 Review
Review

Affiliation:
1Department of Hospital Medicine, Hartford Hospital, Hartford, CT 061061, USA
Email: Mohamed.elnaggar.md@gmail.com
ORCID: https://orcid.org/0000-0003-4415-2789
Affiliation:
1Department of Hospital Medicine, Hartford Hospital, Hartford, CT 061061, USA
ORCID: https://orcid.org/0000-0003-0759-1010
Affiliation:
2Department of Gastroenterology, University of Kentucky, Lexington, KY 40508, USA
ORCID: https://orcid.org/0000-0003-0156-8119
Affiliation:
3Digestive Diseases Section, Department of Internal Medicine, Yale School of Medicine, New Haven, CT 06510, USA
Email: james.boyer@yale.edu
ORCID: https://orcid.org/0000-0002-8959-6036
Explor Immunol. 2024;4:658–678 DOI: https://doi.org/10.37349/ei.2024.00165
Received: May 16, 2024 Accepted: September 28, 2024 Published: October 29, 2024
Academic Editor: Jochen Mattner, FAU Erlangen-Nürnberg and UKER, Germany
Primary biliary cholangitis (PBC) is a rare immune-mediated disease, commonly affecting women in their 40s, and ultimately progressing to liver failure. The incidence and prevalence of the disease are increasing worldwide, possibly due to better diagnostic tools. This review will focus on its epidemiology, pathophysiology, diagnosis, prognosis, and new developments in therapy.
Primary biliary cholangitis (PBC) is a T-lymphocyte mediated slowly destructive process of the small intrahepatic bile ducts eventually resulting in cirrhosis and progression to liver cell failure. The name was changed from primary biliary cirrhosis a few years ago, to describe the disease and its natural progress more accurately since most patients are diagnosed before histological evidence of cirrhosis develops.
PBC is a rare “orphan” disease with a prevalence of 19 to 402 cases per million persons depending on the geographic location [1, 2]. Earlier studies suggested that women were mainly affected (90 to 95 percent), mostly between the ages of 30 to 65 years, commonly in their 40s or 50s, with the least reported age as young as 15 years and as old as 93 years in women [3, 4]. The incidence and prevalence of the disease increases with age [5]. Newer studies suggest that the female-male ratio may be closer to 4:1–6:1 [6, 7]. Male PBC patients have a higher rate of progression to cirrhosis and HCC, are diagnosed at a later stage, and respond less well to ursodeoxycholic acid (UDCA) [8, 9].
Geographically, PBC is present throughout the world and in all races and ethnicities. It is more common in North America (Northern Midwest regions of the US) and Northern Europe. The disease burden is increasing over time, particularly in Asian populations, possibly due to better diagnostic tools rather than a true rising incidence [10, 11]. PBC prevalence is 100 times higher in first-degree relatives than in the general population and occurs in twins, indicating genetic susceptibility. In a study of over 1,000 patients, a history of urinary tract infections, past smoking, or use of hormone replacement therapies were also significantly associated with an increased risk of having PBC [12].
Adaptive innate immunity and allelic variations in major histocompatibility complex (MHC) class II (D.R., D.Q.) are associated with susceptibility to PBC [13].
The pathogenesis of PBC is the result of combinations of genetic and environmental factors occurring in a genetically susceptible host [14]. GWAS studies have identified a number of susceptibility genes [15]. In summary, PBC is an autommune disease of unknown etiology resulting in the destruction of the intermediate-sized intralobular biliary ducts, due to loss of immune tolerance, often in association with histologic evidence of granulomas and which slowly progresses over several decades to cirrhosis and liver failure, ultimately requiring a liver transplant.
Several theories emphasize the role of bacterial infections as important environmental factors especially E. coli or Novosphingobium aromaticivorans infections. These infections may result in a loss of tolerance to mitochondrial autoantigens in PBC and induce disease-specific antimitochondrial antibodies (AMA) [16]. Many case series and case-control studies confirm a high incidence of bacterial infections in patients with PBC [17, 18].
Antibiotics, heavy metals like cadmium, and certain cosmetics are other candidate environmental triggers.
Another hypothesis is a defective “biliary HCO3– umbrella” [19]. Normally cholangiocytes (and hepatocytes) secrete bicarbonate into the bile duct lumen. This creates a protective alkaline barrier so that bile salts remain in a polar, anionic state impermeable to the bile duct membrane. However, defects in the apical HCO3– secretion will result in a decrease in the pH of bile and compromise the alkaline barrier, consequently causing bile salts to be protonated and conjugated to glycine instead of taurine. The resulting glycine-conjugated bile acids become apolar and can cross the cholangiocyte membrane by diffusion, independent of bile salt transporter activity [20]. This results in the induction of apoptosis in cholangiocytes [20, 21]. In PBC, expression of cholangiocyte anion exchanger-2 (AE2) is defective which together with the apical Cl–/HCO3– exchanger and type III inositol trisphosphate receptor (InsP3R3), are crucial for normal biliary HCO3– secretion [22–25].
