 Review
Review

Affiliation:
1Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
2Federal State Autonomous Educational Institution of Higher Education I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University), 119048 Moscow, Russia
ORCID: https://orcid.org/0000-0001-6767-2571
Affiliation:
1Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
ORCID: https://orcid.org/0000-0003-3521-5995
Affiliation:
1Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
ORCID: https://orcid.org/0000-0001-8619-9687
Affiliation:
1Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
ORCID: https://orcid.org/0000-0003-2679-231X
Affiliation:
1Institute of Fundamental Medicine and Biology, Kazan Federal University, 420008 Kazan, Russia
2Federal State Autonomous Educational Institution of Higher Education I.M. Sechenov First Moscow State Medical University of the Ministry of Health of the Russian Federation (Sechenov University), 119048 Moscow, Russia
3Shemyakin-Ovchinnikov Institute of Bioorganic Chemistry, Russian Academy of Sciences, 117997 Moscow, Russia
Email: chembio.kazan@gmail.com
ORCID: https://orcid.org/0000-0003-2961-0032
Explor Immunol. 2024;4:837–852 DOI: https://doi.org/10.37349/ei.2024.00176
Received: July 26, 2024 Accepted: November 22, 2024 Published: December 15, 2024
Academic Editor: Cunte Chen, South China University of Technology, China
The efficacy of chimeric antigen receptor (CAR)-T therapy may not match initial expectations due to the influence of multiple circumstances, some of which cannot be predicted. CAR-T treatment groups include high-risk patients, particularly those with TP53 mutations. A significant body of research has demonstrated that mutations in the TP53 gene play a pivotal role in cancer development and progression. Any aberration in the TP53 gene in cancer is invariably associated with complications and a poor prognosis. Moreover, mutations in the TP53 gene have been observed to correlate with resistance to conventional chemotherapy, prompting the use of alternative therapeutic approaches, including CAR-T therapy. However, there is a possibility that abnormalities in the TP53 gene may affect patients after CAR-T cell administration reducing the efficacy of therapy. This review examines the link between TP53 mutations in cancer and the efficacy of CAR-T therapy, as well as the potential implications of this aspect in therapeutic planning.
A significant advancement in the treatment of recurrent/refractory (R/R) hematologic malignancies has been the introduction of chimeric antigen receptor (CAR)-T cell therapy. CAR-T therapy has demonstrated remarkable efficacy for this type of cancer, which is often less responsive to alternative therapeutic approaches. The safety and efficacy of CAR-T therapy have been demonstrated to be superior to conventional chemotherapy. Moreover, CAR-T cells, which are classified as advanced therapy medicinal products (ATMPs), have the potential to be approved as orphan pharmaceuticals. At the present time, CAR-T cells have already successfully completed the preclinical trial stage and are currently being used in clinical settings to treat a number of hematologic and rare diseases for which they have been specifically approved (Table 1) [1]. However, their efficacy in treating the majority of solid tumors remains uncertain, and their wide utilization is significantly limited. There are several challenges associated with the infiltration of CAR-T cells into solid tumors and their subsequent activation within these complex environments. The immunosuppressive nature of the tumor microenvironment (TME) further complicates the use of CAR-T cells in the treatment of malignant neoplasms. The selection of antigens represents a pivotal and intricate process in ensuring the optimal functioning of CAR-T cells. This is due to the fact that tumor cells possess the capacity to downregulate antigens, a phenomenon known as antigen escape, through the application of CAR-T cell selection pressure [2].
