 Review
Review

Affiliation:
1Psychosomatic Laboratory, GIFT Institute of Integrative Medicine, 56126 Pisa, Italy
2Aplysia APS, GIFT Institute of Integrative Medicine, 56126 Pisa, Italy
Email: ciarantogift@gmail.com
ORCID: https://orcid.org/0000-0001-8456-3430
Affiliation:
3Department of Medicine, Surgery and Neuroscience, University of Siena, 53100 Siena, Italy
ORCID: https://orcid.org/0000-0003-0868-418X
Explor Immunol. 2024;4:722–742 DOI: https://doi.org/10.37349/ei.2024.00168
Received: February 15, 2024 Accepted: October 08, 2024 Published: November 04, 2024
Academic Editor: Giustino Varrassi, Fondazione Paolo Procacci, Italy
The article belongs to the special issue Immunology and Pain
The narrative review aims to shed light on the influence of inflammation in the comorbid chronic pain and major depressive disorder (MDD). This connection is known to be multifactorial, with a dynamic interaction between genetic and epigenetic factors. However, a growing body of evidence has shown that the co-presence of MDD and pain is underlain by immune mechanisms involved in the persistence of the inflammatory process. In particular, the cytokines released following activation of the innate immune system during inflammation cause changes at the endocrine level that result in glucocorticoid resistance, as well as altering the synthesis and metabolism of some central nervous system (CNS) mediators. Cytokines appear to generate neuroinflammation by activating normally protective microglia. Various other mechanisms, including changes in the function of the glutamatergic, GABAergic, and serotonergic systems are also implicated, but inflammation-induced reduction of BDNF (brain-derived neurotrophic factor) appears to be the deciding factor. In turn, neuroinflammation leads to sickness behavior, which is characterized by anhedonia and social withdrawal. This review explored these mechanisms, which may be at the root of comorbid pain and MDD. Although intriguing, however, most available evidence comes from animal studies, and rigorous clinical exploration is warranted.
Chronic pain and major depressive disorder (MDD) are both widespread and prevalent, and often comorbid [1]. Several factors may be involved in this comorbidity, including inflammatory, genetic, epigenetic, and other mechanisms. This narrative review aimed to shed light on the role played by inflammation.
The immune system is a complex system of biological structures and processes that has evolved to protect us from disease through two lines of defence: innate immunity and adaptive immunity. Innate immunity is rapid and non-specific, and provides protection from viruses, bacteria, and other foreign invaders by means of physical barriers and leucocytes [2]. Adaptive immunity, on the other hand, is an actively acquired second line of defence, developing slowly and involving antibodies, which build up a memory of pathogens or foreign substances encountered [3]. There is a great deal of synergy between the adaptive immune system and its innate counterpart.
The primary function of innate immunity is the recruitment of immune cells to sites of infection and inflammation via cytokines. Cytokines are small proteins involved in cell-cell communication, and their production leads to the release of antibodies and other proteins and glycoproteins that activate the complement system. The cells involved in the innate immune response are phagocytes (macrophages and neutrophils), dendritic cells, mast cells, basophils, eosinophils, natural killer (NK) cells, and T-cell lymphocytes. There are two main types of phagocytes: neutrophils and macrophages. Both of these cell types share a similar function, specifically to engulf (phagocytose) microbes. They also contain granules that, when released, assist in the elimination of pathogenic microbes [4].
Following infection or physical injury, the immune system reacts through local and general innate immunity responses. The paladins of circulating immune surveillance are the macrophages, dendritic cells, and neutrophils; the activation of their receptors [toll-like receptors (TLRs)] by pathogens sets in motion an intracellular cascade, with the transcription of two main intracellular factors: nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and interferon (IFN) regulatory factors [2, 3, 5, 6]. These transcription factors enable the expression of pro-inflammatory response genes such as tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1), leading to the production of cytokines, the effectors of the inflammatory response [5, 7]. Cytokines coordinate responses among cells, and modify neuroendocrine and neurophysiological processes, with effects on physiology and behavior [8]. IL-1, IL-6, TNF-α are pro-inflammatory cytokines that activate chemokines. These are involved in immune surveillance, functioning to identify pathogens and attract other cells to the site of infection via chemoattraction [9]. In summary, cytokines have a local pro-inflammatory action, which determines the classical picture of heat, swelling, redness, pain, and impaired function. At a general level, some cytokines (e.g., IL-6), together with C-reactive protein (CRP), cause fever, increased heart rate and respiratory rate, and promote the social withdrawal behavior that both helps the organism recuperate through isolation at rest and reduces the risk of spreading the infection [6, 9].
Inflammatory activity is also regulated by brain processing, preparing the body for lesions in advance. This involves activation of the host defence response, an anticipatory (pre-injury) redistribution of immune cells from the innate immune system to sites of possible injury so as to promptly repair the damage and ensure survival [10, 11]. The pro-inflammatory reaction is therefore extremely useful when limited in time, and in acute situations, while it loses its homeostatic function when prolonged. In fact, there are various chronic pathologies (e.g., asthma, rheumatic pathologies, diabetes, etc.) sustained by a continuation of the pro-inflammatory phenomenon.
The innate immune system is also activated when an individual is exposed to adverse conditions like social isolation, conflicts, rejection, and exclusion [12]. In a sort of phylogenetic evolution, the genome contains our ancestral defence system, which is activated, in humans as in primates, by a social threat. This is functional acutely but stressful when chronic (when it is associated with overactivation of the pro-inflammatory system).
Two systems are involved in converting social/environmental adversity into pro-inflammatory transcription: the sympathetic nervous system (SNS) and the hypothalamic-pituitary-adrenal (HPA) axis [13]. The parasympathetic nervous system, on the other hand, seems to act as a modulator of the immune response at a regional level, preventing an excessive inflammatory response [14]. The SNS regulates the cytokine-mediated pro-inflammatory response (IL-1, TNF, and IL-6) in the primary and secondary lymphoid organs and perivascular and vascular structures by modulating the gene transcription response, linked mainly to β-adrenergic receptors [12, 15]. Under normal conditions, the HPA axis suppresses transcription, pro-inflammatory, and antiviral genes (e.g., via transcription regulator type I of the IFN gene) as it activates the adrenal gland to produce the most powerful anti-inflammatory (cortisol).