PBC is classified as an auto-immune mediated disease based on its association with HLA determinants. However immunosuppressive agents are ineffective in PBC. Nevertheless, T cell dysfunction plays an important role in the pathogenesis of PBC as does the humoral immune response and proinflammatory cytokines.
CD4+ and CD8+ T-cells constitute the primary immune response, CD8+ lymphocytes which are cytotoxic are increased around small bile ducts and target bile epithelial cells in early stages of PBC by 3 main pathways: Fas/FasL, T cell receptor (TCR)/MHC, and Fas/Fas receptor signaling pathways [26, 27].
Proinflammatory cytokines are also involved in the immune response in PBC, via activating NK cells which stimulate dendritic cells which not only activate Th1 and T17 phenotypes but also attrack B cells leading to the production of AMA, which target PDC E2 antigen produced from apoptotic bile duct epithelial cells leading to production of antigen/antibody complex that may also lead to cell damage [28].
Other cytokines that play a role are:
IL12 differentiates Th0 cells into Th1 cells which stimulates T cell growth function.
IL23, which mediates the transformation of CD4 T cells into Th17 cells which produce IL6, IL17 and TGFB.
IL17 not only mediates inflammation in PBC patients but also activates stellate cells which leads to a progression of liver fibrosis to liver cirrhosis.
Both IL23 and IL17 are significantly elevated in portal areas of PBC patients.
The clinical presentation of PBC varies. Fifty to sixty percent of patients are asymptomatic and are diagnosed based on an abnormal alkaline phosphatase and a positive AMA. In a middle-aged female, the most common symptom is fatigue followed by pruritus. Other symptoms include dry eyes, jaundice, and abdominal pain [29–31]. PBC may also be associated with other autoimmune disorders including hypothyroidism, rheumatoid arthritis, Sjogren’s disease, and celiac disease.
Fatigue: Fatigue is manifested by tiredness and excessive daytime sleepiness. Fatigue is often difficult to distinguish from depression and affects the quality of life in these patients. The mechanism is not known although muscle mitochondrial dysfunction has been proposed resulting in extreme acidosis after exercise [32, 33].
Pruritus: This important symptom can vary from mild to severe and is present in up to 50–70% of patients. The pathogenesis has been debated for years. Recent studies indicate that certain bile acids activate mas-related G-protein-coupled receptors in sensory nerve endings. Autotoxin and endogenous opiates have also been implicated [34]. Symptoms are worse at night, and with humid and dry weather or after a hot shower. Symptoms of pruritus can precede jaundice and do not correlate with the severity of the disease.
Right upper quadrant discomfort: has been reported in 8% of patients [35].
Malabsorption and steatorrhea: Patients with advanced PBC may develop malabsorption and steatorrhea resulting in fat-soluble vitamins deficiencies (vitamins A, D, E, and K).
Physical examination: Asymptomatic patients usually have a normal examination. In more advanced cases, jaundice and cholesterol deposits in the skin can be seen as well as features of cirrhosis.
Skin: In ~40% of patients [36] findings include hyperpigmentation due to melanin deposition but not due to Jaundice [37, 38]. Xanthomata, due to hypercholesterolemia, occur in less than 5 percent of patients and are typically late in the disease course; however, xanthelasma occurred in approximately 10% of patients prior to available treatment. Other skin findings include dry skin, excoriations from pruritus, and jaundice which typically occurs later in the disease course. Spider nevi may be present as cirrhosis develops.
Hepatosplenomegaly: This can be a sign of cirrhosis and portal hypertension, ascites, temporal and proximal limb muscle wasting, and lower extremity edema are late manifestations and signs of decompensation in the disease.
Patients with PBC and resultant cirrhosis are at increased risk of HCC. A meta-analysis of 17 studies found that PBC has a significantly higher risk of overall cancer (relative risk 1.55, 95% CI) and HCC (relative risk 18.80, 95% CI) compared to the general population. In a large cohort study of 4,565 patients, 123 patients developed HCC, yielding an incidence rate (IR) of 3.4 cases/1,000 patient-years. Biochemical non-response to UDCA after 12 months was associated with future risk of HCC (hazard ratio 4.52, p < 0.0001), especially in male patients [39, 40].
Chronic cholestasis can lead to deficiencies in fat-soluble vitamins, including vitamin D, which is crucial for bone health. The majority of patients with PBC experience osteopenia, and 20–44% develop osteoporosis. It is essential for patients with PBC to undergo bone mineral density testing at the time of diagnosis, with the frequency of subsequent tests determined by the initial results [41].