Approved CAR-T cell therapies
| Trade name (generic name) | Target | Indication | Approval year | Company | Clinical study |
|---|---|---|---|---|---|
| Kymriah (tisagenlecleucel) | CD19 | Patients under the age of 25 with B-cell ALL that is refractory or in second or subsequent relapse | 2017 | Novartis | ELIANA trial [3] |
| Adults with R/R LBCL after two or more lines of systemic therapy (including DLBCL-NOS, HGBCL, and DLBCL arising from FL) | 2018 | JULIET trial [4] | |||
| Adults with R/R FL after two or more lines of systemic therapy | 2022 | ELARA trial [5] | |||
| Yescarta (axicabtagene ciloleucel) | CD19 | Adults with R/R LBCL (including PMBCL, HGBCL, and DLBCL arising from FL) after two or more lines of systemic therapy | 2017 | Gilead Sciences | ZUMA-1 trial [6] |
| Adults with R/R FL after two or more lines of systemic therapy | 2021 | ZUMA-5 trial [7] | |||
| Patients with R/R LBCL and within a maximum of 12 months following first-line chemoimmunotherapy | 2022 | ZUMA-7 trial [8] | |||
| Tecartus (brexucabtagene autoleucel) | CD19 | Adults with R/R mantle cell lymphoma | 2020 | Gilead Sciences | ZUMA-2 trial [9] |
| Adult patients with R/R B-cell precursor ALL | 2021 | ZUMA-3 trial [10] | |||
| Breyanzi (lisocabtagene maraleucel) | CD19 | Adults with R/R LBCL who have previously received 2 or more systemic therapies | 2021 | Bristol Myers Squibb | TRANSCEND NHL 001 trial [11] |
Adults with R/R LBCL:
| 2022 | TRANSFORM trial [12] | |||
| Adults with R/R CLL and SLL, who have received at least 2 prior therapies, including a BTK inhibitor and B-cell lymphoma 2 inhibitor venetoclax | 2024 | TRANSCEND CLL 004 trial [13] | |||
| Carteyva (relmacabtagene autoleucel) | CD19 | Adults with R/R LBCL after two or more lines of systemic therapy | 2021 | JW Therapeutics | RELIANCE trial [14] |
| Abecma (idecabtagene vicleucel) | BCMA | Patients with R/R multiple myeloma after three or more previous lines of therapy including a proteasome inhibitor, an immunomodulatory drug, and an anti-CD38 antibody | 2021 | Bristol Myers Squibb | KarMMa trial [15] |
| Carvykti (ciltacabtagene autoleucel) | BCMA | Patients with multiple myeloma:
| 2022 | Janssen Pharmaceuticals | CARTITUDE-1 trial [16] |
| ARI-0001 | CD19 | Patients aged > 25 years with R/R ALL | 2021 | Hospital Clinic of Barcelona | CART19-BE-01 trial [17] |
ALL: acute lymphoblastic leukemia; BCMA: B-cell maturation antigen; BTK: Bruton tyrosine kinase; CAR: chimeric antigen receptor; CLL: chronic lymphocytic leukemia; DLBCL: diffuse large B-cell lymphoma; FL: follicular lymphoma; HGBCL: high-grade B-cell lymphoma; LBCL: large B-cell lymphoma; NOS: not otherwise specified; PMBCL: primary mediastinal B-cell lymphoma; R/R: recurrent/refractory; SLL: small lymphocytic lymphoma
Furthermore, a study on mice showed that TP53 mutations in chronic lymphocytic leukemia (CLL) lead to a high tumor burden, reduce T cell transplantation efficacy, and lead to diminished effectiveness of anti-CD19 CAR-T cell treatment [18]. Normally, the wild-type p53 protein functions as an oncosuppressor by inhibiting tumor growth. However, often disruption in its activity leads to protein malfunction and inactivation. In cancer, TP53 gene mutations have been repeatedly linked to a worse prognosis, more aggressive disease development, and resistance to treatment. Multiple studies have provided evidence of the correlation between mutations or deletions in the TP53 gene in CLL and the development of resistance to chemoimmunotherapy, along with a significantly deteriorated prognosis of the disease [19–21]. Consequently, the objective of this study is to assess the efficacy of CAR-T therapy in individuals with TP53 mutations.
Approved CAR-T cell products contain a CAR consisting of an antibody-like antigen binding domain, a surface co-stimulatory factor, a T cell receptor (TCR), and a co-receptor signaling domain [22]. In a conventional CAR, the antigen-binding domain is typically composed of an antibody-derived scFv (single chain variable fragment). Nevertheless, there exist alternative options, such as nanobodies, which are truncated versions of scFv and consist of heavy-chain variable domains (VHHs), native receptors, and small peptides, including peptide-centric [23] and specific peptide enhanced affinity receptor (SPEAR) [24]. The functions of the antigen-binding domain are influenced by various factors such as the total number of CAR domains, which can vary depending on the specific construct design (e.g., dual CAR-T cells have two domains). In addition, important parameters include the type of protein used to derive the antigen-binding domain, its affinity and/or avidity for the antigen, and the combined affinity of several various CAR constructs on a single CAR-T cell surface. Each of these features results in different cell properties and behaviors which together may affect the safety and efficacy profile of CAR-T cells in patient’s body [25].
The architecture of CARs is continuously being revised and is now in its fifth generation. The first iteration of CAR comprises solely an extracellular domain that selectively identifies antigens and an intracellular CD3ζ signaling domain. However, its ability to target tumors is severely restricted due to the lack of co-stimulatory signals. Second-generation CARs have incorporated an intracellular motif containing a co-stimulatory receptor signaling domain into their structure (Figure 1). Second-generation CAR-T cells are capable of proliferating and releasing cytokines to exert anti-tumor effects even without the presence of external co-stimulatory molecules. Third-generation CAR is equipped with two co-stimulatory domains, specifically engineered to enhance the cytotoxicity of CAR-T cells. Fourth-generation CARs include additional molecular components to facilitate the expression of functional transgenic proteins, such as interleukins or suicide molecules, thereby improving the cytotoxicity and safety of CAR-T cells. The advanced fifth-generation CARs employ an adaptor-specific recognition domain to substitute the tumor-specific scFv extracellular structural domain used in earlier generations of CAR-T cells. This recognition domain binds to an adapter molecule targeting a tumor-specific target, such as the split, universal, and programmable (SUPRA) CAR system [26] and the biotin-binding immune receptor (BBIR) CAR [27].