There are at least three mechanisms by which this suppression occurs: 1) interruption of pro-inflammatory gene expression by glucocorticoid (GC) receptor binding to gene promoter sequences; 2) inhibition of transcription of pro-inflammatory factor NF-κB by induction of anti-inflammatory gene transcription; and 3) antagonism of gene transcription through a protein-protein interaction between pro-inflammatory transcription factors [NF-κB, activator protein-1 (AP-1)] [13]. However, the suppressive function of the HPA axis transforms into an inflammation-increasing function when the two concomitant phenomena (suppressive and pro-inflammatory) are adaptive. For example, when a real and perceived danger persists, or under conditions of acute stress or social threat that coexist with physical danger [16, 17], resistance to GCs occurs as cells become less sensitive to their anti-inflammatory effects [18]. The fact that acute stress alters GC sensitivity has been demonstrated in both animal and human experimental studies [19], and HPA hyperactivity with altered GC sensitivity has been found in MDD; 46% of subjects with the former display a flattening of circadian variations in cortisol, explained by altered GC sensitivity [20] (Figure 1).
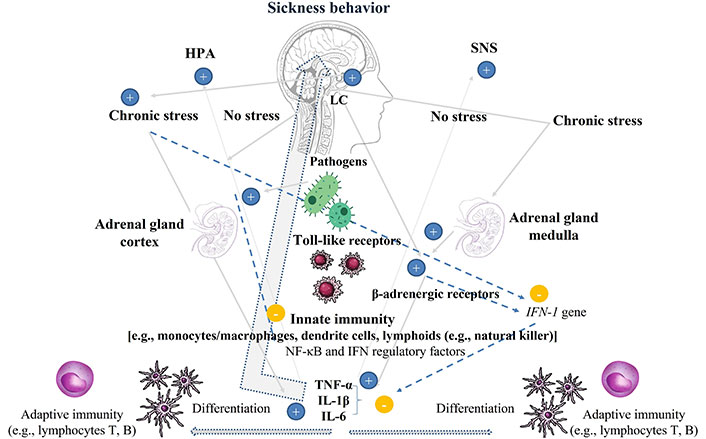
Endocrine and immune systems in physiological homeostasis, after infection, and in reaction to chronic stress. HPA: Under physiological conditions the HPA axis has anti-inflammatory properties, inhibiting the cytokines IL-1β, TNF-α, and IL-6. Under chronic stress, however, HPA hyperactivity is pro-inflammatory. SNS: SNS hyperactivity exerts pro-inflammatory actions via β-adrenergic receptors in lymphoid cells inhibiting the antiviral IFN type I (IFN-1). Pathogens: activate the innate immunity with production of cytokines. Cytokines (IL-1β, TNF-α, and IL-6) activate microglia and the HPA and SNS producing sickness behavior; cytokines stimulate the differentiation of dendritic cells presenting the antigen to adaptive immunity. +: promote; –: suppress. HPA: hypothalamic-pituitary-adrenal; IFN: interferon; IL-1β: interleukin-1β; LC: locus coeruleus; NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; SNS: sympathetic nervous system; TNF-α: tumor necrosis factor-α
Note. Partly resourced from Canva (https://www.canva.com/). © 2024 Canva Pty. Ltd.
In rats, an increase in plasma cortisol levels has been detected with the onset of muscular hyperalgesia and a prolongation in the production of the inflammatory factors that induce it (a form of nociceptor neuroplasticity termed hyperalgesic priming) [21, 22]. This mechanism, from a translational perspective, could be considered as the basis of the onset of chronic pain conditions like fibromyalgia and widespread pain [23]. Environmental stressors could impact the maturation of the nociceptive system, indicating that the onset and persistence of pain and the dysregulation of the HPA axis are strongly linked. For example, stressful conditions in the first days of life, even if not attributable to major trauma but to everyday situations such as neonatal stress from limited bedding, are associated with hyperalgesia [21, 24], owing to alterations in the HPA system. However, the evidence from experimental studies showed that this relationship is more certain in males than in females. In fact, only in male rats is hyperalgesia blocked by administering an antisense for GC receptors [21].
Childhood maltreatment, considered one of the most important environmental stressors, has been acknowledged to contribute to the development of MDD [25]. While in acute stress the HPA axis responds by first increasing and then reducing the production of cortisol, in recurrent or chronic stress cortisol levels remain high, overcoming its typical circadian rhythm. Persistent high cortisol levels under conditions of chronic stress lead to GC resistance, and the physiological ability to maintain the levels of GCs, catecholamines, and cytokines in a homeostatic balance (called allostasis) is lost; this allostatic overload may leave an individual unable to cope with the stressful event [26].
Resistance and vulnerability to stress depend on individual differences inducing different neurobiological and neuroimmune responses as a result of the ability to cope [27, 28].
HPA hyperactivity and consequent resistance to GCs appear to be implicated in the genesis of MDD, diabetes, and metabolic syndrome [29]. An increasing body of evidence suggests that stressors interact with the genome leading to stable changes in DNA structure, gene expression, and behavior. These epigenetic changes mainly concern GC signaling [e.g., nuclear receptor subfamily 3 group C member 1 (NR3C1), FK506-binding protein 5 (FKBP5)], serotonergic signaling [e.g., solute carrier family 6 member 4 (SLC6A4)], and neurotrophin (NT) genes [e.g., brain-derived neurotrophic factor (BDNF)] [25].
Pain has been defined as, ‘an unpleasant sensory and emotional experience associated with, or resembling that associated with, actual or potential tissue damage’ [30]. Pain is always a personal experience that is influenced to various degrees by biological, psychological, and social factors, whereas nociception is the neural process of encoding and processing noxious stimuli [30]. The nociceptor is a peripherally localized neuron preferentially sensitive to noxious stimuli, or to stimuli that would become noxious if prolonged, and capable of encoding stimulus intensities within the noxious range. It is highly modifiable in response to injury to its axon and on exposure to inflammation and can interact with the environment.
