bile ducts, with the development of ductulopenia and cholestasis, in the terminal stage of which liver cirrhosis develops [1, 2]. Damage to small biliary epithelial cells (BECs) by bile acids underlies the pathogenetic mechanism of PBC development. The imbalance between aggressive factors (bile acids) of the bile and protective factors [biliary bicarbonate (HCO3–) “umbrella”] of the BECs triggers this process [2]. The imbalance develops as a result of insufficient HCO3– supply to the bile ducts due to decreased activity of inositol-1,4,5-triphosphate receptor type 3 (InsP3R3) and chloride/HCO3– anion exchanger 2 (AE2) caused by increased activity of microRNA 506 (miR-506) in BECs lining the bile ducts [3]. The factors that induce the increased expression of miR-506 gene remain unknown. The absence of significant concordance indices between monozygotic twins in PBC strongly suggests that epigenetic changes, which do not affect DNA structure, are involved in the initiation of the pathological process [4]. Damage to small BECs is accompanied by a progressive inflammatory ductal reaction and fibrosis, ultimately leading to small bile duct ductulopenia and cholestasis [5].
The cornerstone of the diagnosis of PBC is the detection of antimitochondrial autoantibodies (AMAs) and/or PBC-specific antinuclear autoantibodies (ANAs) in patients presenting with cholestatic liver diseases [6]. The specificity of the presence of AMAs in plasma reaches more than 95% [7]. It is one of the defining characteristics of a pathological process in small BECs [7]. A 2014 meta-analysis comprising 24 studies demonstrated that the combined sensitivity and specificity of AMAs in the diagnosis of PBC were 84.5% and 97.8%, respectively [8]. AMAs are detected in patients already in the asymptomatic stage of PBC, indicating their involvement in the mechanism of disease development. The presence of AMAs and ANAs served as the basis for considering PBC as a prototype of autoimmune disease and for describing it as a “paradigmatic model of autoimmune disease” [4, 9–11]. The foundation of modern diagnosis for the majority of autoimmune disorders is the identification of autoantibodies directed against the body’s own tissue antigens [12]. Autoantibodies are frequently referred to as “witnesses of the autoimmune process” [12]. It is postulated that the presence of high titers of AMAs in the majority (95%) of patients with PBC and ANAs in approximately 50% of these patients strongly suggests a loss of tolerance of B-cells to their own antigens [13]. Therefore, since approximately the mid-1960s, PBC has been described as an autoimmune liver disease [14]. Moreover, PBC is described as a chronic cholestatic liver disease that is characterized by immune-mediated destruction of BECs of small and medium intrahepatic bile ducts [1, 15]. PBC is considered to be an autoimmune disease due to its shared characteristics with other autoimmune diseases. This disease primarily affects women, and autoreactive T cells play a role in the pathological process [16–20] (Table 1).
Presence and absence of characteristic signs of autoimmune disease in PBC
| Autoimmune disease signs | The presence or absence of a sign in the PBC | Explanation | References |
|---|---|---|---|
| Immune dysfunction leading to loss of tolerance to certain tissues and proteins | In doubt | AMAs persist after liver transplantation but do not correlate with disease recurrence | [1, 9, 10] |
| Autoimmune autoantibodies | Yes | АМАs, 95% (+); 5% (–) | [7, 21] |
| Approximately 50% of patients with PBC have antinuclear autoantibodies: anti-gp210, anti-Sp100, anti-p62 | [22] | ||
| Development of immune complex-mediated inflammation; damaging effect of immune complexes on healthy tissues | In doubt | AMAs are formed in experimental models of PBC after stimulation with recombinant E2 pyruvate dehydrogenase protein without causing inflammation or bile duct destruction | [1] |
| Systemic (multiple organ) dysfunction of the internal organs | No | Local lesion of BECs of small bile ducts | [9] |
| There is a correlation between disease severity and antibody titers | No | Antibody titers are not correlated with disease severity | [1, 4] |
| Identifying the antigen has not led to a clear understanding of the primacy origin of AMAs and the disease | |||
| Women more likely to be affected | Yes | Female to male ratio 9:1 | [23] |
| Chronic course of disease | Yes | Chronic, gradually progressive course of the disease | [1, 10, 23] |
| Spontaneous remissions are possible, alternating with exacerbations | No | Disease course without remissions and exacerbations of disease activity | [4] |
| May occur during the childhood years of life | No | PBC has no analogs in children | [10] |
