 Review
Review

Affiliation:
1Department of Immunology, School of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
ORCID: https://orcid.org/0000-0002-6651-5977
Affiliation:
1Department of Immunology, School of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
ORCID: https://orcid.org/0009-0001-2334-9294
Affiliation:
2Department of Internal Diseases Medicine, School of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
Affiliation:
1Department of Immunology, School of Medicine, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
3Cancer Research Center, Cancer Institute, Avicenna Health Research Institute, Hamadan University of Medical Sciences, Hamadan 6517838736, Iran
Email: gh.solgi@umsha.ac.ir
ORCID: https://orcid.org/0000-0001-8929-5658
Explor Immunol. 2024;4:640–657 DOI: https://doi.org/10.37349/ei.2024.00164
Received: March 21, 2024 Accepted: September 23, 2024 Published: October 21, 2024
Academic Editor: Nitin Saksena, Victoria University, Australia
The impaired function of regulatory T (Treg) cells and the imbalance of Treg/Th17 cells play a central role in developing autoimmune diseases such as systemic lupus erythematosus (SLE). Treg cells are crucial for maintaining immune homeostasis and tolerance to self-antigens. One of the most important transcription factors that regulate the differentiation and function of Treg cells is the FOXP3 protein. Aberrant epigenetic modifications affecting FOXP3 gene expression and consequently dysregulated function of Treg cells have been implicated in the pathogenesis of SLE. Therefore, understanding the intricate interplay between FOXP3 expression pattern in Treg cells and epigenetic regulatory mechanisms (e.g., DNA methylation, histone modifications and non-coding RNAs such as microRNAs and long non-coding RNAs) is crucial for unravelling the underlying mechanisms of SLE. Moreover, targeting these epigenetic pathways may offer novel therapeutic strategies for restoring immune balance and ameliorating autoimmune pathology. This review report aimed to provide an update on the epigenetic controlling of FOXP3 gene expression in SLE disease.
Autoimmunity arises from a dysregulated immune response wherein the body’s defense system mistakenly targets its antigens, leading to chronic inflammation and tissue damage due to the loss of immune tolerance. This phenomenon is influenced by various factors including gender, age, race, and geographical location [1, 2]. Notably, gender significantly impacts the prevalence of autoimmune diseases, with rates being 80–90% higher in women, potentially attributed to hormonal fluctuations throughout their lifecycle [3, 4]. Autoimmune diseases typically do not directly cause mortality but, they profoundly impact individuals’ quality of life. With over 100 identified types, autoimmune diseases are broadly categorized into organ-specific and systemic forms based on the extent of the autoimmune response against self-antigens. In organ-specific conditions like multiple sclerosis (MS), type 1 diabetes (T1D), and autoimmune thyroiditis (AITD), the immune system targets specific autoantigens within a particular tissue or cell line [5].
Conversely, systemic autoimmune diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis (RA) involve an autoimmune response against multiple autoantigens expressed across various tissues or cell types. While the immunological mechanisms driving different autoimmune diseases share similarities, the primary initiating factor remains unclear [6, 7]. A complex interplay of genetic, epigenetic, and environmental factors contributes to disease development. Genetic predisposition, including variations in HLA genes and non-HLA gene polymorphisms, influences susceptibility to autoimmune diseases. Environmental factors like viral infections and dysbiosis of the intestinal microbiota can trigger autoimmune responses, potentially through mechanisms such as molecular mimicry. Additionally, epigenetic factors play a crucial role in modulating autoimmunity by regulating the expression of numerous proteins involved in immune function [8].
Autoimmune responses typically result from disturbances in central and peripheral tolerance mechanisms, particularly in those genetically susceptible individuals, exacerbated by environmental and epigenetic factors [9]. A key player in preserving tolerance and immune homeostasis is regulatory T (Treg) cells. They are naturally produced in the thymus [natural Treg (nTreg): CD4+CD25+FOXP3+ T cells], constituting the primary Treg cells population, and can also be induced in peripheral tissues [induced Tregs (iTregs)] following immune activation. Based on their transcription factors and secreted cytokines, iTreg cells can be classified into three subsets: IL-10+ FOXP3– T cells (TR1 cells), TGF-β+FOXP3+ Treg cells (Th3), and CD8+ Treg cells [10, 11].
The FOXP3 transcription factor, encoded by the FOXP3 gene, plays a crucial role in Treg cells differentiation both in the thymus (nTreg) and peripheral tissues (iTreg) [10]. Epigenetic factors, particularly microRNAs (miRNAs), can downregulate FOXP3 expression. This downregulation not only decreases Treg cells frequency, which is implicated in the immunopathogenesis of autoimmune diseases but also reduces the expression levels of key genes involved in immune regulation such as CTLA-4 and IL-10 by Treg cells. Conversely, FOXP3 deficiency in Treg cells leads to the upregulation of genes associated with effective immune responses, including tumour necrosis factor-alpha (TNF-α), interferon-γ (IFN-γ), and IL-17 [12, 13]. This review makes focuses on the role of epigenetic factors in regulating FOXP3 gene expression and its impact on the differentiation and function of Treg cells concerning the immunopathogenesis of SLE.
SLE is a systemic autoimmune disease characterized by its multi-faceted pathological effects. Initially targeting a single tissue, but progressively affects various organs including the skin, joints, blood, kidneys, and central nervous system. The name “SLE” originates from the distinctive butterfly-shaped rash often observed on the faces of affected individuals. Among the complications of SLE, kidney disorder stands out as particularly severe [14, 15]. However, the spectrum of symptoms and clinical manifestations is wide, encompassing fatigue, fever, loss of appetite, discoid rashes, and neurological disorders. These arise from the deposition of immune complexes in multiple tissues such as joints, bones, muscles, skin, and peripheral and central nerves [14]. While SLE can manifest at any age, it typically emerges post-puberty, with hormonal influences, particularly estrogen that plays a significant role. Gender is a crucial factor in SLE development, with incidence rates approximately nine times higher in women compared to men. Moreover, women aged between 15 and 45, typically within their reproductive years, exhibit a heightened susceptibility to SLE [3].