The soma of the nociceptors is small, resides in the dorsal root ganglion (DRG), and sends its myelinated (Aδ) and unmyelinated (C) fibres to both the periphery, where stimulus detection and impulse generation occur, and to the central nervous system (CNS). Even in the absence of peripheral stimulation, there is a basal activity in the nerve fibre that is greater than that in the dorsal root, where “tonic GABA (gamma-aminobutyric acid) filtering” occurs. The DRG cells express the major proteins necessary for GABA synthesis, and sensory neurons release GABA in response to depolarization. Depolarization of GABAergic DRG neurons produces a net inhibitory effect on the action potential transmission due to the filtering effect at nociceptive fibre T-junctions. In vivo, focal infusion of GABA or GABA reuptake inhibitor to sensory ganglia dramatically reduces acute peripherally induced nociception and alleviates neuropathic and inflammatory pain. On the other hand, focal application of GABA receptor antagonists to sensory ganglia triggers or exacerbates peripherally induced nociception [31].
Inflammatory cells, namely the mast cells and lymphocytes, in addition to activating the pain associated with peripheral nociceptors, also interact with the cell body, as they are located at the level of the DRG neurons. Since inflammatory cells modify protein synthesis by DRG neurons, in conditions of inflammation, their proliferation generates long-term sensitization and affects plasticity [32]. Hence, the onset and persistence of pain do not depend only on peripheral hyperalgesia, but also on central sensitization (responsible for chronic pain) due to both the continuous discharge of presynaptic neurons (those of the DRG) and post-synaptic neurons (interneurons).
After an injury or during chronic pain, nociceptor neurons express and release inflammatory mediators in their central nerves in the spinal cord, specifically cytokines, calcitonin gene-related peptide (CGRP), growth factors, adenosine triphosphate (ATP), and enzymes that activate the microglia. Being of myeloid lineage (like macrophages), the microglia produce TNF-α, IL-1β, and prostaglandin E2 (PGE2), but also generate NTs, including some isoforms of BDNF. Some NTs sensitize primary nociceptor neurons and second-order pain-mediating interneurons, thereby contributing to the production of central sensitization [32]. During inflammation, T cells also infiltrate the spinal cord. It seems that the microglia and T cell-induced central sensitization mechanisms are gender-specific, the microglia being more involved in male rats (BDNF modification-induced mechanic hyperalgesia), and the T cells more involved in females [33].
Pain is one of the cardinal signs of inflammation, as demonstrated by the effectiveness of non-steroidal anti-inflammatory drugs (NSAIDs) in reducing pain [34]. NSAIDs act by inhibiting cyclooxygenase, which is involved in the synthesis of PGE, in particular PGE2, which appears to be an activator of inflammatory pain [35]. PGE2 sensitizes the nociceptive neuron, not directly but through proximal ion channels, and also induces persistent hyperalgesia via PKA (protein kinase A) and PKC-mediated activation of NF-κB in DRG neurons [36]. During inflammation, the threshold for nociceptor activation is reduced, leading to pain sensitivity or “hyperalgesia”.
The aetiopathogenesis of pain in humans and animals also involves an interaction between the gut microbiome and the immune system, with associated behavioral modifications. The gut-associated lymphoid tissue (GALT) detects pathogens, and is tolerant to gut flora species, but not to other microorganisms. The intestinal microbiome contributes to maintaining homeostasis by defending the intestinal epithelium via the production of cytokines and antibodies [37]. The gut microbiota also plays an important role in the catabolism of dietary fibre, which is not completely hydrolyzed by the host enzymes during digestion.
The main products of gut bacterial fermentation of dietary fibre are short-chain fatty acids (SCFAs), including acetate, propionate, and butyrate, which can be used for de novo synthesis of lipids and glucose, the main sources of energy for the host [38]. SCFAs also play a vital role in intestinal physiology, as they help maintain epithelial cell integrity, immune cell function, and hormone secretion [39]. SCFAs are important factors in visceral sensation [40, 41], and butyrate has demonstrated analgesic properties in a model of acute visceral pain [42]. However, it may also elicit visceral hypersensitivity by increasing nerve growth factor (NGF) secretion by enteric glial cells [43].
In the gut lumen, human and bacterial proteases are both present at high concentrations and regulate intestinal barrier permeability. Proteases are enzymes that catalyze the degradation of proteins into smaller peptides or amino acids, which bind to protease-activated receptors (PARs), a family of GPCRs (G protein-coupled receptors) that includes PAR1, PAR2, PAR3, and PAR4. Activation of these receptors is believed to be linked to chronic inflammatory diseases and pain [44].
Inflammatory pain results from mast cell proteases activating transient receptor potential ankyrin 1 (TRPA1)-positive DRG neurons through binding to PAR2 [44]. Moreover, following infection or stress, mast cells induce colonocytes to release tryptase, a serine protease that cleaves and activates PARs to regulate tight junction assembly in the gut epithelium [45]. Inflammation, costimulation, and immune priming are induced, via activation of TLR4, by lipopolysaccharide (LPS) from gram-negative bacteria, which drives mechanical hypersensitivity. This suggests that the intestinal microbiome is involved in both inflammatory pain and neuropathic sensitization [46].
In the Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition (DSM-5) [47], MDD [which involves a single or recurrent major depressive episode (MDE)], is grouped with the depressive disorders, separate from bipolar disorders. Dysthymia, a milder but long-lasting form of depression, is now termed persistent depressive disorder (PDD). However, both PDD and MDD are characterized by one or more MDEs, of which those with melancholic and atypical characteristics are particularly relevant to differences in the immunity and HPA axis activity.
With the 5th edition of the DSM, both the melancholic and atypical forms of MDEs are included in the specifiers [47]. The prevalence of the atypical form is reported as being 18–36%, while the melancholic form is 35.2% [48]. The atypical form of depression is characterized by mood reactivity to environmental contexts, with hypersomnia, carbohydrate cravings, leaden paralysis, and strong sensitivity to rejection, which results in significant social or occupational impairment. In contrast, melancholia, described by Kraepelin, is characterized by severe anhedonia without mood reactivity, psychomotor disturbance, or neurovegetative symptoms (e.g., insomnia, loss of appetite, and weight) [49, 50]. Symptoms of melancholic depression peak in the morning, whereas patients suffering from atypical depression display a worsening of symptoms over the course of the day [51].
The relevant role of cortisol and HPA axis alteration in some psychiatric disorders, in particular in MDD with melancholic manifestations, was first described by Bleuler in [51] and then by Carroll [52]. The latter found that administering dexamethasone (1 mg p.o. at 11 p.m.), a powerful GC with a long half-life, to subjects with MDD with melancholic features resulted in a failure to suppress the secretion of the 11-hydroxycorticosteroids (e.g., cortisol). In other words, subjects with melancholic depression lack the normal inhibitory GC HPA feedback in the production of cortisol.