Hypercholesterolemia is seen in ~75–95% of PBC patients, due to cholestasis which decreases the production of bile acids from cholesterol. Despite high cholesterol levels, PBC-related hyperlipidemia is generally not associated with increased atherosclerotic risk unless metabolic syndrome is present [42–44]. However, statins are often recommended.
The diagnosis of PBC hinges on a combination of the clinical presentation (middle-aged female with an elevated alkaline phosphatase) and serological markers, particularly AMA and imaging studies. Advanced diagnostic tools like transient elastography (T.E.) are emerging as valuable non-invasive methods for assessing liver fibrosis, complementing traditional liver biopsy, and enhancing clinical management and treatment planning for PBC patients [45].
Diagnostic criteria: Criteria established by the American Association for the Study of Liver Diseases (AASLD) for PBC in 2018 require the fulfillment of two out of three conditions: biochemical evidence of cholestasis with elevation of ALP activity, the presence of AMA, or liver biopsy results showing chronic inflammation and damage to the small or medium bile ducts. A liver biopsy may not be required as biochemical testing is usually sufficient for diagnosis in most cases [46].
AMA are the most prevalent biomarker in PBC found in about 95% of patients. In clinical settings, enzyme-linked immunosorbent assay (ELISA) or immunofluorescence are used to detect the AMA. Traditionally, at least nine AMA subtypes have been identified, and four of them (M2, M4, M8, and M9) seem to be associated with PBC [47]. The titer of the AMA does not correlate with disease progression or the clinical course of the patients and is not pathogenic. AMA are detected in less than 1% of healthy individuals with normal liver biochemistry [48, 49] and some will develop PBC in the future.
The predominant AMA subtype is anti-M2. Despite the diagnostic value of the marker having high sensitivity and specificity for the diagnosis of PBC, the presence of these autoantibodies is not pathognomonic for PBC; they have been reported in various other liver conditions, including chronic active hepatitis and cryptogenic cirrhosis, which occasionally leads to false positives in PBC diagnosis [50, 51]. To ensure accurate diagnosis, international guidelines use diagnostic criteria as the ones adopted by the AASLD [45].
Approximately 50–70% of patients with PBC have positive antinuclear antibodies and may also have diagnostic significance, particularly the nuclear pore complex protein gp210 and the anti-Sp100, which have high specificity but low sensitivity for PBC. They are present in over 30% of PBC patients who are AMA-negative. Some studies suggest that Sp100 and gp210 are associated with more advanced disease and a poor prognosis [52, 53]. Sp100 titers change over time with disease progression [52, 53]. In addition, anti-gp210-positive patients typically show a poorer response to UDCA and have more severe histopathologic features compared to anti-gp210-negative patients [54].
Elevated levels of immunoglobulin M (IgM) are commonly found in autoimmune diseases as a response to an underlying antigen response. Elevated IgM is found in approximately 80% of patients with PBC [55]. Although IgM is not part of the diagnostic criteria for PBC, studies have shown that IgM improves with the treatment of PBC [56]. Thus, IgM may be of clinical relevance in some patients to monitor response to therapy.
In all patients with evidence of cholestasis, national society guidelines recommend imaging of the liver and biliary tree to rule out other causes of biliary obstruction [45]. Magnetic resonance cholangiopancreatography (MRCP) is the preferred imaging procedure when there is a suspicion of biliary obstruction but is not part of the initial diagnostic workup. Typically, imaging to rule out extrahepatic biliary obstruction involves a right upper quadrant ultrasound or MRCP, with endoscopic retrograde cholangiopancreatography (ERCP) considered if the suspicion for mechanical obstruction is high.
T.E. (fibroscan): Measures liver stiffness (pKa) and has an increasing role in monitoring the response to medical therapy. A recent systematic review and meta-analysis evaluating the effectiveness of T.E. versus liver biopsy in staging liver fibrosis in PBC patients concluded that T.E. exhibited excellent performance offering a non-invasive alternative that could potentially replace liver biopsy in clinical practice, particularly for stages F3 and F4 of fibrosis [57].
There are several predictive models based on clinical presentation and laboratory data. The two commonly used models are the UK-PBC score and the GLOBE score [45, 58, 59].
UK-PBC score: The UK-PBC score includes serum alkaline phosphatase, bilirubin, and aminotransferases after 12 months of UDCA therapy, platelet count, and baseline albumin. This model evaluates the risk of liver transplantation (L.T.) or liver-related death.
GLOBE score: This is based on five variables: serum bilirubin, albumin, alkaline phosphatase, platelet count after one year of UDCA treatment, and age at the start of therapy. This score estimates the duration of transplant-free survival.