The prevalence of SLE varies across different populations. In American white and Northern European populations, it affects around 53 and 40 cases per 100,000 people, respectively, whereas the prevalence is higher among black individuals, at approximately 159 cases per 100,000 people [16]. In Iran, according to data from the Rheumatology Research Center, SLE affects around 40 cases per 100,000 people, with women of reproductive age constituting around 90% of patients [17].
Although the precise etiology of SLE remains incompletely understood, it shares similarities with other autoimmune disorders in being influenced by environmental factors. These include exposure to ultraviolet (UV) rays, inadequate intake of vitamin D, viral infections, smoking, gut microbiota dysbiosis, and sex hormones, particularly estrogen. Family-based studies have shown that the incidence of SLE among relatives of patients ranges from 5% to 12%, highlighting the genetic component in disease development [18].
Among the predisposing genes to SLE, polymorphisms in DRB1 and DQB1 are prominent. Studies have indicated that alleles such as DRB1*03:01 and DRB1*15:01 confer a 2–3 times increased risk of developing SLE. While earlier investigations implicated direct roles of DRB1*03~DQB1*02 and DRB1*15~DQB1*06 haplotypes in SLE pathogenesis, recent findings both in human studies and in mouse models, suggested that DRB1 molecules primarily contribute to the production of SLE-specific autoantibodies. Research conducted on Iranian SLE patients has revealed associations between specific DRB1 alleles and the production of distinct autoantibodies. For instance, DRB1*03 is linked to the production of anti-SSA/Ro, anti-SSB/La, and anti-coagulant autoantibodies, while DRB1*04 and DRB1*15 are associated with the production of anti-cardiolipin and anti-Sm antibodies, respectively [19, 20]. Moreover, the distribution of DRB1 and DQB1 alleles has been correlated with patterns of clinical manifestations in SLE patients. For example, DRB1*03 may induce skin and kidney symptoms while inhibiting pulmonary symptoms, whereas DRB1*04, DRB1*08, and DRB1*10 alleles may induce cardiovascular, pulmonary, and thrombotic symptoms, respectively. Conversely, the presence of DRB1*07 and DRB1*13 alleles may decrease the likelihood of joint and skin symptoms associated with SLE [21]. In addition to HLA genes, polymorphisms in non-HLA genes such as complement genes (C1q, C2, and C4), IRF5, PTPN22, and STAT4 have been implicated in increasing susceptibility to SLE [22]. These genetic factors collectively contribute to the complex pathogenesis of SLE, influencing disease onset, progression, and clinical manifestations.
Furthermore, epigenetic alterations such as DNA methylation mediated by DNA methyltransferases, contribute to the development of SLE [23]. Studies on T lymphocytes from SLE patients have revealed a decrease in DNA methyltransferase 1 and 3 levels. Additionally, histone modifications in IL-17 gene have been linked to an increased IL-17 expression, whereas in IL-2 gene it was related to a decreased expression in SLE patients [24]. miRNAs, a type of non-coding RNA involved in post-transcriptional regulation of gene expression, also play a significant role in SLE pathogenesis. Studies have uncovered distinct patterns of miRNA expression in SLE patients [25]. For instance, miR-3148 downregulates the expression of Toll-like receptor 7 (TLR7), contributing to heightened inflammatory responses in SLE patients. Conversely, upregulation of miR-155 targets suppressor of cytokine signaling 1 (SOCS1), leading to enhanced type 1 interferon signaling and inflammatory responses in macrophages. Moreover, increased levels of miR-30a stimulate B cell activation in SLE patients [26]. Long non-coding RNAs (LncRNAs) represent another class of non-coding RNAs implicated in SLE. Although fewer LncRNAs have been studied in SLE, research has shown significant reductions in linc0949 and linc0597 levels in SLE patients compared to healthy individuals [21]. These findings underscore the complex interplay of epigenetic mechanisms involving DNA methylation, histone modifications, and non-coding RNAs in the pathogenesis of SLE, suggesting potential avenues for therapeutic intervention and biomarker discovery.
Upon neutrophil cell death during NETosis or tissue cell death triggered by environmental stimuli like UV rays, nuclear autoantigens, such as nucleic acids, become exposed to the immune system. B lymphocytes recognize these released nucleic acids, leading to the production of various autoantibodies including anti-dsDNA, anti-C1q, anti-cardiolipin, anti-Ro, anti-La, and anti-Sm antibodies. While these autoantibodies are commonly utilized as diagnostic markers for SLE, they also contribute to the formation of immune complexes [27, 28]. Additionally, the release of HMGB1 molecules from deceased cells binds to pre-existing immune complexes (antibodies and nucleic acids), impeding their clearance by macrophages and dendritic cells. Plasmacytoid dendritic cells (pDCs) then uptake these immune complexes via Fc receptors (FcRs) and recognize them through TLR7 and TLR9 present in the endosome membrane. This recognition triggers the production of IFN-α from pDCs. IFN-α, in turn, activates dendritic cells, facilitating the presentation of autoantigen epitopes to autoreactive CD4+ T cells. These CD4+ T cells assist autoreactive B lymphocytes in producing autoantibodies via the CD40/CD40L signalling pathway and IL-21 production. Moreover, dendritic cells and macrophages influence B cells by secreting pro-inflammatory cytokines such as TNF-α, IL-6, and B-cell activating factor (BAFF). This cytokine milieu promotes further production of autoantibodies and induces the recruitment of immune cells, exacerbating tissue damage (Figure 1). Notably, more than 50% of SLE patients exhibit increased levels of BAFF, leading to the consideration of anti-BAFF monoclonal antibodies for SLE treatment [29]. Furthermore, in SLE patients, mutations in the RAG gene may result in the production of mutated B-cell receptors (BCRs), ultimately leading to the generation of autoreactive B cells. This mutation may also contribute to a decline in the surrogate light chain, increased proliferation and differentiation of plasma cells, elevated frequencies of memory B cells, and heightened cytokine production related to B cells, such as IL-6 and IL-21. These findings indicate a complex interplay between various immune cells and molecules in the pathogenesis of SLE, providing potential targets for therapeutic intervention [30, 31].