This evidence was so groundbreaking that the dexamethasone test was considered the first biological state-dependent marker; it is able to diagnose melancholic depression with a sensitivity of 67% and a specificity of 95% [53, 54]. In addition, in severe depression elevated cortisol levels have been found in the serum, saliva, and cerebrospinal fluid, alongside raised corticotropin-releasing factor (CRF) levels [53]. The frequent association with Cushing’s syndrome, and vice versa, has led to a differentiation between exogenous from endogenous depression, which is characterized by hypercortisolaemia [55]. In the latter, an increase in basal cortisol has been demonstrated during the day, especially between 11:30 p.m. and 12 a.m. [55, 56].
Although several studies have shown that a positive dexamethasone suppression test (DexST) (≥ 3.5 μg/dL threshold) is an indicator of severe depression with recurrent episodes and a greater prevalence of suicidal behavior [56, 57], some other studies appear to have invalidated Carroll’s results, reporting a specificity of the non-DexST test of 15% in non-psychiatric control subjects, and 37% in other psychiatric disorders [58]. Furthermore, as previously mentioned, depression can follow chronic stress (exogenous factor), in which hypercortisolaemia may occur [59], and the distinction between exogenous and endogenous is therefore questionable. However, the differentiation between MDD with melancholic features and atypical depression is borne out by their different responses to psychopharmacological treatments [60]. In addition, it is possible that maladaptive sickness behavior and associated circulating inflammatory markers, may not play the same key role in each subtype of MDD.
Atypical depression is 2–3 times more frequent in women, generally has a chronic course, and arises earlier than melancholic depression. Its early onset and greater interpersonal sensitivity lead to the hypothesis of a personality origin characterized by emotional dysregulation and self-consolation (e.g., hyperphagia, hypersomnia) [60]. The HPA activity of atypical depression is antithetical to the melancholic form; cortisol levels are low, and there is downregulation of the HPA axis [60]. That the two clinical forms are diametrically opposed is also demonstrated by the fact that in melancholic depression the symptoms worsen in the morning, when there are higher levels of cortisol, while in the atypical form, the symptoms worsen in the afternoon, when the levels of cortisol are lower [61].
An insufficient stimulation of the adrenal gland is confirmed by the low CRF values in the blood and in the cerebrospinal fluid [62]. Both the medial prefrontal cortex (MPFC) and the anterior cingulate cortex (ACC) have numerous GC receptors, and in turn, appear to influence the modulation of the HPA axis under stress. In particular, it seems that the dorsal ACC (dACC) inhibits HPA activity under stress, while the ventral infralimbic region seems to stimulate it [62]. In a sample of women, endotoxin administration appeared to bring about an increase in IL-6 and dACC hyperactivity [63], associated with an increase in depressed mood and social isolation [62]. Moreover, basal hypocortisolism and reduced HPA activity have been found in post-traumatic stress disorder (PTSD) and other stress-related syndromes such as somatic symptom disorder [60, 64], especially in those who have been directly exposed to trauma and who have more severe PTSD symptoms [65]. Another distinguishing feature of the atypical form of depression is that it is frequently found in bipolar II disorder [66], and in addition to being associated with psychiatric syndromes, it has also been described in chronic pain [67] (Table 1).
Stress and inflammation in depressive episode subtypes and pain
| Features | Melancholic episode | Atypical episode | Pain |
|---|---|---|---|
| Mood | Flat, anhedonia | Reactive | - |
| Worsening of mood | Morning | Evening | - |
| Insomnia | Early awakening | Hypersomnia | Insomnia |
| Eating behavior | Hyporexia | Craving for carbohydrates | - |
| Activity | Slowdown | Leaden paralysis | Fatigue |
| Innate immunity | ↑ | ↑ | ↑ |
| Adaptive immunity | ↑ | ↑ | ↑ |
| TNF-α | ↑ | ↑ | ↑ |
| IL-1 | ↑ | ↑ | ↑ |
| IL-6 | ↑ | - | ↑ |
| CRP | ↑ | - | ↑ |
| BBB | Decreased | - | - |
| DexST | Suppressed | Normal | Normal |
| SNS | ↑ | - | ↑ |
| CRF | ↑ | ↓ | - |
| ACTH | ↑ | ↓ | Normal stress stimulus response |
| Cortisol | ↑ at basal level | Reduced production | Blunted stress stimulus response |
| KYN | ↑ | - | ↑ |
| 5HT | ↓ | ↓ | ↓ |
| GABA | ↓ | - | ↓ |
| BDNF | ↓ | ↓ | ↓ |
↑: hyperactivity or increased level; ↓: hypoactivity or decreased level; -: no date. TNF-α: tumor necrosis factor-α; IL-1: interleukin-1; CRP: C-reactive protein; BBB: blood-brain barrier; DexST: dexamethasone suppression test; SNS: sympathetic nervous system; CRF: corticotropin-releasing factor; ACTH: adrenocorticotropic hormone; KYN: kynurenine; 5HT: serotonin; GABA: gamma-aminobutyric acid; BDNF: brain-derived neurotrophic factor
Microglia are the most important exponents of the immune system in the CNS, sharing some functions with macrophages. Under normal conditions, microglia are involved in the modulation of synapses (e.g., synapse pruning and neurogenesis) [68, 69]. Nevertheless, activation of the microglia is the most important substrate of the central neuroinflammatory process, determining sickness behavior (a set of adaptive behavioral changes that develop in ill individuals during the course of an infection) in MDD [62, 70]. Indeed, microglia, together with astrocytes, contain enzymes responsible for serotonin (5HT) metabolism, such as IDO (indoleamine 2,3-dioxygenase) and TDO (tryptophan 2,3-dioxygenase). It has been seen that, while physiologically the microglia express more TDO, during inflammation the greatest expression pertains to IDO, which converts tryptophan into kynurenine (KYN), reducing the substrate for the synthesis of 5HT [62].