Studies of life expectancy in PBC before the use of UDCA showed a mean survival of 6.9 years with asymptomatic patients having a survival rate similar to that of age and sex-matched controls [60]. A ten-year follow-up of a large cohort indicated that patients who were asymptomatic at the time of diagnosis had a median predicted survival that was twice as long as that of symptomatic patients (16 years vs. 7.5 years, p < 0.0001) [61]. However, it was observed that asymptomatic patients eventually developed symptoms. While some studies have questioned the strength of this association [29, 31, 62, 63], it is likely that the better prognosis seen in asymptomatic patients is due to lead-time bias, meaning these patients were diagnosed earlier in the disease course.
Outcomes in patients with PBC have improved significantly since treatment with UDCA was available. Long-term studies have determined that UDCA improves transplant-free survival no matter what stage of disease exists when UDCA is begun or the biochemical response [64] establishing UDCA as the first line of therapy. Factors associated with a worse prognosis include symptoms at the time of diagnosis, higher alkaline phosphatase and bilirubin levels, more advanced histologic stage, presence of antinuclear antibodies, cigarette smoking, and specific genetic polymorphisms [65].
These two liver function tests are associated with transplant-free survival. In a meta-analysis of 15 studies with 4,845 patients, increased alkaline phosphatase and bilirubin levels were associated with worse outcomes after one year of enrollment [65]. Alkaline phosphatase level > 2 times the upper limit of normal was associated with decreased 10-year transplant-free survival as opposed to alkaline phosphatase level ≤ 2 times the upper limit of normal (62 percent vs. 84 percent). Similarly, a bilirubin level > 1 time the upper limit of normal was associated with lower 10-year transplant-free survival compared with a bilirubin level ≤ 1 time the upper limit of normal (41 percent vs. 86 percent). The association of alkaline phosphatase and bilirubin levels with transplant-free survival was seen in patients who had received UDCA as well as those who had not.
PBC progresses through 4 histological stages. Stage 1, portal inflammation with or without granulomas; Stage 2, periportal fibrosis; Stage 3, bridging fibrosis between portal triads; and Stage 4, a fully developed biliary type cirrhosis. However, treatment with UDCA may slow progression through these stages. One study (before UDCA was available), reviewed 916 biopsy specimens from 222 patients with PBC and reported that cirrhosis developed within four years in 31 and 50 percent who presented initially with stage 1 or 2 disease, respectively [66].
Stage 4 disease (cirrhosis) has the worst prognosis and is associated with complications including ascites, esophageal variceal bleeding, hepatic encephalopathy, and hepatocellular carcinoma [67, 68].
Antinuclear antibodies (particularly, anti-gp210, anticentromere antibodies, and anti-Sp100) may be associated with an increased risk of progression and liver cell failure [58, 59]. Patients with AMA-negative PBC are thought to have a similar clinical course and response to treatment as patients with AMA-positive PBC [69, 70].
An association between PBC and cigarette smoking has been suggested in epidemiologic studies [12, 71]. Two studies suggest that cigarette smoking is associated with more advanced fibrosis [71, 72]. One study showed that patients who never smoked were less likely to have advanced fibrosis (METAVIR fibrosis score of 3 or 4) compared to patients who were smokers in the past or were current smokers (16 percent vs. 33 percent) [72]. There was a 5 percent increase in the likelihood of advanced fibrosis associated with each pack-year increase in smoking.
Certain polymorphisms of genes involved in immunity [SLCA2 (exchanger anion, AE2) and TNF-alpha] were associated with prognosis and response to UDCA therapy in a study of 258 patients [73]. More studies are needed to understand the relevance of these observations to natural history models and the choice of therapy.
PBC medications slow disease progression and complications, reduce symptoms, and delay liver transplants (Figure 1). UDCA is a first-line U.S. Food and Drug Administration (FDA) approved therapy for patients with PBC and abnormal liver enzymes [45]. Ocaliva (OCA or obeticholic acid) was the only FDA-approved drug for second-line therapy for those with inadequate response to UDCA in the USA until recently when two PPAR agonists were also approved (elafibrinor and seladelpar). Fibrates (fenofibrate) off-label and bezafibrate (available in Asia and Europe) are also alternatives for those with insufficient response to UDCA. The use of OCA and fibrates is discouraged for those with decompensated liver disease. Black box warnings advise against OCA use in patients with evidence of cirrhosis and portal hypertension [74].