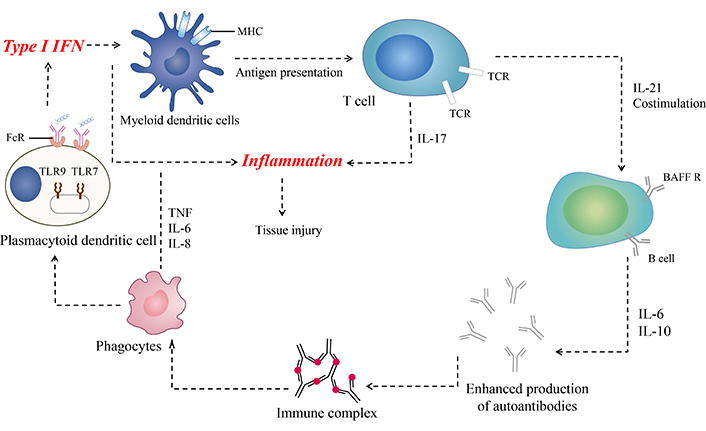
Immunopathogenesis of systemic lupus erythematosus (SLE). After exposure to nuclear antigens, autoreactive B cells produce autoantibodies, and immune complexes are formed. Plasmacytoid dendritic cells (pDCs) produce IFN-I after recognizing immune complexes through FcR or nuclear particles (e.g., ribonucleoprotein and U1snRNP) through Toll-like receptors (TLRs). IFN-I affects myeloid dendritic cells and activates them to present autoantigen-derived peptides to autoreactive T cells. It also promotes the differentiation of activated B cells into plasmablasts and antibody-secreting plasma cells. IL-17, along with co-stimulatory molecules from autoreactive T cells and BAFF from myeloid dendritic cells, enhances autoantibody production by autoreactive B lymphocytes. Autoantibodies can bind to nuclear antigens, form immune complexes, and activate innate immune cells, creating a positive feedback loop that amplifies the pathogenic processes in SLE [32]. MHC: major histocompatibility complex; FcR: Fc receptor; TNF: tumour necrosis factor; BAFF: B-cell activating factor; BAFF R: BAFF receptor
Recent evidence suggests a significant role for IL-10 in the development and progression of SLE [33]. Studies have consistently demonstrated elevated serum IL-10 concentrations and IL-10 mRNA levels in SLE patients. Consequently, targeting IL-10 or its receptors may offer promising therapeutic avenues for SLE treatment [34]. Despite its important role in maintaining peripheral tolerance, inhibiting cytokine production by Th1 cells, inducing apoptosis in B cells, and regulating antigen presentation, its effects in antibody-mediated autoimmune diseases like SLE appeared to be complex. Excessive IL-10 levels have been implicated in autoreactive B cell activation and plasma cell differentiation, leading to heightened autoantibody production (Figure 1). Furthermore, IL-10 can suppress the production of TGF-β from T cells, monocytes, and natural killer cells, which are protective against SLE development. Overall, these findings highlight the potential involvement of regulatory B cells (Bregs) through IL-10 and its receptor (IL-10R) in the pathogenesis of SLE. Understanding the intricate balance of IL-10 signalling in autoimmune diseases like SLE may offer insights into novel therapeutic strategies aimed at modulating immune dysregulation and alleviating disease burden [35].
Early investigations into SLE suggested a predominance of Th2 cells, credited to the elevated levels of cytokines related to the Th2 subset in SLE patients. However, recent studies have yielded contradictory results regarding the distribution of Th1 and Th2 cell populations, indicating variability depending on the stage of the disease. Generally, Th2 cells are involved in the production of autoantibodies, while Th1 cells contribute to heightened inflammation in SLE. The dynamic interplay between Th1 and Th2 subsets underscores the complexity of immune dysregulation in SLE and highlights the need for further research to elucidate their exact roles in disease pathogenesis and progression [36, 37].
In addition, other helper T cell subsets such as Th9 and Th22 have emerged as potential contributors to the pathogenesis of SLE. Studies indicated a significant increase in the frequency of CD4+IL-9+ T (Th9) cells in the peripheral blood of SLE patients compared to healthy individuals. Moreover, a positive correlation between the proportion of Th9 cells frequency, IL-9 serum levels, and the SLE Disease Activity Index (SLEDAI) has been reported [38]. The role of Th9 cells in SLE pathogenesis is underscored by their production of IL-9, which may enhance autoantibody production through the induction of B cell proliferation. Although most studies suggest a decline in serum IL-22 concentrations in SLE patients, there are conflicting findings regarding the frequency of CD4+IL-22+ T (Th22) cells. Some studies indicated an increase in Th22 cells frequency in SLE patients with skin manifestations, while others reported a decrease in those with nephritis symptoms. Overall, Th22 cells frequency may serve as valuable indicators for delineating tissue involvement in SLE patients [39]. These findings highlight the potential involvement of Th9 and Th22 cells in the complex immunopathogenesis of SLE, providing insights into novel therapeutic targets and diagnostic strategies for this autoimmune disorder.