Microglia are also involved in the synthesis of dopamine, and responsible for creating a glutamatergic imbalance when activated by a peripheral increase in KYN. This induces the activation of IDO, transforming KYN into quinolinic acid (QUIN), an agonist of the N-methyl-D-aspartate (NMDA) receptor (QUIN) [62]. QUIN can trigger glutamate release and is capable of inhibiting glutamate uptake in glial cells, causing a higher concentration of extrasynaptic glutamate overall and changing network connectivity [62].
The increase in glutamate in the dACC, nucleus accumbens, and MPFC seems to underlie the relationship between anhedonia and motor slowing and the increased activity of microglia, astrocytes, circulating CRP, and IFN-α [62]. The increased extrasynaptic (rather than intersynaptic) NMDA receptor activity is attributed to a reduction in the synthesis of BDNF, essential for cell survival and synaptogenesis [62].
BDNF is a member of the NT family, which includes NGF, NT3, and NT4. BDNF is involved in the development, morphology, synaptic plasticity, and function of the brain [71]. It is present in the periphery, being accumulated in platelets and in the CNS, neurons, astrocytes, and microglia. Brain BDNF concentrations are linked to synaptic plasticity, neuronal atrophy, and neuronal survival and differentiation. Highly represented in the hippocampus, BDNF is found in smaller concentrations in the cerebral cortex and brainstem.
Several factors can modulate BDNF expression or function. Early life stress, physical exercise, and environmental factors can modulate the function of BDNF and the effect of inflammation on the level of BDNF [72]. A decline in BDNF levels has been found with age and in age-related neurodegenerative diseases like Alzheimer’s [72]. BDNF is also considered a crucial biomarker for the pathogenesis of depression, being used to distinguish between healthy individuals and those with MDD. A number of studies suggested that individuals carrying the val66met polymorphism of BDNF are more vulnerable to developing depression, suicidality, and resistance to antidepressant treatment [72].
Studies on mice have shown that a reduction in BDNF levels is correlated with increased inflammatory factor levels and dysfunction in the BDNF-TrkB (tropomyosin-related kinase B) system that underlies sickness behavior [73]. A growing body of evidence demonstrated a relevant involvement of BDNF in the action of antidepressants. Increased expression of BDNF mRNA in the prefrontal cortex and hippocampus is seen during the response to electroconvulsive therapy (ECT), tricyclics, and SSRI (selective 5HT reuptake inhibitor) treatment [74]. In rodents, the infusion of low doses of BDNF leads to antidepressant-like behavior after 10 days, sustaining a plasticity mechanism that mediates long-term antidepressant effects [75].
While delivering BDNF to the brain seems to be a promising treatment, it poses challenges due to the limited ability of large protein molecules to cross the blood-brain barrier (BBB). BDNF is also prone to oxidative damage in vivo. This ready degradation of BDNF can, however, be overcome by the use of molecules such as BDNF carriers or dietary intake of antioxidants. Zinc supplements in particular have antioxidant properties. Zinc forms part of the enzyme superoxide dismutase (SOD), and in obese subjects increases BDNF levels and reduces depressive symptoms [72]. Furthermore, quercetin, a natural antioxidant, has been suggested to protect BDNF from oxidation. Indeed, recent studies highlight BDNF protein delivery via quercetin-alginate nanocarriers as a promising antidepressant treatment [73]. The antidepressant action of quercetin nanogels appears to be exerted not only on BDNF propagation but also on the inhibition of TNF-α and IL-6 mRNA expression in RAW 264.7 macrophage cells [73]. BDNF also seems to be involved in the action of another new treatment for resistant depression, namely ketamine (a non-competitive NMDA-receptor antagonist). Ketamine acts rapidly and is associated with both an increase in hippocampal BDNF production and cerebral plasticity. In fact, the administration of low-dose ketamine results in a calcium channel blockade. The consequent reduction of intracellular calcium entry leads to inhibition of spontaneous cellular hyperactivity (in the absence of depolarization) [76].
Central neuroinflammation appears to be mediated by both microglia and macrophages, the latter generally located at the interface between the brain parenchyma and blood vessels. Under physiological conditions, the BBB prevents access to immune cells and other substances circulating in the blood (e.g., pathogens—a protection greater than 98%). But under certain circumstances, membrane permeability is established, granting entry to circulating immune cells such as T cells. These, together with chemokines, lead to greater intercellular adhesion and parenchymal infiltration, and may be responsible for the autoimmune processes underlying neurodegenerative and neuropsychiatric disorders such as MDD [70]. In fact, increased BBB permeability has been associated with suicidal risk in psychiatric disorders [77]. A reduction in astrocyte and oligodendrocyte numbers is also found in MDD, and this seems to be responsible for greater BBB permeability, allowing monocytes to infiltrate the brain parenchyma [78].
Cytokines, like immune cells, reach the CNS via three pathways: 1) through weaker regions (whether incomplete, as in circumventricular sites, or permeable, as in the organum vasculosum of the lamina terminalis) or disease-induced disruptions of the BBB; 2) through a neural pathway via cytokine receptors on afferent nerve fibres that transmit the signal to the brain parenchyma; and 3) through infiltration of the meninges and brain parenchyma by immune cells, attracted by a chemokine [69].
IL-6 represents the ‘state indicator for acute exacerbation’ in melancholic patients [79, 80], while CRP and TNF have been associated with MDD severity. Mild-moderate inflammation has also been associated with treatment-resistant depression and poor response to antidepressant treatments [69]. The cytokines produced by the microglia (but apparently also by astrocytes) directly promote the production of norepinephrine, 5HT, and dopamine, being directly involved in processes that modify behavior. In fact, by altering the sleep-wake cycle, they can cause a reduction in activity and interest in socialization, care, mating, and pleasurable activities during the daytime. Such behavior is defined as sickness behavior, whose symptoms and behaviors are comparable to a depressive episode [79].
Considering that stress can lead to MDD and that viral infection is a stressful condition that induces sickness behavior, with symptoms typical of a depressive episode, it follows that a viral pandemic, as seen with coronavirus-19 (COVID-19), could be followed by a higher incidence of MDD. Indeed, a 2020 USA study comparing a COVID-19 sample of 1,441 participants (43.0% aged 18–39 years; 50.2% male; 64.7% non-Hispanic White) with a pre-COVID-19 sample of 5,065 participants (37.8% aged 18–39 years; 48.6% male; 62.9% non-Hispanic White) showed a higher prevalence of depressive symptoms in every category of the former. A greater risk of depressive symptoms (investigated using the Patient Health Questionnaire-9) during COVID-19 was associated with having lower income (odds ratio, 2.37 [95% CI, 1.26–4.43]), less than $5,000 in savings (odds ratio, 1.52 [95% CI, 1.02–2.26]), and exposure to more stressors (odds ratio, 3.05 [95% CI, 1.95–4.77]) [81].