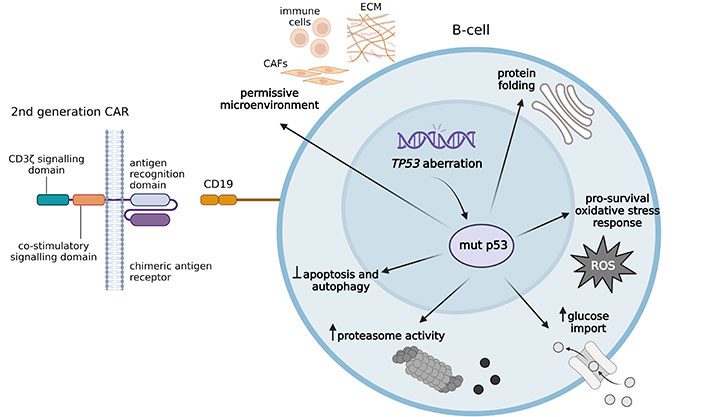
Second-generation CAR-T recognizes CD19 on the target B-cell. The presence of mut p53 protein promotes tumor progression, increases tumor aggressiveness, and is likely to reduce the efficacy of CAR-T cells. mut p53 induces a pro-survival response to oxidative stress, assists in protein folding, and increases proteasome activity. mut p53 protein resists autophagic cell death and prevents apoptosis. It promotes anabolic growth by increasing glucose uptake and facilitating the Warburg effect. mut p53 protein influences the environment by stimulating angiogenesis and triggering the release of proinvasive secretome. CAFs: cancer-associated fibroblasts; CAR: chimeric antigen receptor; ECM: extracellular matrix; mut p53: mutant p53; ROS: reactive oxygen species. Created in BioRender. Mirgayazova, R. (2024) https://BioRender.com/r03q792
In summary, incremental changes in CAR configurations that happened with each successive generation have led to significant improvements of CAR-T cell therapy safety and efficacy. Currently, second-generation CAR-T cells, which exhibit compromised efficacy and side effect profiles, are employed in clinical practice. The growing number of CAR-T cell therapies approved by regulatory authorities over recent years (Table 1) is indicative of the successful application of these therapies.
CAR-T cell immunotherapy has already demonstrated significant clinical efficacy for a number of hematological malignancies, including acute lymphoblastic leukemia (ALL), diffuse large B-cell lymphoma (DLBCL), LBCL, and multiple myeloma (MM). CAR-T cell-based therapy has been approved by relevant organizations for the treatment of the aforementioned and some other diseases (Table 1). Most recently, in March 2024, the Food and Drug Administration (FDA) approved Breyanzi for the treatment of patients with R/R CLL and small lymphocytic lymphoma (SLL) who have received at least two prior therapies, including a Bruton tyrosine kinase (BTK) inhibitor and the B-cell lymphoma 2 inhibitor venetoclax [13].
However, in contrast to the impressive clinical outcomes demonstrated in hematological malignancies, the effectiveness of CAR-T treatment in solid tumors has been mostly disappointing so far. There is currently a limited number of suitable antigens that can be effectively targeted by CAR-T cells in solid tumors. Overexpressed proteins such as mesothelin (MSLN) and epithelial cell adhesion molecule (EpCAM) are prominently present on the surface of tumor cells, including breast, prostate, and stomach cancers. These proteins could serve as promising targets for the development of CAR-T cell therapies against solid tumors [28].
TME contains immunosuppressive components that impede the infiltration and efficacy of CAR-T cells. These include tumor-associated macrophages (TAMs), regulatory T (Treg) cells, myeloid-derived suppressor cells (MDSCs), and tumor-associated fibroblasts (TAFs). Moreover, various cytokines such as transforming growth factor beta (TGF-β), IL-4, and IL-10 can facilitate migration of immune cells that suppress the immune response, thereby indirectly inhibiting CAR-T cells. In addition, several inherent features of tumors, such as faulty blood vessels and a dense extracellular matrix (ECM) that contains cancer-associated fibroblasts (CAFs) and inappropriately expressed adhesion molecules, contribute to the insufficient movement and penetration of CAR-T cells within the tumor. While several stroma-targeting drugs have been reported [29], it is important to note that these targets may also be present in healthy tissues. CAR-T therapy specifically targeting CAFs has been demonstrated to recognize and attack malignant multipotent bone marrow stromal cells (BMSCs) [30]. However, this therapy has been associated with severe bone toxicity and cachexia, leading to deadly outcomes.