| PBC is more common between 40 and 60 years | |||
| Positive effects of immunosuppressive therapy and genetically engineered biologics (targeted therapy) | No | Immunotherapy is not effective | [24–26] |
| Occasionally seen associated with other autoimmune diseases | Yes | The probability of association with extrahepatic autoimmune diseases in patients with PBC is over 60% | [27–29] |
| The most common combination is PBC with Sjögren’s syndrome |
+: presence of antimitochondrial autoantibodies; –: absence of antimitochondrial autoantibodies. PBC: primary biliary cholangitis; AMAs: antimitochondrial autoantibodies; E2: dihydrolipoyl transacetylase; BECs: biliary epithelial cells
The antigen for AMA production in PBC is the dihydrolipoyl transacetylase (E2) of the pyruvate dehydrogenase complex (PDC, E2 PDC), which localizes to the inner mitochondrial membrane. Despite the clear definition of the antigen to which AMAs are produced [9, 10, 13, 30], the question of the factors that trigger the mechanism of AMA formation and their significance in the development of the pathological process in PBC remains open. It is unclear how the E2 PDC antigen on the inner mitochondrial membrane is targeted by immunocompetent cells. The PDC is known to consist of three subunits: pyruvate dehydrogenase (E1), E2, and dihydrolipoyl dehydrogenase (E3). It is not known why PBC produces autoantibodies against only one E2 subunit. When experimental animals are immunized with E2 as a recombinant polypeptide, AMAs formation occurs. However, no damage to cholangiocytes is observed. Therefore, the question arises whether AMAs are the factor that triggers the destruction of BECs. There is also no answer to the question of why only the E2 antigen of PDC of small BECs is involved in this process. The responses to these questions are important for comprehending the involvement of the immune system in the pathogenesis of PBC.
Autoimmune diseases are a broad class of diseases, heterogeneous in their clinical manifestations, that develop due to disorders of the immune system, accompanied by loss of tolerance to certain tissue antigens with subsequent pathological production of autoimmune autoantibodies. It is believed that all autoimmune diseases have similar pathogenic mechanisms [12]. The resulting antigen-antibody complexes are capable of mediating the development of inflammation and target tissue damage. It has been shown that damage to target tissues of the macroorganism is a common basis for triggering new immune responses [12]. At the same time, the products of destruction of the body’s own cells and tissues act as adjuvants (enhancing the immune response). All this leads to the development of systemic (multi-organ) disorders of internal organ functions. The persistence of immune dysfunction and tissue destruction, accompanied by the subsequent recognition of tissue antigens by antigen-presenting cells, serves to support the chronic course of the disease. In this instance, the course of autoimmune diseases may be accompanied by spontaneous remission of the pathological state, which alternates with exacerbation. The severity of autoimmune diseases and the dysfunction of internal organs are typically correlated with the titer of autoantibodies.
Despite the appearance of AMAs already in the asymptomatic stage of the disease, the autoimmune theory of PBС is not entirely convincing, since many signs characteristic of autoimmune pathology are absent or not clearly confirmed in these patients (Table 1).
Signs of PBC that do not fit the criteria of the autoimmune theory of the disease:
Virtually all investigators note that the disease is not systemic in nature, but is limited to localized damage to the small intrahepatic bile ducts [1, 9];
PBC does not fit into the pattern of inflammation mediated by immune complexes [1];
The mitochondrial autoantigen E2 PDC, to which AMAs are formed, is located on the inner mitochondrial membrane, is not tissue specific, and has a high tolerance to the immune system [9];
E2 PDC is abundant in the mitochondria of all nucleated cells. However, only small BECs are the target of autoimmune attack in PBC [1];
Neither the titer nor the type of AMAs correlates with the clinical presentation, severity, and progression of the disease [10];
AMA titers or changes in their dynamics are not prognostic markers for PBC progression [31–33];
As a chronic liver disease, PBC is generally characterized by a gradual, slow progression without flares and remissions of activity typical of autoimmune diseases;
Unlike other autoimmune diseases, PBC has no pediatric analog [10];
Immunosuppressive therapy and genetically engineered (targeted) biologic drugs are unable to halt the progression of PBC [24–26].