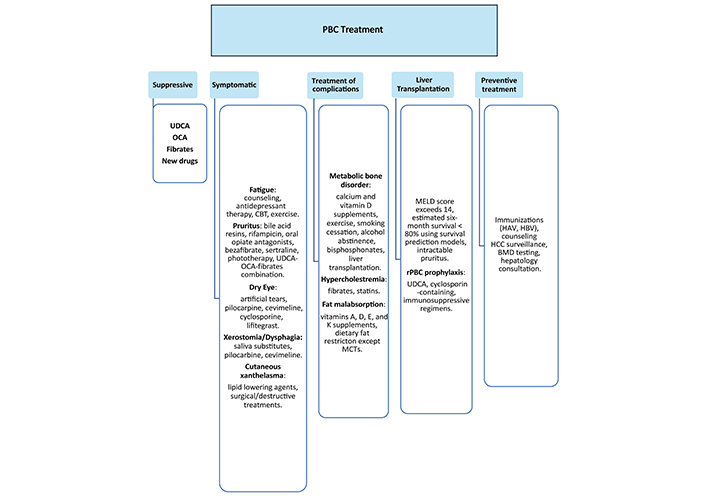
PBC treatment. BMD: bone mineral density; CBT: cognitive behavioral therapy; MCTs: medium-chain triglycerides; OCA: Obeticholic acid; PBC: primary biliary cholangitis; rPBC: recurrent primary biliary cholangitis; UDCA: ursodeoxycholic acid
UDCA was first approved by the FDA for gallstone disease. FDA approved the use of UDCA for patients with PBC in 1997. A biochemical response with UDCA is associated with delayed disease progression [72, 75], lower rates of complications, all-cause, and liver-related mortality, reduced need for liver transplant, and 10-year cumulative liver transplant-free survival [64, 73, 76].
A dose of 13–15 mg/kg per day orally is recommended. UDCA is generally well tolerated. Common side effects are hair loss, weight gain, flatulence, and diarrhea [77].
Recent studies showed that most patients with PBC benefit from UDCA but about 40% have an inadequate response [78]. Early monitoring and modification of treatment in those with insufficient response were found to significantly increase the response rate 12 months after treatment modification [79].
OCA, a potent agonist of the farnesoid X receptor, was approved by the FDA as second-line therapy for PBC in 2016. OCA monotherapy is also associated with a greater reduction of ALP compared to placebo [80].
The phase III POISE trial’s (a combination of UDCA and OCA) primary end-point consisted of a reduction of the alkaline phosphatase < 1.67 ULN, and > 15% overall reduction, with the maintenance of a normal bilirubin level and was met in 47% of patients in the 10 mg/day dose [81].
A dose of 5–10 mg/day of OCA is recommended for PBC patients with inadequate response to UDCA. This dose should be adjusted to 5 mg/week with a maximum dose of 10 mg twice weekly for those with cirrhosis (Child-Pugh class B/C) but is contraindicated in advanced and decompensated disease [82].
The most reported side effects of OCA are pruritus and fatigue. Other side effects like esophageal varices and ascites are less common and were mostly observed in those with pre-existing advanced liver disease [81, 83].
A recently published systematic review and meta-analysis showed that 5 mg/day of OCA may be sufficient to achieve biochemical remission and was associated with a lower risk of pruritus compared to 10 mg/day [84].
A retrospective cohort study of PBC patients with inadequate biochemical response to dual therapy showed that triple therapy with UDCA, OCA, and fibrates can attain adequate biochemical response and improve pruritus in patients with difficult-to-treat PBC [85]. At the time of this writing, OCA had only received conditional approval by the FDA and European authorities. A review of evidence of long-term efficacy failed to confirm this outcome.
Fibrates have been used as off-label alternatives for PBC patients with inadequate response to UDCA. Fenofibrate was approved by the FDA for the treatment of hypertriglyceridemia but has been shown to significantly reduce ALP concentrations in retrospective cohorts [86]. Several studies have shown that fibrates are associated with improved liver biochemistry in treatment naïve patients and those with inadequate biochemical response to UDCA [87, 88].
In the BEZURSO trial, which combined UDCA with bezafibrate, the POISE criteria were met in 31% of patients compared to 0% in the placebo group and 67% normalized their alkaline phosphatase at 24 months compared to 2% in the placebo group [89]. Bezafibrate is widely used as second-line therapy in Japan and Europe.
A Japanese nationwide 10-year retrospective cohort study showed that the UDCA-bezafibrate combination was associated with a lower risk of all-cause and liver-related mortality and reduced need for liver transplant [90]. Both bezafibrate and fenofibrate are associated with improvement in pruritus and bezafibrate also improved the fibroscan assessment of liver fibrosis [89]. Common side effects of fibrates are myopathy, elevations of creatinine, and drug-induced liver injury. Dose reductions often reverse these findings. Rhabdomyolysis is a rare complication of fibrate therapy.
Shu et al. [91] developed a predictor index that can accurately predict biochemical nonresponse to UDCA treatment with a predictive value of 0.886, sensitivity of 82.80%, and specificity of 84.4%.