Further investigations on cytokine levels associated with different subsets of T cells have revealed a dysregulation in SLE patients [40]. Elevated levels of IL-17 suggest a deviation in the balance of T lymphocyte frequencies toward Th17 cells in SLE. Additionally, an increase in IL-6 levels has been observed, promoting Th17 cell differentiation while inhibiting Treg induction, thus disrupting the Th17/Treg balance and exacerbating the inflammatory response in SLE. Studies have consistently demonstrated a significantly increased Th17/Treg ratio in SLE patients compared to healthy controls. Moreover, there exists a positive correlation between the Th17/Treg ratio and disease severity in SLE [41]. While some studies demonstrated a rise in the Th17/Treg ratio, contradictory findings indicated a significant increase in Treg cells in SLE patients compared to healthy individuals [42]. This discrepancy may suggest impaired Treg cell function in modulating Th17 differentiation in SLE. These findings underscore the intricate balance between Th17 and Treg cells in SLE pathogenesis, offering potential targets for therapeutic intervention aimed at restoring immune homeostasis and alleviating disease severity [43].
Treg cells primarily differentiate in the thymus (nTreg) upon recognition of self-epitopes-HLA complexes with high affinity. Functionally, Treg cells are crucial for maintaining immune homeostasis and tolerance to self-proteins. They exert regulatory effects through various mechanisms, including the production of anti-inflammatory cytokines and the expression of molecules such as granzymes A/B, perforin, as well as galectin-9 in mice, to induce apoptosis in target cells, disrupt metabolic pathways, and suppress pathways involved in antibody production. By modulating immune responses in this manner, Treg cells play a pivotal role in preventing autoimmune responses and maintaining immune tolerance [32, 44].
Numerous studies have consistently shown a significant decrease in Treg cells proportion in autoimmune diseases, particularly in SLE patients compared to healthy individuals. This decrement in Treg cells frequency inversely correlates with disease severity [45]. Additionally, there is evidence suggesting that the suppressive functions of Treg cells may be impaired in SLE patients. Therefore, enhancing the number or function of these cells is crucial for controlling autoimmune diseases. Recent studies have identified Treg cells as promising therapeutic targets. Strategies to bolster Treg cells function include the use of drugs such as rapamycin and N-acetylcysteine, which have been shown to increase their suppressive capabilities. Moreover, approaches involving the production of Treg cells with recombinant chimeric antigen receptors (CARs) on their surface or the induction of polyclonal Treg cells hold promise for more effective inhibition of autoreactive lymphocytes [10, 46]. Overall, Treg cells which constitute 5–15% of CD4+ T cells, are characterized by high expression of the transcription factor FOXP3 in their nucleus. They also express high levels of IL-2Rα (CD25) and CTLA-4 (CD152) which bind to IL-2 and B7 co-stimulatory molecules, respectively, contributing to their regulatory function. Additionally, Treg cells exhibit low expression of CD127 on their surface, further distinguishing them from other T cell subsets [47].
Studies have elucidated the underlying mechanisms for dysregulation of Treg cells in SLE patients. One such mechanism involves impaired production of IL-2, a critical cytokine for Treg cells differentiation and function. This disruption leads to a decline in Treg cell frequency in SLE patients, contributing to immune dysregulation [48]. Another mechanism involves OX40L-dependent signaling in Tregs, which may explain the downregulation of FOXP3, a transcription factor crucial for Treg cell development and function, in SLE patients. The OX40L/OX40 axis has been identified as an important inflammatory loop in SLE, promoting the differentiation of effector and memory T cells into follicular T lymphocytes while simultaneously blocking the suppressive function of Treg cells. Multiple studies have demonstrated an inverse correlation between the downregulation of FOXP3 in SLE patients and the proportion of dendritic cells expressing OX40L, indicating a dependency of FOXP3 expression reduction on the strength of OX40L/OX40 signaling. Consequently, blocking OX40L signaling with anti-OX40L monoclonal antibodies has been shown to restore the suppressive function of Treg cells by inhibiting FOXP3 downregulation. These findings underscore the potential therapeutic utility of targeting the OX40L/OX40 axis to restore Treg cells function and mitigate immune dysregulation in SLE patients [49].
The transcription factor FOXP3, a member of the FOX and FORK-protein family, is encoded by the FOXP3 gene located on the X chromosome and plays a pivotal immunological role by regulating Treg cells differentiation and function [50]. Human FOXP3 comprises 431 amino acids with a molecular weight of 47.25 kDa. It consists of four main domains, each with distinct functions: a repression domain, a zinc finger, a leucine zipper motif, and FKH domain. FOXP3 is capable of binding to over 2,800 genomic loci, exerting both positive and negative regulation on gene expression at both transcription and post-translation levels. Three conserved non-coding sequences (CNS) within the FOXP3 gene participate in regulating FOXP3 gene expression at the transcriptional level (Figure 2) [47, 51]. The first CNS (CNS1) plays a crucial role in activating FOXP3 expression in response to signalling mediated by TGF-β. TGF-β facilitates the binding of transcription factors such as Runx1, Runx3, SMAD3 and NFAT to CNS1 of the FOXP3 gene promoter, thereby inducing FOXP3 expression. Notably, SMAD3 binding to CNS1 is critical for inducing FOXP3 expression during the differentiation of FOXP3+-iTreg cells outside of the thymus. Studies in mice with CNS1 defects have shown defective iTreg induction, leading to an inability to maintain tolerance in peripheral tissues such as the intestine (Figure 2) [51].