In this context, there are currently doubts as to whether PBC is a true autoimmune disease [1]. To answer this question, it is important to have a clear understanding of the mechanism of AMA formation. Therefore, the efforts of many researchers are aimed at identifying the factors that trigger this process and at elucidating the pathophysiological mechanisms underlying the formation of AMAs [1, 13]. Attempts to prove or disprove the autoimmune theory of AMA formation underlie these studies.
In this regard, it is important to understand what leads to the formation of AMAs:
Immune system dysfunction leading to loss of tolerance to the unchanged E2 PDC protein antigen?
The appearance of conformational and/or structural changes in the E2 PDC antigen that result in a change in its immunological properties, which are recognized by the unaltered (healthy) immune system as a foreign antigen?
If the formation of AMAs is the result of an immune system dysfunction leading to loss of tolerance to the unmodified antigen, it should most likely be considered a sign of autoimmune disease. However, if the formation of AMAs is a consequence of the acquisition of new immune properties by the E2 PDC antigen, which is naturally recognized by the “healthy” immune system as a foreign antigen (neoantigen), it would be reasonable to ask whether PBC can be considered a true autoimmune disease.
As previously noted, in patients with the classical course of PBC, AMAs are formed against the antigenic component associated with E2 PDC, which is localized on the inner membrane of mitochondria [9, 10, 13, 30]. The majority of studies conducted a priori postulate that in PBC there is a disruption in the immune system leading to an altered tolerance of the immune system to E2 PDC [1, 13, 34]. Researchers are directing their efforts to understand the mechanism of development and impairment of this tolerance. The loss of immune tolerance to E2 PDC in PBC is believed to be the initial event in the mechanism of AMA formation and plays an important role as a triggering factor for cholangiocyte damage [1, 13, 34]. The view of immune-mediated destruction of small and medium intrahepatic bile ducts is discussed by many researchers [1, 15, 35]. At the same time, clear evidence of the damaging effect of the immune complex AMA-E2 PDC on BECs and the mechanism of this deleterious effect remain unknown.
But if there is an alteration in the immune system’s tolerance to the highly conserved and “ubiquitous” protein E2 PDC, it should lead to the development of disorders, which will be of systemic character. However, in PBC, the alterations are limited to the local damage of small and medium intrahepatic bile ducts. This suggests that in PBC, there are likely modifications to the E2 PDC antigen, which significantly alter its immunological properties to the extent that the unaltered immune system begins to recognize it as a foreign neoantigen. And this happens with the E2 PDC antigen located in the mitochondria of BECs lining only the small and middle bile ducts. And exactly these changes with E2 PDC antigen located in small BECs will serve as the initiating factor to start the synthesis of AMAs. Subsequently, the formed AMAs will react exclusively with neoantigens in BECs of small and middle bile ducts, but will not interact with normal, unaltered E2 PDC in mitochondria of other cells. A review of the available scientific data suggests that the localized nature of the observed damage in BECs lining the small and middle bile ducts in patients with PBC is most likely caused by an alteration in the immunological properties of the E2 PDC antigen of the mitochondria of small BECs only. If this is indeed the case, it is of paramount importance to elucidate the mechanism of the formation of such an immune-altered antigen. Furthermore, it is crucial to identify the factors that can initiate the transformation of E2 PDC into a neoantigen. Since E2 PDC is only one component of the PDC, it is essential to understand and elucidate the mechanisms underlying the acquisition of neoantigenic properties by this particular subunit of the PDC.
Furthermore, the conversion of E2 PDC into an immune-modified neoantigen is not sufficient for the formation of AMAs. It is also important to understand how this neoantigen leaves the inner membrane of mitochondria and cholangiocyte, and how its encounter with immunocompetent cells responsible for autoantibody production occurs. Furthermore, it is imperative to understand the rationale for the involvement of the E2 PDC antigen of BECs, which are exclusively located in small- to medium-sized bile ducts, in this process [10]. The specific triggers and mechanisms that initiate such processes in BECs of small- and medium-sized bile ducts remain unknown. In recent decades, new scientific data have emerged to elucidate the pathogenesis of cholangiocyte injury [3, 36]. These data demonstrate that the damage to BECs is not due to immune-mediated reactions, but rather to a defect in the biliary HCO3– “umbrella”. The latter protects BECs from the detergent action of toxic bile acids under physiological conditions [3, 36]. It has been demonstrated that HCO3– production is reduced in PBC, resulting in the entry and accumulation of bile acids in BECs (the damaged biliary HCO3– “umbrella” theory) [36–38]. These data allowed us to suggest that the gradual accumulation of bile acids in BECs, already in the asymptomatic stage of PBC, may serve as a trigger mechanism for small cholangiocyte damage, the development of ductulopenia, and the formation of AMAs [2]. For this purpose, it is important to represent the mechanisms of small and large BEC protection from the toxic action of bile acids.