The alkaline phosphatase levels have been validated as a marker of severity and progression in PBC [65] and should be monitored every 3–6 months. Improvement in the alkaline phosphatase level is usually observed within 3 months, while normalization may take a year after starting treatment [65]. Multiple scoring systems have been developed to assess biochemical response to UDCA treatment (Table 1) [92].
Multiple scoring systems have been developed to assess biochemical response to UDCA treatment [92]
| Scoring System | Criteria for treatment of non-responsiveness | Time of evaluation from treatment initiation |
|---|---|---|
| Barcelona | ALP decrease ≤ 40% of the initial level and ALP ≥ ULN | 12 months |
| Paris II | ALP ≥ 1.5 × ULN or total bilirubin > 1 mg/dL or AST ≥ 1.5 × ULN | 12 months |
| Rotterdam | Total bilirubin ≥ ULN, albumin < LLN, or both | 12 months |
| Toronto | ALP ≥ 1.67 × ULN | 24 months |
| UK-PBC formula | Uses baseline albumin and platelet count, along with 12-month measurements of total bilirubin, ALP, and AST or ALT | 12 months |
| GLOBE formula | Uses age, total bilirubin, ALP, albumin, and platelet count | 12 months |
Assessment of the response to UCDA for an incomplete response can be done 6 months after initiating therapy although previously 12 months was used as the time before adding a second line drug [45, 82]. A retrospective cohort analysis showed that attaining normal levels of bilirubin or ALP is associated with the lowest risk for L.T. need or death [91]. Therefore, current recommendations are to normalize the ALP if possible and to use second-line therapy at any abnormal level of ALP when UDCA shows an incomplete response.
Most patients with PBC who respond to UDCA monotherapy do so within 4–6 weeks. This raises the question as to whether decisions to begin second-line therapy might be made much earlier both to save time and cost. This would be an example of personalized medicine.
Multiple drugs have been studied previously for the treatment of PBC including colchicine, cyclosporine, methotrexate, penicillamine, and mycophenolate mofetil, but all were shown to have no clinical benefit [74, 93–97]. Below are some drugs in development during the writing of this review, two of which now are FDA-approved and are of benefit to patients with PBC (Table 2).
Summary table of ongoing clinical trials
| Drugs | Trial type | Outcome | Comment |
|---|---|---|---|
| Bezafibrate | UDCA is combined with bezafibrate in 2 years phase III RCT (NCT01654731) | The POISE criteria were met in 31% of patients compared to 0% in the placebo group and 67% normalized their alkaline phosphatase at 24 months compared to 2% in the placebo group | Bezafibrate is widely used as second-line therapy in Japan and Europe |
| Rituximab | A phase II trial (NCT02376335) | Was ineffective for the treatment of fatigue | - |
| Elafibranor | A phase III trial (NCT04526665) ELATIVE trial | 51% met the primary end-point vs. 4% in placebo. 15% normalized the alkaline phosphatase | Elafibrinor has now been FDA conditionally approved (IQIRVO®) |
| Seladelpar | A phase III RCT of seladelpar in patients with inadequate response to UDCA or with UDCA intolerance (NCT04620733) RESPONSE trial | 61.7% met the primary end-point compared to 20% in the placebo group while 25% normalized the alkaline phosphatase | Seladelpar has now been FDA conditionally approved (LIVDELZI®) |
| Volixibat | The VANTAGE trial is assessing efficacy in PBC (NCT05050136) | Trial is on-going | This drug inhibits the uptake of bile acids in the terminal ileum (IBAT inhibitor) |
| Linerixibat | GSK2330672, 28 days phase 2a RCT & GLIMMER, 12 weeks phase 2b RCT (NCT02966834) | This is a novel inhibitor of the apical bile acid transporter with minimal systemic absorption. It has shown promising results in reducing pruritus. | - |
| Probiotics | A phase 2 trial for the combination of probiotics and UDCA in those with inadequate response to UDCA monotherapy (NCT03226067) | Trial is on-going | - |
IBAT: intestinal bile acid transporter; UDCA: ursodeoxycholic acid; -: not applicable
Several open-label trials have shown significant improvement of alkaline phosphatase levels with rituximab therapy at 6, 9, and 12 months [97, 98]. A phase II RCT with clinical trial gov number NCT02376335 found rituximab to be ineffective for the treatment of fatigue [99]. The most significant side effect of rituximab is de novo colitis [100].
In a phase II clinical trial (NCT04526665) 79% met POISE with 120 mg/day, and 21% normalized ALP and pruritus were improved [101]. A phase III trial (NCT04526665) was recently completed [102] and 51% met the primary end-point vs. 4% in placebo, and 15% normalized the alkaline phosphatase. However, the NRS pruritus score did not reach significance compared to the placebo group. This drug (IQIRVO®) has now met FDA conditional approval.