In the phase I clinical research conducted by Ahmed et al. [31], autologous modified virus-specific T (VST) cells that target cytomegalovirus, Epstein-Barr virus, or adenovirus were used to improve the persistence of systemically administered CAR-T cells that target HER2. This clinical trial included a cohort of 17 individuals diagnosed with progressing glioblastoma multiforme (GBM) who were administered with one or several infusions of autologous anti-HER2 CAR-VST cells without preceding lymphodepletion. The presence of CAR-T cells was observed in the peripheral blood for a duration of 12 months, which surpasses the duration observed in another study [32, 33]. The median overall survival (OS) was 11.1 months from the initial T cell infusion and 24.5 months from the time of diagnosis, demonstrating the clinical advantage. In addition, Hegde et al. [34] have effectively developed bispecific CAR-T cells that specifically target IL13Rα2 and HER2. These multi-targeted CAR-T cells have the potential to address the issue of antigen escape and offer promising prospects for the future application of CAR-T in glioma treatment [34]. In phase I clinical trial conducted by Brown et al. [35] three patients with recurrent GBM (rGBM) were administered a total of 12 regional doses of CAR-T cells targeting IL13Rα2 (NCT00730613). Despite the limited sample size, a group of 3 patients who experienced GBM recurrence achieved a median OS time of 10.9 months. In recent preclinical investigations the inclusion of Nivolumab, a monoclonal antibody that targets programmed cell death protein 1 (PD-1), has been found to enhance the ability of anti-GD2 CAR-T cells to destroy cancer cells. Additionally, in new nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice model of GBM, the combination of anti-GD2 CAR-T cells with Nivolumab was shown to increase the animals’ survival [36].
In addition, the activation of CAR-T cells leads to the production of various cytokines, such as IL-1, IL-2, IL-4, IL-6, IL-8, IL-10, and tumor necrosis factor alpha (TNFα) resulting in cytokine release syndrome (CRS)—a dangerous potentially fatal side effect [37].
Some individuals treated with CAR-T therapy may also experience immune effector cell-associated neurotoxicity syndrome (ICANS) [38]. While CAR-T cell activity can be enhanced by inflammatory cytokines like IL-2, it is important to note that systemic IL-2 administration can lead to the development of severe capillary leak syndrome and ultimately result in organ failure. Allen et al. [39] developed therapeutic T cells with engineered cytokine circuits based on tumor-specific synthetic Notch (synNotch) receptor that stimulates the production of IL-2. In immune-excluded tumor models such as pancreatic cancer and melanoma, the use of modified synNotch→IL-2 induction circuits resulted in a strong infiltration of CAR-T or TCR-T cells into TME. These findings open up the potential avenue for using CAR-T therapy to treat solid tumors [39].
The TP53 gene, also known as the tumor protein p53 gene, is located on the short arm of chromosome 17 and encodes a protein р53 that plays a critical role as tumor suppressor. The p53 protein is involved in the regulation of cell cycle, deoxyribonucleic acid (DNA) repair, cellular senescence, maintaining genomic stability, and inducing apoptosis, all of which are important mechanisms for preventing the initiation and progression of cancer. The TP53 gene is activated in response to a variety of cellular stresses and can induce cell cycle arrest for the purpose of DNA repair or, if the damage is irreparable, it can initiate programmed cell death [40].
The TP53 gene consists of 11 exons that encode for a protein with several functional domains, including the transactivation domain, DNA-binding domain, oligomerization domain, and regulatory domain. These domains allow the p53 protein to interact with various cellular components and carry out its tumor-suppressing functions. Upon activation by cellular stress signals, such as DNA damage or oncogene activation, p53 undergoes post-translational modifications that lead to its stabilization and activation. Subsequently, activated p53 translocates to the nucleus and binds to specific DNA sequences to modulate the expression of its target genes.