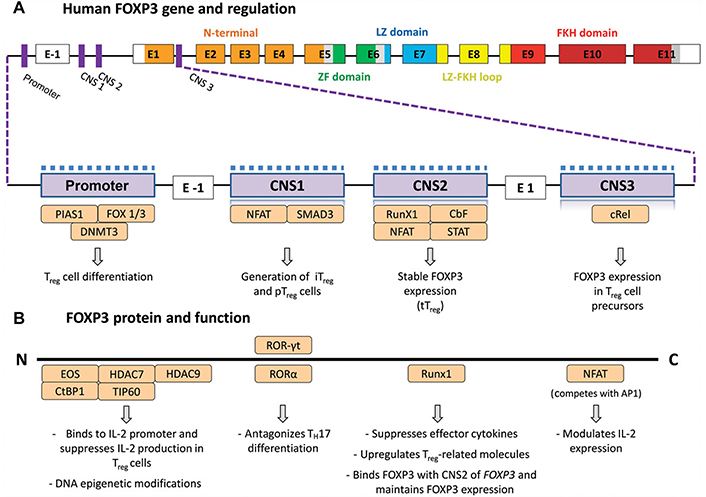
FOXP3 gene structure and FOXP3 protein interactions with other transcription factors. (A) FOXP3 gene structure indicating the modulatory regions (purple), the N-terminal proline-rich (PRR) domain (orange), the zinc-finger (ZF) domain (green), the leucine-zipper (LZ) domain (blue), the LZ-forkhead (FKH) loop (yellow), and the FKH domain (red). The magnified region illustrates the regions that are targets of epigenetic changes that characterize Treg cells, such as demethylation (upper dashed line) and histone modifications (lower grey squares). Several transcription factors (light orange) affect those regions to regulate FOXP3 expression. (B) FOXP3 protein interacts with several transcription factors (light orange) that affect FOXP3 expression. CNS: conserved noncoding enhancer sequence; tTreg: thymus-derived Treg cells; iTreg: induced Treg; pTreg: peripheral Treg cells; TH17: IL-17–producing T cells. Reprinted from Bacchetta et al. study [52]. © 2016 The Authors. CC BY-NC-ND
The second CNS (CNS2) is characterized by CpG-rich islands and is known as a Treg cell-specific demethylated region Located in the first intron of FOXP3. During differentiation, nTreg cells undergo extensive hypomethylation in CNS2. Conversely, in vitro-iTreg cells, which are less stable in terms of FOXP3 expression and suppressive activity, exhibit methylation in the CNS2 region. CNS2 is crucial for maintaining FOXP3 expression in nTreg cells after they exit the thymus. Deletion of CNS2 leads to loss of FOXP3 function. Methyl-CpG-binding domain protein 2 (MBD2) binds to CNS2 and recruits ten-eleven translocation demethylases (TET) to maintain CNS2 hypomethylation. Knockout studies of MBD2 in mice have demonstrated a decrease in Treg cells frequency and suppressive activity. Additionally, DNA methyltransferase 1 (DNMT1), induced by pro-inflammatory cytokine IL-6, leads to methylation in CNS2, suppressing nTreg cells activity. Conversely, exposure to IL-2 mobilizes TET enzymes to maintain CNS2 hypomethylation (Figure 2) [47, 51].
The third CNS (CNS3), located between exon 1 and 2 of the FOXP3 gene, serves as an epigenetic switch region, promoting the accumulation of permissive histone modifications in the FOXP3 promoter. Although CNS3 is essential for initiating FOXP3 expression, it appears dispensable for its maintenance. Investigations on CNS3 knockout mice demonstrated a significantly reduced frequency of nTreg cells and impaired TGF-β-mediated induction of iTregs (Figure 2) [51].
Furthermore, mutations in the FOXP3 gene can lead to the absence of Treg cells, resulting in a severe and rapidly fatal autoimmune disorder known as immune dysregulation, polyendocrinopathy, X-linked enteropathy (IPEX). This condition highlights the critical role of FOXP3 in regulating immune tolerance and preventing autoimmune responses [52]. In addition to mutations, FOXP3 gene polymorphisms have been reported in the various regions of this gene including the promoter, intron, exon, or poly A region. In patients with SLE, polymorphisms in the promoter region, rs3761548 and rs2294021, of the FOXP3 gene have been documented [53]. These polymorphisms may contribute to alterations in FOXP3 expression or function, potentially affecting Treg cells activity and immune homeostasis [54].
Epigenetics encompasses the study of heritable changes in gene expression that do not entail alterations in the DNA sequence itself. These changes are mediated through a variety of molecular mechanisms, including DNA methylation, histone modifications, and non-coding RNA interactions. DNA methylation involves the addition of a methyl group to the 5’ position of the cytosine pyrimidine ring, typically resulting in gene silencing [55]. Histone modifications, such as acetylation, methylation, phosphorylation, and ubiquitination, influence chromatin structure and accessibility, thereby regulating gene transcription [56]. Non-coding RNAs, including miRNAs and LncRNAs, modulate gene expression post-transcriptionally by binding to target mRNAs and affecting their stability or translation [57]. Collectively, these epigenetic mechanisms enable cells to respond to environmental signals, maintain cellular identity, and regulate complex developmental processes. They also play critical roles in disease pathogenesis, particularly in cancer and autoimmune diseases [58].
Recent studies have highlighted three main epigenetic regulatory mechanisms governing the expression of FOXP3 in Treg cells: DNA methylation, histone modification, and non-coding RNA (Figure 2). These epigenetic changes play a critical role in maintaining the phenotype and function of Treg cells, which are essential for immune homeostasis and tolerance. Aberrant epigenetic modifications affecting FOXP3 gene expression have been implicated in the pathogenesis of autoimmune diseases, particularly in SLE. Dysregulation of these epigenetic mechanisms can disrupt the balance of immune cells and their responses which may subsequently induce autoimmunity. Understanding the intricate interplay between epigenetic regulation and FOXP3 expression in Treg cells is crucial for unravelling the underlying mechanisms of autoimmune diseases like SLE. Moreover, targeting these epigenetic pathways may offer novel therapeutic strategies for restoring immune balance and ameliorating autoimmune pathology [59–61].