That being said, self-assessment questionnaires do not allow us to diagnose depressive episodes with certainty. In fact, symptoms related to the infection (asthenia, slowness, etc.) could be confused with depressive symptoms [82]. The aetiopathogenesis of MDD is multifactorial and dependent on internal and external or contextual factors, but an Italian study investigating the biopsychosocial perspective of impact of COVID-19 on mental health (MH) indicated that the restrictions and isolation related to the COVID pandemic worsened MH, especially in young people. Independently of age, that study showed that the worse MH is a consequence of lower positive self-appraisal of general health, and an increased perceived interference of the pandemic with life and life satisfaction in subjects forced into isolation by the COVID-19 lockdown [83].
Another Italian study, using brain imaging, investigated 42 COVID-19 survivors without brain lesions during the infection. About 3 months after infection, they displayed alterations in the structure of the grey matter (GM) in the prefrontal cortex, ACC, and insular cortex. These brain modifications were inversely correlated with the score on the Zung scale for depressive symptom severity. Worse self-rated depression was associated with increased systemic immune-inflammation index (SII), which reflects the immune response and systemic inflammation based on peripheral lymphocyte, neutrophil, and platelet counts, and inversely correlated with GM volumes in the ACC and insula [84]. These findings indicated that these structures may be implicated in the role immunity plays in the onset of depression.
As with the chicken and the egg debate, the question of whether pain causes depression or vice versa will likely never have a satisfactory answer. What we do know is that 65% of patients with depression report pain and 61% of pain patients have depression [85], with the outcome and management of each being worsened in the presence of the other. Antidepressants and mood stabilizers are effective in both [86, 87], and chronic pain has been proposed to be a variant or somatization of major depression [88]. Although several neuroimaging studies have shown that depression and pain are sustained by the same neurophysiological systems, very few have properly measured patients in the grip of a depressive episode and (chronic) pain. Nonetheless, in an activation likelihood estimation (ALE) brain imaging meta-analysis, it has been reported that an alteration in MPFC-amygdala connectivity occurs in subjects with pain and concomitant depression [60]. In contrast, subjects with depression and concomitant pain show a reduction in the activity of the dorsal prefrontal cortex (DPFC), responsible for both cognitive reappraisal and emotion regulation, underlying the increased intensity of painful stimuli [85].
In addition, one study found that around 60% of subjects with chronic pain also had a mood spectrum disorder over the course of their lifetime, with 61.41% of those cases being unipolar depression (without lifetime manic or hypomanic episodes and without hyperthymic and cyclothymic temperaments) [88, 89]. Pain intensity and cognitive pain component scores are higher in unipolar depression than in bipolar disorder, even though patients with the latter have more suicidal ideation, more melancholic depressive episodes, and a longer history of psychiatric disorder before pain onset than those with the former [89]. Moreover, a cross-sectional study comparing 269 subjects with melancholic depression versus 144 with atypical depression and 401 controls revealed greater musculoskeletal pain in subjects with melancholic as opposed to atypical depression [90]. What is more, an observational cohort study, albeit with a small sample (total n = 76, of which 40 atypical and 27 melancholic), found a greater association between fibromyalgia and atypical, as compared to melancholic, depressive episodes [67]. Some studies consider fibromyalgia to be a functional somatic syndrome with an increased prevalence of trauma [91, 92], and a diathesis-stress model has been suggested to underlie its pathogenesis [93]. This body of evidence could explain a hypoactivity of the HPA system in fibromyalgia as we find in atypical depression and PTSD. On the contrary, there are studies on the fibromyalgic syndrome (FMS) concomitant with depression that showed a higher blood cortisol levels [94]. It has therefore been suggested that, rather than measuring blood cortisol alone, therefore, it would seem to be necessary to explore the reactivity of the HPA system to stressful stimuli [95]. Indeed, the presence of circulating pro-inflammatory cytokines appears to generate an interaction between the HPA system and 5HT levels. There is extensive literature on the aetiopathogenic role of 5HT (5-hydroxytryptamine) deficiency in the onset of pain [95]. In particular, high levels of the 5HT metabolite 5-hydroxyindoleacetic acid (5-HIAA) in the urine have been noted in patients with migraine [96], which appears to confirm a link between 5HT and pain.
To explore this connection, recent studies have investigated the role played by pro-inflammatory factors in 5HT metabolism [97], in particular, the amino acid tryptophan [2-Amino-3-(lH-indol-3-yl)-propanoic acid (TRP)], a precursor of the neurotransmitter 5HT, which performs various brain functions. The brain concentration of 5HT increases when TRP is administered, whether orally, intraperitoneally, or subcutaneously, to adult animals [98]. In addition, chronic low-grade inflammation has been shown to activate the metabolism of TRP [99], whose catabolism is mainly expressed (95%) as the transformation of TRP into KYN [100]. The metabolite of the KYN pathway (KP) of TRP catabolism is variously active, and its rate-limiting step has been identified [98]; the first metabolite of tryptophan is N-formyl-L-KYN, and this reaction is limited by the enzymes IDO-1, IDO-2, and TDO [99]. KYN, the central metabolite of TRP, derives from the transformation of N-formyl-L-KYN thanks to the activity of the enzyme KYN formamidase [100]. Other catabolites of KYN have been found to include kynurenic acid (KynA) [irreversible transamination via KYN aminotransferase (KAT) isoenzymes] [101].
If, on the one hand, the pro-inflammatory cytokines IL-1β, IL-2, IL-6, IFN-γ, and TNF-α activate the enzymes IDO-1 and IDO-2 (generally expressed in non-hepatic tissues) [102], on the other hand, during inflammation the cortisol production by the adrenal gland activates TDO [103]. During inflammation, therefore, there is a shift in the tryptophan pathway leading to its degradation, rather than making it accessible for the synthesis of 5HT. Consequently, a surge in the production of KYN lowers the availability of TRP for 5HT synthesis [104]. This alteration in the TP negatively impacts the TRP-5HT pathway and contributes to the development of pain and depression [105]. A large meta-analysis has highlighted that the shift of TRP from 5HT to KYN is implicated in several psychiatric pathologies, including MDD, bipolar disorder, and schizophrenia [106]. Furthermore, alterations in the TRP-5HT pathway have been found in both chronic pain [107] and comorbid chronic pain and depression [108, 109].