In addition, the TP53 gene has the highest frequency of genetic alterations in human tumors [41]. TP53 mutations primarily lead to the inactivation of normal p53 functions, which is advantageous for cancer development because it prevents cells from undergoing natural tumor-suppressor responses such as senescence and apoptosis. In addition, cancer cells appear to acquire advantageous properties by exclusively retaining the mutant variant of the p53 protein. Mutant p53 has the ability to reorganize the transcriptomes and proteomes of tumor cells by forming new associations with transcriptional regulators, enzymes, and other cellular proteins [42, 43]. Specific missense mutations in p53 have been found to disrupt key cellular pathways, promote cancer cell growth and survival, facilitate invasion, migration, metastasis, and resistance to chemotherapy [44, 45]. Some reports suggest that mutant p53 plays a role in adapting to stressful conditions that occur during tumor development. These conditions include DNA damage caused by excessive cell growth, oxidative and proteotoxic stress, changes in nutrient levels, physical constraints, signals from surrounding tissues, and the body’s immune response against the tumor [46]. Mutant p53 has been shown to improve protein folding [47] and increase proteasome activity [48] in human breast, lung, and pancreatic cancer cell lines. It also resists autophagic cell death in breast cancer [49] and inhibits therapy-induced apoptosis in head and neck cancer [50]. The mutant protein increases glucose uptake, which contributes to the Warburg effect in mice lacking the TP53 gene [51]. In addition, mutant p53 has been found to regulate the extracellular environment by promoting angiogenesis in breast cancer [52], increasing cancer-promoting inflammation in the colon of p53 knockout mice [53], and stimulating the production of a proinvasive secretome in human lung tumors and their derived cell lines [54] (Figure 1).
The presence of mutated p53 in tumors has been shown to significantly alter the TME, promoting immune evasion through the modulation of myeloid cells and T cells. It has been demonstrated that mutant p53 can influence the polarization of TAMs, skewing them towards an M2-like phenotype that supports tumor growth and suppresses anti-tumor immunity [55]. This M2 polarization is associated with increased production of immunosuppressive factors such as IL-10 and TGF-β, which further impede T cell activation and proliferation. Furthermore, mutant p53 has been shown to enhance the recruitment of MDSCs, which inhibit T cell function and promote an immunosuppressive microenvironment [56].
Ghosh et al. [57] demonstrated that mutant p53 has the effect of disrupting the activity of the cytoplasmic DNA sensing pathway, specifically the cyclic GMP-AMP synthase-stimulator of interferon genes-TANK-binding kinase 1-interferon regulatory factor 3 (cGAS-STING-TBK1-IRF3) axis, which is crucial for triggering the innate immune response. In contrast to wild-type p53, mutant p53 binds to TBK1, preventing the formation of a vital trimeric complex comprising TBK1, the endoplasmic reticulum-resident STING, and IRF3. This complex is essential for the activation, nuclear translocation, and transcriptional function of IRF3. The impairment of innate immune signaling caused by mutant p53 leads to altered cytokine production, which in turn facilitates immune evasion. Furthermore, mutant p53 impairs IRF3-induced apoptosis. It is worth noting that reactivating TBK1 signaling can circumvent the effects of mutant p53, resulting in the revival of immune cell functionality and the elimination of cancer cells. These findings have significant implications for the development of new treatments. Strategies that reactivate TBK1 could potentially restore immune surveillance and target tumors with mutant p53.
It has been demonstrated that TP53 mutations are more prevalent in cancerous tissues than in healthy ones. In the context of cancer, it is crucial to recognize that mutations and deletions that arise in the TP53 gene are common genetic events that are associated with adverse clinical manifestations and a poor prognosis. It has been observed that TP53 mutations are found in some high-risk metastatic malignancies, including esophageal adenocarcinoma, ovarian serous carcinoma, and small cell lung carcinoma. These mutations occur with a high frequency (85–98%) in these cancers [58]. Additionally, a comparative study of primary and metastatic samples demonstrated that TP53 mutations are significantly more frequent in secondary and metastatic lesions [59]. These observations are in excellent agreement with earlier studies that have noted that the development of high-grade glioblastoma from a previous low-grade astrocytoma is associated with the specific growth of individual TP53-mutant cells that were already present in the previous cancer [60].
Somatic TP53 mutations in cancer are found in all regions of the gene but with a marked concentration at specific sites. DNA-binding and transactivation domains carry markedly more mutations in hotspots (the TP53 Database R20, July 2019: https://tp53.isb-cgc.org) [61].
In addition, several single nucleotide variations (SNVs) have been identified in a significant number of patients, ranging from hundreds to thousands. It has been observed that germline TP53 mutations are present in individuals diagnosed with Li-Fraumeni syndrome, a disorder characterized by an increased susceptibility to cancer throughout life.