Multiple studies have elucidated significant differences in the function of Treg cells and the methylation status of the FOXP3 promoter, particularly in the context of SLE. The promoter region of the FOXP3 gene exhibits hypermethylation, especially in the CpG islands in Treg cells of SLE patients [62]. Conversely, in healthy individuals, this region of the FOXP3 gene is demethylated in Treg cells. Moreover, a non-coding region of the FOXP3 gene known as the binding site for NFAT and SMAD transcription factors and termed the TGF-β-sensitive sequence, undergoes methylation changes in SLE [22]. Additionally, the third non-coding conserved region referred to the specific demethylated region of Treg cells, demonstrates significant differences in methylation levels between SLE patients and healthy controls. This region, crucial for the stability and maintenance of FOXP3 expression in Treg cells, exhibits a distinct methylation pattern in SLE patients compared to healthy individuals. Of note, the most pronounced difference in methylation pattern is observed in this specific demethylated region. While this region is completely demethylated in Treg cells, its methylation status is intricately linked to the stability and regulation of FOXP3 expression. These findings underscore the importance of epigenetic regulation, particularly DNA methylation, in modulating FOXP3 expression and Treg cells function, with implications for immune dysregulation and autoimmune disease pathogenesis such as SLE [59–62].
Histone modifications in the FOXP3 gene represent another crucial epigenetic mechanism involved in the regulation of Treg cells differentiation and development. Studies have revealed distinct patterns of histone acetylation and trimethylation in histones H3 and H4 between Treg cells and other T cell subsets [63]. Specifically, histone modifications in the promoter region of the FOXP3 gene are orchestrated by histone methyltransferases and histone acetyltransferases. These enzymes dynamically regulate the chromatin structure and accessibility of the FOXP3 promoter, thereby influencing the transcriptional activity of the gene [64]. The balance between histone acetylation and methylation plays a pivotal role in shaping the transcriptional landscape of the FOXP3 gene and consequently modulating Treg cell function. Dysregulation of histone modifications in the FOXP3 locus can disrupt the differentiation and stability of Treg cells, contributing to immune dysregulation and autoimmune disease pathogenesis. Understanding the complex interplay between histone modifications and FOXP3 expression in Treg cells is essential for deciphering the epigenetic regulation of immune tolerance and autoimmune responses. Also, targeting histone-modifying enzymes may offer promising therapeutic strategies for modulating Treg cells function and restoring immune homeostasis in autoimmune disorders, especially in SLE [59–61].
miRNAs are small single-stranded, non-coding RNAs comprising 20–23 nucleotides in length. They have garnered significant attention for their pivotal role in post-transcriptional regulation of gene expression by binding to specific regions of target mRNA, typically the 3’-UTR. Changes in the expression pattern of miRNAs in Treg cells, play a crucial role in modulating the function and differentiation of these cells. Dysregulation of miRNA-mediated pathways can disrupt the frequency and function of Treg cells, particularly nTreg cells, contributing to various immunological diseases, particularly autoimmune disorders [65, 66]. Several studies have elucidated the role of miRNAs in regulating FOXP3 expression in Treg cells (Table 1). Through their ability to target specific mRNA transcripts, miRNAs can fine-tune the expression of FOXP3, a master regulator of Treg cells development and function. Dysregulation of miRNA-mediated FOXP3 regulation may perturb the balance of Treg cell populations and exacerbate autoimmune responses [67, 68].
Non-coding RNAs that affect FOXP3 gene expression
| Non-coding RNAs | FOXP3 gene regulation | Targeting effects | Autoimmune diseases | Refs |
|---|---|---|---|---|
| miRNAs | ||||
| miR-16 | +ve | Rheumatoid arthritis | [69] | |
| miR-34a | –ve | Systemic lupus erythematosus | [70] | |
| miR-23a-3p | +ve | Targeting SIRT1 | Graves’ disease | [71] |
| miR-106-5p | –ve | Targeting the NR4A3/FOXP3 pathway | Immune thrombocytopenic purpura | [72] |
| miR-124a | +ve | Neuropathic pain | [73] | |
| miR-133a/b | –ve | IgA nephropathy | [74] | |
| miR-155 | +ve | Targeting SIRT1 | Neuropathic pain | [73] |
| miR-210 | –ve | Psoriasis vulgaris | [75] | |
| miR-219-5p | +ve | Regulatory mechanisms on Th17/Treg cells | Ulcerative colitis | [76] |
| miR-223-3p | –ve | Inhibition of HCAECs cells | Kawasaki disease | [77] |
| LncRNAs | ||||
| MEG3 | –ve | Directly interacts with miR-125a-5pUpregulation of MEG3 promoted the expression of FOXP3 | Immune thrombocytopenic purpura | [78] |
| HOTTIP | –ve | HOTTIP directly binds to miR-1908-5pUpregulation of HOTTIP leads to an increase in the Th17/Treg cells ratio | Rheumatoid arthritis | [79] |
| Flicr | –ve | Rheumatoid arthritis | [80] | |
| RP11-340F14.6 | –ve | An increase in Th17/Treg ratio | Juvenile idiopathic arthritis | [81] |
| DQ786243 | +ve | Associated with Crohn’s disease severity | Crohn’s disease | [82] |
| circular RNAs | ||||
| circETS1 | –ve | Increases the severity of SLE by affecting the miR-1205 | Systemic lupus erythematosus | [83] |
+ve: positive; –ve: negative
Xie et al. [70] conducted a seminal study to elucidate the effects of miR-34a on Treg cell differentiation and their function in patients with SLE. They reported that miR-34a acts as an inhibitor for FOXP3 expression. Moreover, they indicated that IL-6 and TNF-α can induce the upregulation of miR-34a via NF-kB activation, subsequently leading to the downregulation of FOXP3 expression. Consequently, the repression of FOXP3 expression results in a decrease in the frequency of Treg cells in SLE patients. Studies have demonstrated that miR-10a stimulates the differentiation of Treg cells by modulating the activity of the Bcl-6 transcriptional repressor, consequently enhancing the upregulation and stability of the FOXP3 gene. Moreover, the increased expression of miR-95 has been found to induce FOXP3 expression in Treg cells, albeit through a mechanism that remains to be fully understood. Conversely, elevated levels of miR-15a-16 and miR-24 in Treg cells have been linked to a significant reduction in the expression of both FOXP3 and CTLA-4, which is indicative of Treg cell dysfunction [84]. Due to the paucity of similar studies on SLE, the definition of the exact link between miRNAs and FOXP3 expression in this autoimmune disease needs further investigation.