5HT, in addition to carrying out its action as a neurotransmitter, also has hormonal properties. The synthesis of 5HT occurs through hydroxylation (via tryptophan hydroxylase) of TRP, and subsequently through decarboxylation (5-hydroxytryptophan decarboxylase). TRP hydroxylase has two isomeric forms, one of which is found in the brain and the other in the intestine. 5HT is also a precursor of melatonin, and plays a role in the regulation of sleep, the circadian rhythm, and the immune system, but also modulates the HPA axis [24]. The shift of TRP from 5HT synthesis to the KYN production pathway does not appear to have only negative effects on pain, but could also play a protective role in the production of KynA. KynA appears to have affinity for three ionophore receptors: the kainic acid receptor, the strychnine-insensitive glycine-binding site of the NMDA receptor, and the α-amino-2,3-dihydro-5-methyl-3-oxo-isoxazolpropionic acid (AMPA) receptor [110]. In micromolar concentrations, KynA inhibits the AMPA and NMDA receptors, which are responsible for the genesis of pain and depression [111, 112], while this does not occur at nanomolar dosages [113–115].
In the presence of inflammation, on the other hand, the increased catabolism of TRP produces QUIN, which increases glutamate levels with hyperactivity of some areas such as the ACC involved in the genesis of anhedonia but also in the affective dimension of pain [116]. Many limitations in the search for new treatments for comorbid pain and depression arise from the fact that the evidence comes from animal research, while few human studies have been conducted. Nevertheless, trials of the new generation of drugs are yielding promising results, demonstrating that they can be of great help in controlling the pain-depression association. An example is the steroid allopregnanolone, which has been approved for the treatment of postpartum depression. During pregnancy, this neuroactive hormone performs its function on the GABAAR complex (ionotropic GABAA receptor), and the drop in its levels at the end of pregnancy appears to be responsible for the onset of postnatal depression [116]. As previously mentioned, GABA plays an important role in the modulation of both inflammation and pain [117]. Furthermore, an enhancement of GABA also appears to follow 2-day administration of ketamine. The action of the latter seems to occur by decreasing DMN (default mode network) connectivity and reducing the reactivity of amygdala-hippocampal circuity in response to emotional stimuli [116]. It is known that both the amygdala and the hippocampus are rich in GC receptors, and that the amygdala, in turn, has an excitatory influence on the HPA axis [118].
The GABA inhibitory filter has been implicated in depressive disorder, with a reduction in GABA values being found in the cerebrospinal fluid. MR spectroscopy has also revealed a reduction in GABA levels in some brain areas in subjects with MDD, with normalization during the remission of symptoms [115]. An anti-inflammatory action has also been suggested for GABA, as it prevents the release of IL-6 by inhibiting p38 mitogen-activated protein kinase (MAPK) in rat glioma cells [119]. The spinal production of p38 MAPK depends on the presence of TNF-α in the spinal cord, and in turn induces efferent signals that directly mediate the inflammatory response, increasing the level of IL-1, IL-6, and matrix metalloproteinase 3 (MMP3) [119]. In the rheumatoid arthritis (RA) model, it seems that GABA downregulates p38 MAPK activity, reducing the production of pro-inflammatory cytokines in joints affected by RA [119].
The role played by 5HT deficit, alterations in BDNF and cytokine levels due to inflammation could therefore constitute the target for new neuromodulatory treatments for depression or other psychiatric disorders. In fact, a recent study showed that the percentage of change in the score of negative symptoms of the PANSS (the positive and negative syndrome scale) correlated with the level of some cytokines (IL-27 and IL-6) at baseline. The PANSS change was obtained after 2 months of treatment with second-generation antipsychotic olanzapine (with properties on 5HT1A and 5HT2A receptors) in subjects with first-episode psychosis [120].
The contribution of immunity to comorbid pain and MDD is expressed through different mechanisms. First and foremost, it should be noted that the cascade of inflammatory factors activated after injury, infection, or disease may lead to both pain and depressive symptoms. The proinflammatory reaction is extremely useful in the short term but if prolonged it delays the anti-inflammatory activity. The cytokines IL-6, IL-1β, and TNF-α and PCR released during inflammation are responsible for the onset of sickness behavior, a depressive-like behavior. This occurs because fever increases heart and respiratory rates, generating a tendency towards social withdrawal, designed to counter the spread of infection, and because some cytokine receptors are directly involved in certain symptoms of depression (e.g., IL-1β in anhedonia). Furthermore, it seems that IL-1β plays an important role in the alterations in the sleep-wake cycle and in the cognitive and memory processes seen in MDD. In chronic conditions, neuroinflammation is also associated with an increase in nociceptive neuron sensitization due to release of ATP and CGRP, but also TNF-α, IL-1β, and PGE2 resulting from the activation of the microglia. Nociceptor sensitization is the basis of both inflammatory and neuropathic pain.
Another mechanism underlying comorbid pain and MDD is an alteration in the tonic regulation mechanisms of the GABA system. As reported above, administration of GABA or a GABA reuptake inhibitor reduces neuropathic and inflammatory pain due to the action on GABAergic DRG cells, which convey pain. Therefore, the loss of the inhibitory filter action of GABA may underlie both pain and depression, in which common aetiopathogenic mechanisms due to inflammation can be included.
Glutamate and NMDA receptor activity could also underpin comorbid pain and MDD. Ketamine (an NMDA antagonist) is an anaesthetic used especially for neuropathic pain, and recent studies have shown it has antidepressant action in drug-resistant MDD. The dysfunction of this metabotropic receptor seems to be triggered by inflammatory reduction of BDNF and its the action on glutamate activity. The inflammatory activation of the IDO enzyme leads to production of QUIN, a toxic metabolite for the brain that increases glutamate release and reduces glutamate uptake from the glia. This leads to a rise in extracellular glutamate, contributing to neuroinflammation due to increased microglial activity. The increase in extrasynaptic NMDA receptor activity is associated with a decrease in the synthesis of BDNF, which is essential for synaptogenesis. Glutamatergic hyperactivity generates neuronal sensitization, which leads to both pain and the changes in brain connectivity underlying MDD.