BECs are classified into two categories, based on their size and location within the small- and large-size bile ducts [39]. Bile acids have a toxic effect on both small and large BECs. However, both types of BECs have different defense mechanisms to counteract this toxicity. Known cholangiocyte defense factors include the production and secretion of mucin and HCO3– by BECs [40].
Under physiological conditions, the main function of BECs is the biliary secretion of HCO3– [41]. Scientific evidence suggests that HCO3– plays a critical role in protecting BECs (protective “umbrella”) from the toxic effects of bile acids [37, 38]. HCO3– accounts for 25% to 40% of the total volume of bile excreted [37, 42, 43]. In humans, bile secretion of HCO3– maintains a physiologically neutral or slightly alkaline pH level in the lumen of the bile ducts [37, 42, 43]. It is well known that HCO3– is produced by both small and large BECs that line the bile ducts throughout the biliary tree. Nevertheless, the mechanisms of HCO3– production and secretion by small and large BECs are carried out in disparate manners, which are discussed in detail in the article [44] and the review [2].
Mucin glycoproteins are produced by the peribiliary glands (PBG) [45]. The latter are located in the wall of the large intra- and extrahepatic bile ducts and are in direct communication with their lumen. Experimental data show that glycosylated mucins covering the apical surface of the membranes of large BECs not only protect large BECs from the toxic effects of bile acids but also stabilize the biliary HCO3– “umbrella” [38]. The mucin produced by PBG serves to protect the BECs of the large bile ducts only [40]. Consequently, BECs of the large intra- and extrahepatic bile ducts are doubly protected: first by the mucin produced by PBG and second by HCO3–. It is noteworthy that PBG are absent in small bile ducts. As a consequence, mucin production is absent at the level of the small and intermediate bile ducts [45]. Consequently, HCO3– is the sole factor responsible for the defense of small BECs in the intralobular, interlobular, and septal bile ducts.
The different protective mechanisms against the toxic effects of bile acids in small and large BECs play an important role in the pathogenesis of damage to small BECs in PBC.
The defective biliary HCO3– “umbrella” theory is based on a number of clinical and experimental studies demonstrating inadequate synthesis and delivery of HCO3– to the bile ducts in PBC [46, 47]. As a result, the hepatic bile is acidified and the pH within the small BECs is alkalized. The disruption of HCO3– secretion and pH changes in bile ducts and small BECs has been linked to the involvement of InsP3R3 and chloride/HCO3– AE2 [48]. It has been demonstrated that in liver biopsy specimens and blood mononuclear cells of patients with PBC, the expression of the InsP3R3 and AE2 genes is reduced, indicating their dysfunction and involvement in the pathogenesis of diseases [46, 47]. A reduction in the activity and an impairment of the secretory function of InsP3R3 and AE2, have been associated with miR-506 [49]. An increase in the amount and activity of miR-506 has been observed in BECs of patients with PBC [50]. The X-linked nature of miR-506 may account for the higher incidence of this disease in women (Figure 1) [2, 51].