This is also a recent FDA conditionally approved PPAR-delta agonist (LIVDELZI®). A proof-of-concept phase II trial showed that seladelpar is associated with a significant reduction of ALP levels compared to placebo, and improvement of pruritus and sleep [103]. A phase III RCT of seladelpar in patients with inadequate response to UDCA or with UDCA intolerance (ENHANCE trial, NCT03602560) [103]. 61.7% met the primary end-point compared to 20% in the placebo group while 25% normalized the alkaline phosphatase. Significant improvement in the NRS score for pruritus was also seen in seladelpar and together with elafibranor are significant advances for second-line therapy for PBC [104].
A multicenter, open-label, single-arm study to evaluate the efficacy and safety of saroglitazar in patients with PBC was terminated because of lack of enrollment although saroglitazar resulted in rapid and sustained improvement in ALP levels [105].
Tropifexor, cilofexor, and EDP-305 are being studied with phase 2 clinical trials after showing positive results in mouse models. The cilofexor trial was stopped due to futility.
This is a JAK1 and JAK2 inhibitor that modulates signaling pathways of interleukins, interferons, and growth factors. It was approved for adults with moderate to severe active rheumatoid arthritis and is being evaluated in PBC patients with inadequate responses to UDCA.
This intestinal bile acid transporter (IBAT) inhibitor interrupts the enterohepatic circulation of bile acids and lowers the bile acid pool. The VANTAGE trial (NCT05050136) is assessing efficacy in PBC. IBAT inhibitors have been FDA-approved for pediatric patients with pruritus’ from Alagille syndrome and PFIC patients. The primary end-point is improvement in pruritus.
Linerixibat (GSK2330672, NCT02966834), is a novel inhibitor of the apical bile acid transporter and has shown promising results in reducing pruritus in the GLIMMER phase 2b trial [106]. It is now approved for a phase 3 trial in PBC.
Changes in the gut microbiome lead to alterations in the bile acid pool [107]. Probiotics were shown to induce bile acid synthesis and downregulation of farnesoid X receptor [108]. A phase 2 trial for the combination of probiotics and UDCA in those with inadequate response to UDCA monotherapy is underway (NCT03226067).
Autoimmune hepatitis (AIH)-PBC overlap syndrome is usually classified into 2 categories: 1) Patients who are seropositive for PBC (AMA positive) and have histological features of AIH, and 2) Patients who are often seronegative for PBC (AMA negative) with histological features of AIH and have anti-smooth muscle antibodies (SMA) and/or antinuclear antibodies. There are no randomized, controlled trials that indicate the best treatment of PBC/AIH overlap syndrome. Current AASLD guidelines recommend that treatment should be targeted at the predominant histological pattern of injury [45]. EASL guidelines also recommend combined therapy with UDCA and corticosteroids in patients with PBC-AIH overlap [109].
The International Autoimmune Hepatitis Group (AIHG) agrees that patients with PBC with features of AIH should be considered for immunosuppressive treatment [110].
Fatigue is the most reported symptom of PBC. However, there is no evidence-based recommended treatment for fatigue resulting from PBC. A phase 2, double-blind, randomized, controlled trial showed that rituximab is ineffective for the treatment of fatigue in patients with PBC [98]. A novel home-based exercise program is being evaluated for use with refractory fatigue associated with PBC [111]. Some studies reported that fatigue improves after liver transplant, while others reported no improvement or even worsening fatigue. The different findings can be related to the complex and variable posttransplant treatment regimens, which can be associated with fatigue apart from the pre-existing liver condition [112, 113]. Physicians should provide frequent counseling for their patients regarding these challenging symptoms and consider anti-depressant therapy, cognitive behavioral therapy, and exercise.
Mild pruritus usually responds to warm baths, emollients, or antihistamines. Anion-exchange resins cholestyramine (powder), questran (pills), or colesevalam (pills) are the initial therapy for patients with PBC who suffer from more clinically significant pruritus and should be given p.o. 20 minutes before each meal in 4 g amounts [45]. Rifampicin, oral opiate antagonists, or sertraline can be tried for pruritus refractory to anion-exchange resins [45].
UDCA and other medications should be given at least 1 hour before or 4 hours after the anion-exchange resin. Side effects of anion-exchange resins include fat malabsorption, fat-soluble vitamins deficiency, bloating, constipation and/or diarrhea, and decreased absorption of multiple drugs [114]. Anion-exchange resins were found to decrease the absorption of UDCA, digoxin, warfarin, and thiazide diuretics.