Early stress and the inability to cope with it also appear to influence the association between MDD and pain, which have been linked with overproduction of pro-inflammatory factors and an alteration in HPA axis activation. An example is the worse MH seen in young people after the COVID infection. The age of stress exposure can influence the type of depressive episode that can arise. In fact, we have seen that the atypical form arises earlier than the melancholic form, and the former is more frequently associated with nociplastic forms of pain (arising through altered nociception), as in FMS. On the other hand, an age-related decline in BDNF levels could be responsible for MDD in the elderly. This often has melancholic features and is frequently comorbid with chronic pain.
Furthermore, chronic alteration of the HPA axis activity affects the centres that regulate both pain and MDD. ACC hyperactivity, which appears to be involved in both the genesis of anhedonia and the affective dimension of pain, also warrants further investigation. Higher levels of IL-6 and dACC hyperactivity occur upon administration of endotoxin leads, accompanied by an increase in depressed mood and social isolation.
Pain and depression may also result from a disruption in the intestinal commensal flora upon invasion by a pathogenic microorganism, leading to an imbalance in both the innate and adaptive immune systems. These can more easily produce neuroinflammation due to the fact that the pathogen itself is able to modify the permeability of the BBB to pro-inflammatory cytokines, responsible for the hyperactivation of microglia with the onset of sickness behavior.
Another mechanism that may underlie the relationship between pain and mood depression is the role of neuroinflammation in altering 5HT metabolism. Deficits in 5HT levels are at the basis of the aetiopathogenetic mechanisms behind the onset of both pain and MDD. In the process of 5HT synthesis by tryptophan, increased microglial expression of IDO has been found during inflammation. This limiting enzyme converts tryptophan into KYN, reducing the substrate for the synthesis of 5HT. It is possible to envisage the immunomodulatory properties of some psychiatric treatments, such as second-generation antipsychotics and serotonergics due to their ability to regulate the levels of BDNF, 5HT, and cytokines.
Last, but by no means least, is the important role played by the HPA axis and sympatho-adrenal medullary (SAM) dysfunction in the inflammatory aetiopathogenesis of both pain and MDD. While under normal conditions the SAM regulates the pro-inflammatory cytokine response and the HPA suppresses transcription, pro-inflammatory, and antiviral genes, in chronic conditions, there is a transformation into an inflammation-increasing function with progressive GC receptor insensitivity. Although HPA activity seems to differ in different clinical forms of depression (increased in the melancholic form and downregulated in the atypical form), the rigidity of the axis with GC receptor insensitivity is the common denominator. Downregulation of the HPA axis is found in stress-related psychiatric disorders and in the atypical form of depression, but has also been found in chronic pain. Interestingly, the dysregulation of the HPA axis appears to be gender dependent, with evidence being more reliable in males than in females.
The wide variability among clinical presentations of MDD and chronic pain is often responsible for contradictory findings, especially in human studies. Hence, even today it is not possible to draw certain conclusions, especially as almost all the evidence is from preclinical studies. Although from a translational perspective, it is possible to transfer the evidence from animal studies into the clinical context, clinical trials are necessary to confirm the results obtained from experimental studies.
This narrative review elucidates how the co-presence of MDD and pain is underlain by immune mechanisms involved in the persistence of the inflammatory process. Several target mechanisms are involved in this process, which to varying degrees can contribute to the association of the two syndromes, but which can be modulated by various factors, among which age, sex, polymorphisms, environment, diet, physical activity, and others.
At the circulatory level, the activation of the innate immune system during inflammation generates the release of cytokines. The cytokines released cause changes at the endocrine level by activating the sympathetic system and the HPA axis, resulting in GC resistance. Cytokines appear to generate neuroinflammation by modifying the activity of microglia (from protective to inflammatory activity) and increasing the permeability of the BBB. Cytokines also affect microglial function, leading to alterations in the synthesis and metabolism of some CNS mediators such as 5HT and GABA, which are involved in the aetiopathogenesis of both pain and MDD.
BDNF appears to be fundamental in brain plasticity due to its role as a myelination and apoptotic factor. Deficits in the synthesis and function of BDNF during inflammation may also have a crucial role, being followed by changes in brain connectivity due to glutamatergic hyperactivity, which underpins the onset of MDD and pain. BDNF activity is labile and subject to oxidative stress. The BDNF-TrkB dysfunction underlies the sickness behavior that is the clinical/behavioral manifestation of central neuroinflammation, although these findings are more certain in male mice. In fact, most studies in this area to date have been conducted on animals, and there is no definitive confirmation. The evidence on the co-presence of pain and MDD requires rigorous clinical exploration.
5HT: serotonin
ACC: anterior cingulate cortex
BBB: blood-brain barrier
BDNF: brain-derived neurotrophic factor
CNS: central nervous system
COVID-19: coronavirus-19
CRP: C-reactive protein
dACC: dorsal anterior cingulate cortex
DRG: dorsal root ganglion
GABA: gamma-aminobutyric acid
GC: glucocorticoid
HPA: hypothalamic-pituitary-adrenal
IDO: indoleamine 2,3-dioxygenase
IFN: interferon
IL-1: interleukin-1
KYN: kynurenine
KynA: kynurenic acid
MAPK: mitogen-activated protein kinase
MDD: major depressive disorder
MDE: major depressive episode
MH: mental health
MPFC: medial prefrontal cortex
NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells
NMDA: N-methyl-D-aspartate
NT: neurotrophin
PARs: protease-activated receptors
PGE2: prostaglandin E2
PTSD: post-traumatic stress disorder
QUIN: quinolinic acid
SCFAs: short-chain fatty acids
TDO: tryptophan 2,3-dioxygenase
TNF-α: tumor necrosis factor-α
TRP: 2-Amino-3-(lH-indol-3-yl)-propanoic acid
AC: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. GC: Writing—original draft, Validation, Writing—review & editing, Supervision. All authors read and approved the submitted version.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

View: 7102
Download: 34
Times Cited: 0
Rucha A. Kelkar ... Giustino Varrassi
Marco Cascella ... The TRIAL Group
Mariateresa Giglio ... Filomena Puntillo