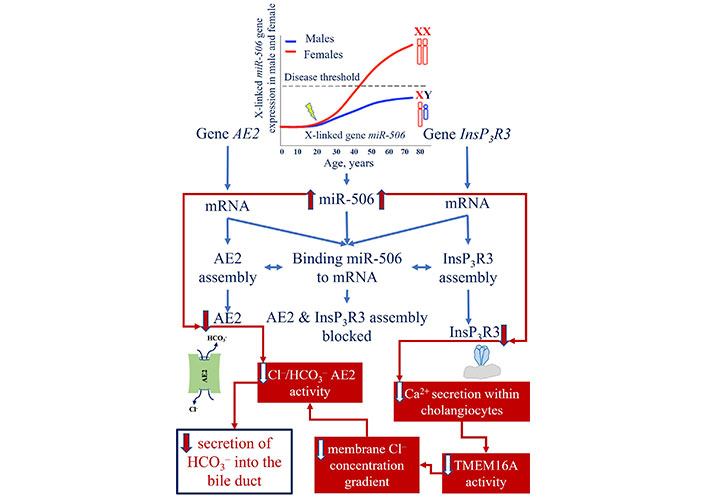
Mechanism of InsP3R3 and AE2 gene expression reduction due to the increase in the amount of miR-506 and its activity. miR-506: microRNA 506; AE2: anion exchanger 2; InsP3R3: inositol-1,4,5-triphosphate receptor type 3; Cl–: chloride; HCO3–: bicarbonate; TMEM16A: transmembrane 16A Cl– channels
Note. Adapted from “New insights into the pathogenesis of primary biliary cholangitis asymptomatic stage” by Reshetnyak VI, Maev IV. World J Gastroenterol. 2023;29:5292–304 (https://doi.org/10.3748/wjg.v29.i37.5292). CC BY-NC.
Changes in hepatic bile pH in PBC facilitate the protonation of bile acids [1, 52]. This allows bile acids to overcome the biliary HCO3– “umbrella” and enter BECs, which ultimately results in their damage [1, 52, 53]. The mechanisms of protonation-deprotonation of bile acids and their subsequent entry into the BECs are discussed in detail in the reviews [2, 54]. It is imperative to underscore that the described mechanism of bile acid intake into BECs in patients with PBC occurs precisely at the level of small BECs. This phenomenon is associated with inadequate HCO3– production by the cells in question and the absence of a mucin component that serves to fortify the protective barrier of small BECs [2, 54].
The destabilization of the biliary HCO3– “umbrella” theory underlies an uncontrolled increase in intracellular bile acid intake into small BECs, which in turn triggers the mechanism of their damage [1, 52, 53]. Upon entering BECs with a slightly alkaline pH, bile acids undergo deprotonation. This promotes their accumulation within small BECs [2]. Bile acids, which possess strong detergent properties, are capable of solubilizing phospholipids and cholesterol from the membrane structures of small BECs. This process results in damage and destruction of the cytoplasmic membrane and membranes of cellular organelles. The disruption of the integrity of the nuclear and mitochondrial membranes of small BECs in PBC results in the release of factors that induce apoptosis. The chronic damaging effect of bile acids on membrane structures has been shown to cause accelerated senescence, apoptosis, and/or necrosis of small BECs lining the intralobular, interlobular, and septal bile ducts [1, 55]. An excessive number of apoptotic cells may result in the formation of a necrotic-like pathway of cell death, which is also known as secondary necrosis [12]. The induction of accelerated cellular apoptosis and secondary necrosis results in the activation of inflammatory and profibrogenic pathways, which in turn contribute to the development and progression of fibrosis [1, 56]. The barrier function of the small BECs of the intralobular, interlobular, and septal bile ducts is compromised. This results in the additional damage, inflammation, and oxidative stress of the small BECs. Cytokines, chemokines, and proinflammatory mediators released by cholangiocytes, in turn, stimulate apoptotic and proliferative responses and activate fibrogenesis [57]. Literature reviews [55, 56] present a description of the conceptual mechanisms underlying these processes. Eventually, ductulopenia develops, which is accompanied by the formation of gradually progressive intrahepatic cholestasis [51, 56]. The disintegration of apoptosis processes induced by bile acids can be considered as a trigger mechanism for the development of ductulopenia of intralobular, interlobular, and septal bile ducts in PBC, already in the asymptomatic stage of the disease [58]. The presented mechanism elucidates the progressive development of senescence, apoptosis, secondary necrosis, and small BEC death, as a result of the intake, accumulation, and damaging effect of bile acids on them. In patients with PBC, this is histologically manifested by progressive damage to the small BECs, with infiltration by mononuclear cells around the BECs of the intralobular, interlobular, and septal bile ducts. The inflammatory cells are typically located in close proximity to the BECs of damaged bile ducts [59].