Zheng et al. [77] study demonstrated a direct association between miR-223-3p and RORγt expression levels in peripheral blood of Kawasaki patients, indicative of the pivotal role of Treg and Th17 imbalances in the development of various immunological diseases, particularly autoimmunity. Also, Heyn et al. [73], investigated the role of miR-124a and miR-155 in the differentiation of Treg cells in patients suffering from neuropathic pain. They observed an increase in the frequency of Treg cells in patients, possibly confining the pain and inflammatory condition. A positive correlation was found between the expression of miR-124a and miR-155 and the upregulation of FOXP3. Moreover, in human CD4+ T cells, miR-124a and miR-155 were identified as direct suppressors of the histone deacetylase sirtuin 1 (SIRT1), potentially leading to increased expression of FOXP3 and subsequent differentiation of T cells towards Treg cells, thus confining inflammation [73]. Chen et al. [85], studied miR-199a-3p expression in acute lung injury (ALI) patients and found that its downregulation activated NLRP1, leading to the induction of pro-IL-1β and pro-IL-18 by caspase-1. The release of IL-1β and IL-18 aggravated the inflammatory response and contributed to the development of ALI. They also demonstrated that the transcription factor FOXP3, along with histone deacetylase 1 (HDAC1) and C-terminal binding protein 2 (CtBP2), formed a transcriptional complex CtBP2-HDAC1-FOXP3 (CHFTC) that specifically bound to the miR-199a-3p promoter and repressed its expression [85]. Likewise, Li et al. [76] elucidated the effects of miR-219-5p in the development of ulcerative colitis in mouse models. They found that the expression of miR-219-5p in the mucosal tissues of the colon was significantly lower in the affected mice compared to the control group. Injection of miR-219-5p mimic using a lentivirus vector significantly downregulated RORγt and STAT3 in the mucosal tissue of the affected mice. Conversely, FOXP3 expression following miR-219-5p injection was upregulated, indicating the alleviation of ulcerative colitis via the regulatory effect of miR-219-5p on Treg/Th17 balance [76]. Zhang et al. [71], investigated the role of miR-23a-3p in the development of Graves’ disease (GD). They found that in GD patients, the frequency of Treg cells was significantly lower, while IL-17+ T (Th17) cells were more frequent. Additionally, they demonstrated that the expression of FOXP3 and miR-23a-3p was significantly decreased, while SIRT1 and RORγt were upregulated in GD patients. This research group showed that Tregs dysfunction in GD patients is mediated by abnormal acetylation of FOXP3, which is regulated by miR-23a-3p through targeting SIRT1 [71]. Li et al. [72], study on the role of miR-106b-5p in the pathogenesis of immune thrombocytopenic purpura (ITP) disease revealed that the serum level of miR-106b-5p was significantly increased in the patients compared to healthy individuals. Further assessments revealed that using a miR-106b-5p mimic significantly reduced the mRNA and protein levels of NR4A3 and FOXP3, indicating miR-106b-5p’s role in inducing an imbalance in Treg/Th17 cells in ITP patients through the NR4A3/FOXP3 pathway [72]. Wu et al. [69], conducted a study indicating the importance of miR-16 in the development of RA. They found a positive correlation between miR-16 expression and mRNA expression levels of RORγt and FOXP3 in both Th17 and Treg cells, respectively. Reduction of miR-16 expression resulted in downregulation of FOXP3 in Treg cells, while an increase in miR-16 expression led to upregulation of RORγt expression. Therefore, miR-16, by regulating RORγt and FOXP3 mRNA expressions, affected the Th17/Treg imbalance in RA patients [69]. Zhao et al. [75], elucidated the role of miR-210 in the immunopathogenesis of psoriasis vulgaris (PV). They found that the expression of miR-210 in CD4+ T cells derived from PV patients was significantly increased compared to healthy individuals. They also observed a negative association between the expression levels of miR-210 and both mRNA and protein levels of FOXP3. Thus, upregulation of miR-210 downregulated FOXP3, leading to compromised Treg cells function in patients with PV [75].
LncRNAs are RNA transcripts with more than 200 nucleotides that not only lack protein-coding capacity but also serve as functional units themselves. LncRNAs play critical roles in numerous cellular processes including cell cycle regulation, differentiation, and metabolism. While LncRNAs were initially thought to be unstable, it has been revealed that most LncRNAs are stabilized through polyadenylation [86, 87]. Several studies have elucidated the role of LncRNAs in the regulation of FOXP3 gene expression in various immunological abnormalities (Table 1).