It is also noteworthy that there are additional genes linked to the TP53 pathway that may exhibit mutations or copy number variations, both of which are similarly correlated with an unfavorable prognosis. The Catalogue Of Somatic Mutations In Cancer (COSMIC) database [62] provides data indicating that 15 genes involved in the TP53 pathway undergo recurrent alterations. These genes include TP53 regulators such as MDM2 and CDKN2A, as well as downstream effectors including CCND1 and PTEN [63]. The function of p53 is suppressed in some virus-associated malignancies. For example, in cervical cancer, the HPV E6 oncoprotein destroys the p53 protein [64]. The p53 protein plays a critical role in these cancers, although the TP53 gene itself is not mutated. Notably, some rare cancers, such as childhood ALL and testicular seminoma, have a high chance of cure despite initial malignancy and metastasis. In contrast to metastatic disseminated adult malignancies, these tumors exhibit the wild-type TP53 gene [65]. The collective data presented herein provide evidence in favor of the argument that a majority of cell types found in advanced or metastatic malignancies exhibit TP53 abnormalities, resulting in the presence of non-functional p53 protein.
Inactivation of p53 functions or its downstream pathway is a common feature of human tumors that often correlates with increased malignancy, poor patient survival, and resistance to treatment [66, 67]. Several studies indicate that TP53 mutations confer chemoresistance to lung cancer cells in vitro and in vivo [68, 69]. In CLL the presence of mutant TP53 variants is associated with poor prognosis, advanced disease stage, rapid progression, chemoresistance, and reduced OS [70].
In their research, Roselle et al. [71] emphasized the substantial impact of the human and great ape-specific p53 isoform, Δ133p53α, in enhancing the efficacy of CAR-T cell therapy for CLL patients who had not responded to previous treatments. Despite lacking a transactivation domain, Δ133p53α is capable of forming heterooligomers with full-length p53, thus influencing the p53-mediated stress response [72]. The study demonstrated that the sustained expression of Δ133p53α enhances the anti-tumor efficacy of CD19-targeted CAR-T cells and mitigates dysfunction in scenarios involving high tumor loads and metabolic stress. The researchers discovered that CAR-T cells expressing Δ133p53α exhibit a robust metabolic profile, enabling them to perform effector functions and proliferate even in nutrient-deprived environments. This is partly due to the upregulation of essential biosynthetic processes and enhanced mitochondrial function. These results highlight the potential advantages of targeting the p53 pathway to enhance CAR-T cell therapies [71].
Mutant TP53 variants have been identified as strong prognostic markers for patients receiving standard first-line treatment in CLL. Preclinical evidence indicates that TP53-mutated CLL leads to early disease onset, high tumor burden, and ineffective T cell engraftment in a mouse model, resulting in suboptimal efficacy of anti-CD19 CAR-T cells [18]. Clinical trials of anti-CD19 CAR-T cells have shown limited complete remissions in cases carrying the del17p gene (TP53 gene locus) [73].
Determining TP53 status may be of a great prognostic value for the optimal decision-making in chemo/radiation therapy. For instance, it is well known that tumors containing the mutant TP53 are more resistant to ionizing radiation than those with the wild-type TP53. The frequent inactivation of TP53 in human tumors suggests that the restoration of the abrogated TP53 pathway is an attractive tumor cell-specific strategy for treating cancers.
Мutations in the TP53 gene lead to genomic instability, tumor cell clonal evolution, growth, and survival [73]. These mutations are also associated with dysregulation of pathways related to CAR-T cell cytotoxicity resulting in poor prognosis and potential drug resistance [74]. In preclinical studies, it has been demonstrated that mutations in the TP53 gene have a negative impact on the duration of CAR-T cell persistence, resulting in a reduction in the long-term efficacy of anti-CD19 CAR-T cell therapy in the context of cancer treatment [18].
The efficacy of treatment with anti-CD19 CAR-T cells in patients with TP53 mutations was evaluated by Zhang et al. [75] in a recent study. A total of 102 patients with B-ALL, of whom 11 had mutations in the TP53 gene, participated in the study. The researchers observed that the OS and leukemia-free survival (LFS) rates at six months were significantly lower in patients with TP53 mutations than in patients without mutations (OS: 51.9% vs. 89.0%; LFS: 42.4% vs. 82.6%; Figure 2). Five out of 11 patients experienced a relapse with a median duration of 56 days, 7 patients died (1 patient due to infection and 4 due to relapse). Patients with TP53 mutations may experience a higher rate and earlier onset of relapse due to rapid mutations in leukemic blasts, enhanced proliferation, and evasion of CAR-T cell cytotoxicity. Authors demonstrate that CAR-T cell treatment followed by consolidative allogeneic hematopoietic stem cell transplantation (allo-HSCT) has superior LFS and OS.