Bezafibrate has been used as an anti-pruritus drug in the FITCH double-blind randomized, placebo-trial in PBC and PSC, and is superior to placebo in improving moderate to severe pruritus in these patients [115]. Phototherapy and plasma exchange are used as an experimental treatment for PBC-associated pruritus when alternative better-studied treatments fail and the pruritus is severe [116]. Liver transplant may be indicated in severe pruritic cases.
Sjogren’s syndrome is often associated with PBC and should be evaluated by an ophthalmologist. Artificial tears should be tried initially. Pilocarpine or cevimeline can be used if symptoms are refractory to artificial tears. Cyclosporine or lifitegrast ophthalmic emulsion can be used under the supervision of an ophthalmologist for those with refractory symptoms.
Over-the-counter saliva substitutes should be tried first, and if the patient remains symptomatic, pillocarpine or cevimeline can be used.
Treatment of cutaneous xanthomas usually begins with lipid-lowering agents. Surgical and destructive therapies can be used as well.
Treatment of metabolic bone disease begins with calcium and vitamin D supplements, exercise, smoking cessation, and alcohol abstinence. Bisphosphonates should be considered if patients are osteoporotic [45]. Oral bisphosphonates should be avoided if patients have esophageal varices, acid reflux, or are unable to adhere to appropriate administration protocol.
L.T. is associated with long-term improvement in bone mineral density [117].
Fibrates were found to reduce total cholesterol, LDL, and triglyceridess in patients with hypercholesterolemia and cholestasis despite UDCA treatment [118].
Statins are preferred for the treatment of hypercholesterolemia as hepatic toxicity is rare [119].
Fat-soluble vitamins K, E, D, and A should be supplemented. Dietary fat restriction is recommended. Medium-chain triglycerides can be substituted in severe cases of steatorrhea.
Patients with manifestations of end-stage PBC should be referred for L.T. when their MELD score exceeds 14 [45]. L.T. is recommended for those with an estimated six-month survival < 80% using survival prediction models [120]. Black and Hispanic patients have an increased rate of transplant wait-list mortality [8].
L.T. is associated with about a 90% survival rate at five years and a 53% survival rate at 20 years after surgery [111, 121]. Higher Kaplan-Meier survival probability compared to nontransplant patients has been shown as early as three months after surgery [120].
Pruritus, hepatic encephalopathy, hepatorenal syndrome, and variceal bleeding are reversible with L.T. Jaundice, ascites, and cutaneous xanthomas slowly resolve over days to a few months, while hepatic osteodystrophy takes the longest time to improve with up to 18 months, even after supplementation with vitamin D and calcium.
PBC recurs in up to one-third of patients after L.T. [121]. Prophylactic use of UDCA after L.T. was associated with reduced risk of recurrence [122] in a recent meta-analysis of 15 retrospective cohort studies including up to 1,727 liver transplanted patients, cyclosporine-containing immunosuppressive regimens are associated with a lower risk of recurrence [123].
Patients with PBC should receive age-specific recommended vaccinations. HAV and HBV vaccines should be administered if the patient does not have immunity to them.
Patients should be counseled about smoking cessation, alcohol abstinence, illicit drug use, adequate calcium and vitamin D supplementation, exercise, fall prevention, and HCC surveillance. Bone mineral density testing is recommended every 2–3 years.
Social work referrals should be considered for those who need help with activities of daily living. Referral to a hepatologist for long-term management is highly recommended.
This comprehensive review article has focused on the importance of early diagnosis, the role of autoimmunity in disease pathogenesis, and recent management updates. Yet despite encouraging therapeutic advances, PBC remains a challenge for the field of hepatology given that its etiology remains unclear, and the long-term prognosis of new second-line therapies is uncertain and thus has had only conditional FDA approval. The progression from asymptomatic stages to decompensated liver disease mandates a thorough screening and tailored management for the individual patient. Recent advancements in the understanding of the disease’s pathogenesis have opened new avenues for targeted therapies and improved patient outcomes. Novel pharmacological agents, alongside established treatments with UDCA, offer new horizons in altering disease progression and are significant advances in enhancing PBC patients’ quality of life.
In summary, while challenges remain, the evolving landscape of PBC treatment and research should provide an optimistic look for better management and understanding of this complex autoimmune liver disease. Ongoing investment in clinical practice and research will be key to achieving significant progress in the fight for the cure of PBC.
AASLD: American Association for the Study of Liver Diseases
AIH: autoimmune hepatitis
AMA: antimitochondrial antibody
IgM: immunoglobulin M
L.T.: liver transplant
OCA: Obeticholic acid
PBC: primary biliary cholangitis
T.E.: transient elastography
UDCA: ursodeoxycholic acid
MBAE, IE, and AAAA: Investigation, Writing—original draft. JLB: Writing—review & editing.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