Pan et al. [88] conducted a study to evaluate the effects of NEAT1 as a LncRNA on cell invasion and migration in hepatocellular carcinoma. They demonstrated that NEAT1, by binding to FOXP3, induces the expression of the pyruvate kinase M2 (PKM2) gene. Inhibition of FOXP3 reverses the effects of NEAT1 on the expression of PKM2, suggesting an association between FOXP3 and PKM2 expression in hepatocellular carcinoma [88]. Also, Zou et al. [89] revealed that the LINC00261 LncRNA expressed in pancreatic cancer tissue inhibits sterol carrier protein-2 (SCP2) by binding to FOXP3 and the increased expression of LINC00261 leads to decreased cell viability, angiogenesis, and tumorigenic potentials [89]. In addition, Liu et al. [90] elucidated the role of LINC00885 LncRNA in the proliferation and invasion of cervical cancer cells in vitro. They found a direct association between FOXP3 and LINC00885 expression in CaSki and HeLa cell lines. Consequently, FOXP3 specifically binds to the LINC00885 promoter and indirectly promotes cancer cell proliferation [90]. Qiao et al. [82] investigated the progressive role of DQ786243 LncRNA in the upregulation of FOXP3 and CREB in patients with Crohn’s disease (CD). They found that DQ786243 may be related to the severity of CD and regulate Treg function by affecting the expression of CREB and FOXP3 [82]. Our study on SLE patients revealed decreased transcript levels of DQ786243 in the patients with HLA risk alleles (HLA-DRB1*03 and DRB1*16) compared to those patients without HLA risk alleles which embody that the contribution of multiple T cell subsets in disease progression as judged by expression analysis of LncRNAs and transcription factors can be inspired by the inheritance of HLA risk/nonrisk alleles in SLE patients [21].
A study by Zemmour et al. [80] assessed the effects of FOXP3 long intergenic noncoding RNA (Flicr LncRNA) on FOXP3 expression. They identified Flicr as a negative regulator of FOXP3 in Treg cells, exerting broad effects on these cells. Thus, modulating Flicr might offer a way to suppress or enhance Treg cell function [80]. Li et al. [78] indicated the role of MEG3 LncRNA in the development of ITP. They found that MEG3 upregulation or miR-125a-5p downregulation promoted the expression of FOXP3 and inhibited the expression of RORγt, potentially modulating the Treg/Th17 ratio involved in ITP immunopathogenesis [78]. Huang et al. [81] demonstrated that RP11-340F14.6 LncRNA was associated with an increase in the Th17/Treg ratio in patients with juvenile idiopathic arthritis (JIA). They hypothesized that RP11-340F14.6 modulates the Th17/Treg ratio by binding to P2X purinoceptor 7 (P2X7R) [81]. Also, Yao et al. [79] elucidated the function of HOTTIP LncRNA in the development of RA. They found that HOTTIP upregulation led to decreased expression of miR-1908-5p, potentially increasing the Th17/Treg cells ratio and exacerbating inflammation in RA mouse models [79].
Initially, circular RNAs (CircRNAs) were thought to be rare isoforms of non-coding RNA produced as a result of splicing errors. However, with the advancement of bioinformatics and high-throughput sequencing, it is now well established that CircRNAs are abundantly expressed and highly conserved across different species. One of the important functions of CircRNAs is to act as sponges for miRNAs, regulating the expression of their target mRNAs [91, 92]. Zou et al. [83] study aimed to elucidate the role of circular RNA, a novel non-coding RNA, in regulating the activity of Treg cells in SLE patients. They observed that transfection with circETS1 led to an increase in FOXP3 expression, while the use of a miR-1205 mimic resulted in a downregulation of FOXP3 (Table 1). This indicated that the miR-1205 mimic reversed the effect of circETS1 upregulation on disease development. In summary, their findings suggested that the upregulation of circETS1 exacerbates the severity of SLE by affecting the miR-1205/FOXP3 axis, thereby disrupting the balance between Th17 and Treg cells [83].
The impaired function of Treg cells and the imbalance of Treg/Th17 cells is one of the important players in the immunopathogenesis of autoimmune diseases and the FOXP3 protein is a key transcription factor that regulates the expression of specific genes concerning Tregs function. Numerous studies on epigenetic regulation of FOXP3 gene expression in autoimmune diseases indicate the fine-tune regulation of FOXP3 and other Treg-specific genes through non-coding RNAs (miRNAs, LncRNAs, CircRNAs) in favour of dysregulation of Treg/Th17 balance to promote autoimmune diseases. Therefore, analysis of the expression patterns of these non-coding RNAs in conjunction with FOXP3 gene may provide a more precise evaluation of Treg cells functions in autoimmunity. Moreover, understanding the intricate interplay between epigenetic regulation and FOXP3 expression in Treg cells is crucial for unravelling the underlying mechanisms of autoimmune diseases like SLE. Finally, targeting these epigenetic pathways may offer novel therapeutic strategies for restoring immune balance and ameliorating autoimmune pathology.
ALI: acute lung injury
BAFF: B-cell activating factor
CD: Crohn’s disease
CircRNAs: circular RNAs
CNS: conserved non-coding sequences
CtBP2: C-terminal binding protein 2
GD: Graves’ disease
HDAC1: histone deacetylase 1
IFN-γ: interferon-γ
ITP: immune thrombocytopenic purpura
iTregs: induced regulatory T
LncRNAs: long non-coding RNAs
MBD2: methyl-CpG-binding domain protein 2
miRNAs: microRNAs
nTreg: natural regulatory T
pDCs: plasmacytoid dendritic cells
PKM2: pyruvate kinase M2
PV: psoriasis vulgaris
RA: rheumatoid arthritis
SIRT1: sirtuin 1
SLE: systemic lupus erythematosus
TET: ten-eleven translocation demethylases
TLR7: Toll-like receptor 7
TNF-α: tumour necrosis factor-alpha
Treg: regulatory T
UV: ultraviolet
PF and AS: Investigation, Writing—original draft. ATR: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GS: Conceptualization, Investigation, Writing—original draft, Writing—review & editing, Validation, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