Herewith protonated conjugated and unconjugated bile acids are not able to overcome the mucin-containing glycocalyx layer that covers the apical surface of large BECs. Mucin glycoproteins serve as a strong defense mechanism against their penetration. The accumulation of bile acids within small BECs results in the solubilization of phospholipids and cholesterol from the outer mitochondrial membrane, thereby increasing its permeability (permeabilization) [60–63]. This can create conditions conducive to the formation of AMAs [64].
The disruption of the outer mitochondrial membrane permeability in small BECs in PBC is accompanied by a loss of membrane potential and the leakage of intermembrane space contents into the cytosol, which is caused by the solubilization of phospholipids and cholesterol by bile acids [63, 65]. The released apoptogenic factors are involved in the apoptotic processes of small BEC [63]. Mitochondrial swelling, which is indicative of disruption to the integrity of the outer mitochondrial membrane, is hereinafter accompanied by damage to the inner mitochondrial membrane, which is the primary target for the formation of AMAs. The solubilization of phospholipids and cholesterol by bile acids from the inner mitochondrial membrane results in their subsequent degradation, accompanied by the release of the PDC. The PDC is comprised of three enzymes (E1 PDC, E2 PDC, and E3 PDC) and two auxiliary proteins [59]. The function of the enzyme depends on the presence of five cofactors: coenzyme A, nicotinamide adenine dinucleotide (NAD), thiamine pyrophosphate (vitamin B1), flavin adenine dinucleotide (FAD), and lipoic acid. It is crucial to highlight the absence of lipids in E1 PDC and E3 PDC, which substantiates their exclusive protein nature. The serological reactivity of serum from patients with PBC to the E1 and E3 components of PDC has been demonstrated to be absent [66].
In the PDC, only the E2 is the lipoprotein due to the presence of lipoic acid, which is covalently bound to lysine at position 173 in the lipoamide domain of the complex [30]. It is noteworthy that the lipoyl-containing immunogenic domain exhibits a high degree of conservation across different species [67]. AMAs target the only immunodominant epitopes of E2 PDC containing lipoic acid [68]. All immunodominant epitopes of E2 PDC include lipoic acid bound to lysine, which is necessary for antigen recognition by T cells [68]. Serum from PBC patients shows serologic reactivity exactly to a specific immunogenic domain containing lipoic acid [69–71].
The data presented above provide the foundation for the hypothesis that the formation of AMAs against E2 PDC in PBC is most likely a consequence of the effect of bile acids on the lipid component of the PDC. Bile acids with pronounced detergent properties are capable of interacting with lipoic acid E2 PDC, resulting in conformational and/or structural alterations in the immunodominant lipoyl epitope. The chemical modification of the lipoyl domain of E2 PDC by bile acids has the potential to alter its immune properties, rendering it a neoantigen. Already in the asymptomatic stage of PBC, autoreactive B lymphocytes are capable of recognizing the immuno-modified E2 PDC neoantigen derived from apoptotic small BECs as a foreign protein. This subsequently stimulates T-cell subpopulations and leads to the formation of specific AMAs [13, 21, 72–75]. In light of the aforementioned evidence, it can be posited that the formation of AMAs in PBC is not primarily associated with immune dysregulation. Rather, it is likely that this phenomenon is driven by the acquisition of neoantigenic properties by the E2 PDC.
The translocation of the E2 PDC neoantigen to autoreactive immune cells can be achieved with the assistance of apoptotic bodies, which are formed as a consequence of the apoptosis of small BECs [13]. The presence of the E2 PDC autoantigen in apoptotic bodies of BECs in PBC was first demonstrated by Lleo et al. [76]. It has also been shown that apoptotic bodies are taken up by macrophages [76]. This process can simultaneously induce the synthesis of local proinflammatory cytokines and facilitate the transfer of the E2 PDC neoantigen into immunocompetent cells, thereby initiating the production of AMAs (Figure 2) [77].