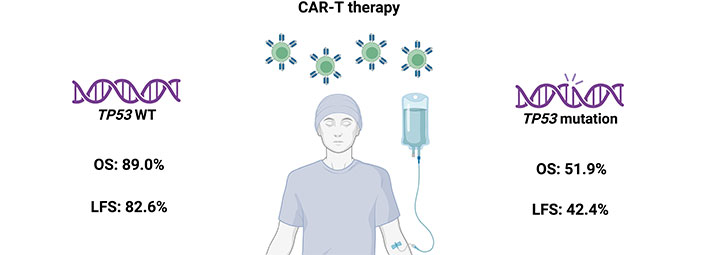
The impact of CAR-T therapy on patient survival. The OS and LFS at 6 months after CAR-T therapy are much lower in the group with TP53 mutation compared to the group with WT TP53 [75]. CAR: chimeric antigen receptor; LFS: leukemia-free survival; OS: overall survival; WT: wild-type. Created in BioRender. Mirgayazova, R. (2024) https://BioRender.com/n41y197
Porpaczy et al. [76] investigated the impact of TP53 mutations on the OS of patients with R/R DLBCL following CAR-T therapy. R/R DLBCL is associated with a poor prognosis, and TP53 mutations in DLBCL are recognized as indicators of poor outcomes and are often associated with treatment resistance. A total of 170 patients with DLBCL and high-grade B-cell lymphoma who were treated at an Austrian medical center between 2000 and 2021 were enrolled in the study. In this study, 29 out of 170 patients underwent CAR-T cell treatment, and TP53 mutations were detected in 10 out of 29 patients (35%) in the CAR-T cell treatment group and in 31 out of 141 patients (22%) in the conventional treatment group. The presence of TP53 mutations was identified as a distinct factor that negatively impacts OS among the 141 patients who did not receive CAR-T cell treatment. However, this correlation was not observed in the cohort of patients who underwent CAR-T cell therapy. The researchers concluded that the presence of TP53 mutations did not influence the clinical outcomes of patients with DLBCL who received CAR-T cell therapy and proposed that a comprehensive assessment in larger cohorts is necessary.
In a retrospective study conducted by Gao et al. [77], the effect of TP53 mutations in R/R DLBCL on the clinical picture after CAR-T therapy was investigated. Authors evaluated the impact of CAR-T cell therapy on patients with TP53 mutations and found that these patients had a median OS of 24.5 months and a median LFS of 6.8 months following CAR-T treatment. There were no significant differences in the objective remission rate (ORR) and LFS between patients with wild-type and mutated TP53 after CAR-T therapy. However, patients with TP53 mutations had significantly poorer OS. Concurrent mutations of chromosome 17 and those found on exon 5 of the TP53 gene were associated with a more unfavorable prognosis. Additionally, individuals with dual mutations in TP53 and DDX3X were identified as a subgroup with a critically poor prognosis.
Xue et al. [78] conducted a retrospective analysis involving 73 patients in China diagnosed with R/R DLBCL who underwent CD19 CAR-T therapy (Axi-cel or Relma-cel) following a chemotherapy regimen of fludarabine and cyclophosphamide. The findings indicated that patients with R/R DLBCL exhibiting both double-expression (MYC > 40% and BCL2 > 50%) and TP53 mutations generally experience a worse clinical outcome post-CAR-T treatment, even when combined with additional therapies. Conversely, the study demonstrated that CAR-T therapy was an effective treatment option for patients with either TP53 mutations or double-expression status, with outcomes comparable to those observed in patients without these genetic alterations.
Eder et al. [79] reported on a 64-year-old male patient diagnosed with LBCL who experienced two relapses after standard-of-care treatment. Next-generation sequencing identified a clonal population with TP53 mutations. The patient received CAR-T cell therapy as a third-line treatment resulting in complete remission. However, this therapy also led to the proliferation of TP53-mutated cells and the development of therapy-induced myelodysplasia with a complex abnormal chromosomal structure. The authors suggest that this case could be considered a typical example of clonal hematopoietic expansion in a patient undergoing CAR-T cell treatment, especially in the presence of TP53-mutated clones.
CAR-T cell therapy has shown promise as a viable treatment for R/R Burkitt’s lymphoma with TP53 mutation, a condition that often has low remission rates when treated with traditional chemoimmunotherapy [80]. Here, the patient with Burkitt’s lymphoma achieved complete remission and prolonged disease-free period after receiving a combination of ant-CD19 and anti-CD22 CAR-T cells, followed by autologous hematopoietic stem cell therapy (ASCT).
Shouval et al. [81] demonstrated that TP53 alterations represent a valuable prognostic indicator in LBCL patients receiving anti-CD19 CAR-T therapy. Gene expression profiling indicates that TP53 mutations are associated with a TME that suppresses the immune system and impairs apoptotic signaling, potentially reducing the effectiveness of CAR-T therapy.