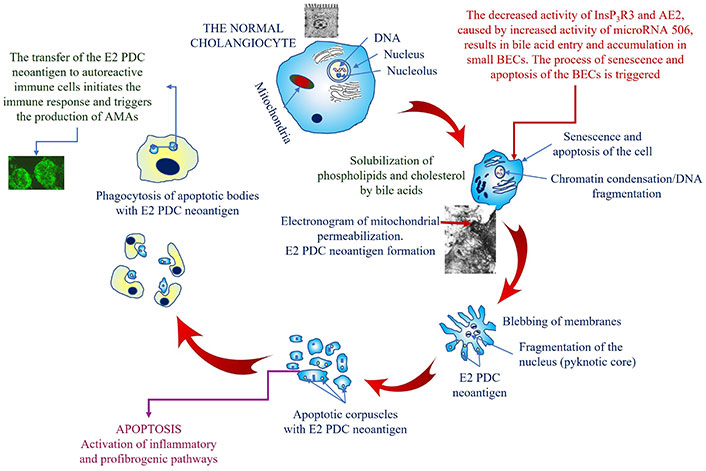
A schematic representation of the mechanism of mitochondrial permeabilization and the AMA formation in PBC. E2 PDC: dihydrolipoyl transacetylase of the pyruvate dehydrogenase complex; AMAs: antimitochondrial autoantibodies; InsP3R3: inositol-1,4,5-triphosphate receptor type 3; AE2: anion exchanger 2; BECs: biliary epithelial cells; PBC: Primary biliary cholangitis
And the pure protein subunits (E1 PDC and E3 PDC) of the PDC are virtually not affected by bile acids. As a result, they will remain intact and no autoantibodies will be produced against them.
The persistent and constant destabilization of the biliary HCO3– “umbrella” in PBC contributes to the continuous accumulation and detergent action of bile acids on small BECs. This process is characterized by the continuous destruction of BECs, which line the intralobular, interlobular, and septal bile ducts. This destruction results in the continuous production of AMAs. Given that AMAs target the immuno-modified E2 PDC neoantigen, they will interact exclusively with BECs that have been damaged by bile acids (i.e., those that are either senescent or apoptotic) and that contain the E2 PDC neoantigen. This contributes to the maintenance of inflammation and fibrosis. Concurrently, AMAs will not exert an immuno-damaging effect on “healthy”, normal cells containing an unchanged E2 PDC [10]. The E2 PDC antigen of other normal nucleated cells remains unchanged and is located on the inner mitochondrial membrane, which also makes it inaccessible to AMAs.
The presented data certainly require further comprehensive studies to substantiate the assumptions made. However, according to our assessment, the available scientific data partially corroborate the hypothetical statements regarding the mechanism by which E2 PDC acquires neoantigenic properties under the influence of bile acids. The data presented below were obtained in the laboratory of Gershwin ME as a result of experimental immunization of animals. The results demonstrated that:
It is not feasible to induce AMA formation with identical reactivity to serum samples from PBC patients through experimental immunization [78]. It is likely that this discrepancy can be attributed to the utilization of native E2 PDC in the immunization of the animals, rather than E2 PDC neoantigen obtained after interaction with bile acids, as observed in PBC.
The experimental data on the immunization of laboratory animals with E2 PDC as a recombinant polypeptide indicate that it leads to the formation of AMAs, but not to cholangiocyte damage [79]. This may serve to corroborate the hypothesis that AMAs are not a trigger factor in the destruction of the small BECs.
It has been demonstrated that octanoic acid derivatives induce the formation of AMAs and the development of liver lesions in guinea pigs and mice [80–83]. These findings highlight the pivotal role of the fatty acid residue and its modification in E2 PDC antigen in the initiation and production of AMAs.
The immunization of rabbits with a xenobiotic (6-bromohexanoate) conjugated to bovine serum albumin was found to induce the production of AMAs without the requirement of the peptide backbone of E2 PDC [84]. This also indirectly suggests that modification of the fatty acid rather than the protein portion of the E2 PDC antigen plays an important role in the production of AMAs. Following the termination of the stimulus, these autoantibodies disappeared [84]. In PBC, the damaging effect of bile acids on the mitochondria of small BECs ensures the constancy of immune modification and the release of E2 PDC, which, in turn, results in the production of AMAs.
It seems a reasonable assumption that the AMA formation and destruction of small BECs are two independent processes. The two processes are unified by their dependence on the damaging action of bile acids, which, due to impaired HCO3– production, enter and accumulate in small BECs. This leads to both cholangiocyte damage and conformational and/or structural changes in the lipophilic domain of the E2 PDC, which is accompanied by the production of AMAs [2, 54].
Some patients with PBC are AMA-negative. Nevertheless, patients who lack AMAs demonstrate comparable characteristics with regard to the course and progression of the disease as those who possess AMAs. It would appear that in 5% of AMA-negative patients, due to some individual characteristics, the accumulation of bile acids in small BECs is not accompanied by immune modification of the E2 PDC antigen. It seems a reasonable assumption that the damage to small BECs associated with the accumulation of bile acids in them will develop by the same mechanism as in AMA-positive patients. This would explain the similar clinical picture, course, and progression of the disease.
A notable limitation of the existing literature is the lack of studies aimed at identifying novel neoantigen properties of E2 PDC. The researches were directed to identify evidence for the mechanisms underlying the loss of immune tolerance to unchanged (normal) E2 PDC antigen [73, 85, 86].
In consideration of the aforementioned facts and considerations, the presence of AMAs in PBC is more likely to indicate an autoimmune-mediated (autoimmune-like) condition than to suggest that PBC is a classic autoimmune disease.
The discovery of AMAs and their successful diagnostic application for PBC testing has facilitated the detection of asymptomatic and early-stage disease, thereby improving the prognosis for patients. Currently, the diagnostic value of AMA in PBC is generally acknowledged. Nevertheless, further research is necessary to gain insight into the mechanisms underlying AMA formation and to determine their significance in the pathogenesis of PBC. The available scientific data permitted the formulation of a hypothesis regarding the potential role of bile acids as contributing factors in the initiation of the mechanism of AMA formation in small BECs in PBC. The hypothesis presented in the review allows us to answer a number of unresolved questions concerning the factors that initiate damage to small BECs, the mechanism of AMA formation, their significance in the development of PBC, and the degree of involvement of the immune system in this process. The evidence presented in the review reveals a number of issues that have remained unclear until now.
It becomes understandable the formation of AMA specifically to the E2 PDC antigen, but not to the E1 PDC and E3 PDC;
The potential possibility for chemical modification of the lipoyl domain of the E2 PDC antigen by bile acids in small BECs with subsequent transformation into a neoantigen was demonstrated. In this instance, the dysfunction of the immune system is excluded;
A potential mechanism for the involvement of bile acids in the formation of access to E2 PDC, located on the inner mitochondrial membrane of small BECs, for immunocompetent cells, which subsequently leads to the formation of AMAs, is presented;
The persistent and continuous damaging effect of bile acids on small BECs is a determining factor in the persistence of E2 PDC neoantigen formation, which, in turn, supports the sustained elevation of AMA titers in the blood of patients with PBC;
The formation of AMAs in response to the E2 PDC neoantigen precludes its cytotoxic action on all other cells containing mitochondria and an unaltered E2 PDC;
The AMA formation in response to the E2 PDC neoantigen of only small BECs determines their diagnostic value but not their prognostic value;
Bile acid involvement as a root cause of small BEC damage defines the course of PBC as a non-purulent destructive cholangitis that does not align with the typical pattern of immune-mediated inflammation;
It seems a reasonable assumption that the involvement of bile acids as the underlying cause of small BEC damage may explain the lack of efficacy of immunosuppressive drugs.
The disclosure of new etiologic factors and detailed pathophysiological mechanisms of PBC may serve as an important basis for the development of new drugs and treatment methods for these patients in the future.
AE2: anion exchanger 2
AMAs: antimitochondrial autoantibodies
ANAs: antinuclear autoantibodies
BECs: biliary epithelial cells
E1: pyruvate dehydrogenase
E2: dihydrolipoyl transacetylase
E3: dihydrolipoyl dehydrogenase
HCO3–: bicarbonate
InsP3R3: inositol-1,4,5-triphosphate receptor type 3
miR-506: microRNA 506
PBC: primary biliary cholangitis
PBG: peribiliary glands
PDC: pyruvate dehydrogenase complex
VIR and IVM: Conceptualization, Formal analysis, Investigation, Writing—original draft, Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

Yuto Sasaki ... Tadashi Matsuda
Aleksandra Kozlova ... Maria Zakharova
Jihye Heo ... Jea-Hyun Baek
Nawal Labiad ... Nardjess Ouikhlef
Omer Okuyan ... Hafize Uzun