Mueller et al. [82] conducted a study to assess the efficacy of CAR-T therapy in treating TP53-mutant acute myeloid leukemia/myelodysplastic neoplasms (AML/MDS), which are known to be resistant to chemotherapy and have a poor prognosis. The study demonstrated that AML cells with TP53 mutations exhibited heightened resistance to CAR-T cell therapy in vitro. The proliferation and viability of CAR-T cells were found to be significantly reduced when co-cultured with TP53-mutant AML blasts, in comparison to those with wild-type TP53. The live-cell imaging results demonstrated that TP53-mutant cells were eliminated at a slower rate, which had an adverse effect on the effectiveness of CAR-T cells. The survival rates of immunocompromised mice with TP53-mutant AML who received CAR-T cell treatment were lower than those of mice with wild-type AML. Gene expression analysis indicated an upregulation of the mevalonate pathway in TP53-mutant cells, alongside a downregulation of the Wnt pathway in CAR-T cells. Pharmacological intervention targeting these pathways restored the susceptibility of TP53-mutant AML cells to CAR-T cell-mediated cytotoxicity. Furthermore, CAR-T cells engineered using CRISPR/Cas9 to enhance Wnt signaling demonstrated improved effectiveness against TP53-mutant AML cells.
Currently, there is a limited number of studies focused on understanding the correlation between patient’s TP53 mutational status and the efficacy of CAR-T therapy due to the fact that the presence of TP53 mutations is not yet universally tested in all cancers. The available clinical information indicates that TP53 mutations have an impact on the outcomes of patients with B-cell malignancies treated with CAR-T cell therapy. Patients with TP53 mutations often exhibit lower survival rates and higher rates of relapse compared to those without the mutation.
Mutations of TP53 gene correlate with poor prognosis following CAR-T therapy. Mutant p53 may be responsible for the poor efficacy of CAR-T cell therapy as a result of alterations to the TME and the influence of key signaling pathways involved in the immune response. The presence of mutant p53 has been observed to enhance the recruitment of Tregs and MDSCs, both of which contribute to an immunosuppressive environment that impairs the efficacy of CAR-T cell therapy. The additional effect of mutant p53 is to disrupt the expression of pro-inflammatory cytokines and chemokines, which further limits the ability of CAR-T cells to infiltrate and persist in tumors.
However, using combination of ant-CD19 and anti-CD22 CAR-T cells, and in some cases following allo-HSCT transplantation allows achieving complete remission for patients with TP53 mutations. Furthermore, pharmacological intervention targeting the mevalonate pathway and the Wnt pathway has been shown to restore the susceptibility of TP53-mutant AML cells to CAR-T therapy. Also, genetically modified CAR-T cells, engineered to enhance Wnt signaling, have shown promising results in treating TP53-mutant tumors. Additional studies and a larger patient sample size are necessary for a more in-depth analysis. Overall, further research is needed to fully understand the impact of TP53 mutations on the response to CAR-T cell therapy. This will enable the development of more efficient treatment protocols and the enhancement of early genetic diagnostics.
ALL: acute lymphoblastic leukemia
allo-HSCT: allogeneic hematopoietic stem cell transplantation
AML: acute myeloid leukemia
CAFs: cancer-associated fibroblasts
CAR: chimeric antigen receptor
CLL: chronic lymphocytic leukemia
DLBCL: diffuse large B-cell lymphoma
DNA: deoxyribonucleic acid
GBM: glioblastoma multiforme
IRF3: interferon regulatory factor 3
LBCL: large B-cell lymphoma
LFS: leukemia-free survival
MDSCs: myeloid-derived suppressor cells
OS: overall survival
R/R: recurrent/refractory
scFv: single chain variable fragment
STING: stimulator of interferon genes
synNotch: synthetic Notch
TAMs: tumor-associated macrophages
TBK1: TANK-binding kinase 1
TCR: T cell receptor
TGF-β: transforming growth factor beta
TME: tumor microenvironment
Treg: regulatory T
VST: virus-specific T
RM: Conceptualization, Writing—original draft, Supervision. RK: Writing—original draft. MF and VC: Visualization. EB: Writing—review & editing, Funding acquisition. All authors have read and agreed to the published version of the manuscript.
The authors declare that they have no conflicts of interest.
Not applicable.
Not applicable.
Not applicable.
Not applicable.
This study was funded by Russian Science Foundation (RSF) grant [22-74-10076] to EB in the decision to submit the paper for publication. This work has been supported by the Kazan Federal University Strategic Academic Leadership Program [PRIORITY-2030]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
© The Author(s) 2024.
Copyright: © The Author(s) 2024. This is an Open Access article licensed under a Creative Commons Attribution 4.0 International License (https://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, sharing, adaptation, distribution and reproduction in any medium or format, for any purpose, even commercially, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.

