
Abstract
In hematological malignancies, autologous immunotherapy with T lymphocytes expressing a chimeric antigen receptor (CAR-T) has been successfully applied. CAR enhances the immuno-cellular effector system directly against cells expressing target antigens. The objective here was to discuss the prospects of applying CAR-T and its variants in autoimmune diseases (AIDs) to deplete pathogenic autoantibodies by eliminating B lymphocytes and plasma cells. B cells play a crucial role in the pathogenesis of AID through the production of autoantibodies, cytokine dysregulation, antigen presentation, and regulatory dysfunction. In AID with numerous autoreactive clones against various autoantigens, such as systemic lupus erythematosus, rheumatoid arthritis, vasculitis, myositis, and systemic sclerosis, CAR-T targeting CD19/CD20 and B-cell maturation antigen (BCMA) have shown success in preclinical and clinical studies, representing an innovative option for refractory patients when standard treatments fail. The suppression of B lymphocytes reactive against specific antigens using cytolytic T cells carrying a chimeric autoantibody receptor (CAAR-T) offers a promising approach for managing various AIDs, especially those with characterized pathogenic autoantibodies, such as pemphigus vulgaris, myasthenia gravis, and anti-NMDAR autoimmune encephalitis. CAAR-T allows the elimination of autoreactive B lymphocytes without compromising the general functionality of the immune system, minimizing common side effects in general immunosuppressive therapies, including immunobiologicals and CAR-T. In vitro, preclinical, and clinical (phase 1) studies have demonstrated the efficacy and specificity of CAR-T and CAAR-T in several AIDs; however, extensive clinical trials (phase 3) are required to assess their safety and clinical applicability. These advances promise to enhance precision medicine in the management of AIDs, offering personalized treatments for individual patients.
Keywords
Autologous immunotherapy, chimeric antigen receptor, chimeric autoantibody receptor, cytolytic lymphocytes, autoimmunity, autoantibodiesIntroduction
Autoimmune diseases (AIDs) are pathologies characterized by an imbalance of the immune system, generally due to a failure in the tolerance mechanisms that protect our organism from an exacerbated immune response that reacts against itself. This failure may affect T or B lymphocytes, or both, leading to direct or indirect tissue destruction through direct cellular cytotoxicity, increased proinflammatory cytokines or increased autoantibodies (AAbs) with their neutralizing and opsonizing capacities, as well as facilitators of phagocytosis, cytotoxicity by the complement system or antibody-dependent cytotoxicity processes [1, 2]. There are more than 80 well-characterized AIDs affecting approximately 5% of the human population [3].
Varying from systemic to organ-specific, conventional treatments for AIDs consist of a combination of various immunosuppressants, glucocorticoids, cytostatics, hormonal therapies, polyclonal antibodies, nonsteroidal anti-inflammatory drugs (NSAIDs) and immunobiologics, such as monoclonal antibodies (MoAbs) targeting receptors, checkpoints and circulating molecules [1, 4]. Ideal treatments should seek to eliminate autoreactive cells while simultaneously preserving protective immunity, a delicate balance aimed at restoring equilibrium in the immune system [5]. With traditional therapies, this goal is not yet achieved because they lead to a systematic suppression of the immune system, especially after several rounds of treatment, which is necessary to generate long-lasting immunosuppression over time. Among the most common adverse effects is an increase in opportunistic infections. However, biologic drugs based on MoAbs tend to be more selective and thus decrease these adverse effects.
To achieve a more effective and permanent restoration of immune system homeostasis, cell-based therapies, such as the use of cytolytic T lymphocytes [T-CD8 and natural killer (NK)], regulatory T cell (Treg), tolerogenic dendritic cells (DC) or even mesenchymal stem cells (MSCs), have been heavily invested in during recent years, as these can persist in the patient longer in vivo than therapies based on MoAbs or cytokines [5]. In particular, cell-based treatments that undergo bioengineering procedures are of particular interest [6]. Rapid advances in molecular biology, gene editing, external gene transduction and gene expression control, the so call cell engineering, have helped to create more predictable and precise cell-based therapies [5].
Within this category of “engineered or synthetic cells” are cytolytic lymphocytes modified using different tools, such as the insertion of a chimeric antigen receptor (CAR). One example is chimeric antigen receptor T cells (CAR-T), T cells modified to express a CAR designed to recognize specific antigens. The CAR is a fusion protein composed of an antigen recognition domain, usually the variable region of an antibody, such as anti-CD19 and anti-B-cell maturation antigen (BCMA), an extracellular spacer domain, a transmembrane domain, and an internal co-stimulation and activation domain [4]. The goal is to target T lymphocytes to exert their cytolytic power against cells expressing antigens toward which the CAR is directed.
CAR-T therapy is a therapy developed originally for the treatment of HIV [7], but in the last decade it was applied for the treatment of hematological malignancies, being employed for the first time in this context for the treatment of refractory leukemias, for which it achieved remarkable efficacy [8, 9]. This success inspired the expansion of its application to other pathologies. Since 2017, the FDA (US Food and Drug Administration) has approved six CAR-T therapies [10], all for treatment in hemato-oncologic diseases, including leukemia, myeloma, and various lymphomas (Figure 1). It is called autologous immunotherapy because the cytolytic T lymphocytes are obtained from the patient himself to whom, subsequently after the procedure of transduction of the CAR receptor by genetic engineering techniques, followed by cell expansion, these lymphocytes are reinjected to localize and eliminate the CAR target cells.
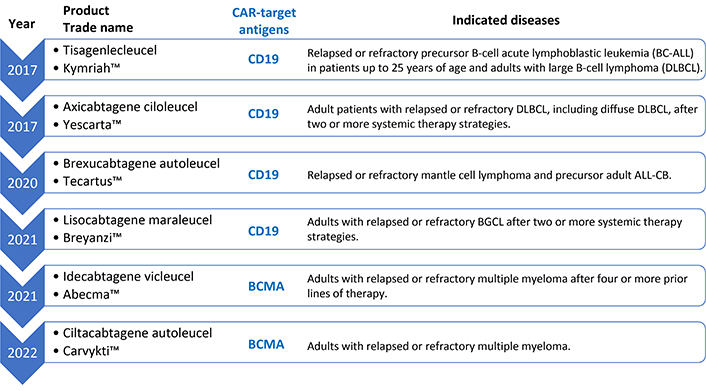
FDA-approved autologous CAR-T-based immunotherapies against cancer. CAR-T: chimeric antigen receptor T cells; BCMA: B-cell maturation antigen
A limitation to the widespread use of CAR-T therapy is its potential toxicity, primarily due to the development of cytokine release syndrome (CRS), as well as neurotoxicity and opportunistic infections resulting from the depletion of healthy B cells [8, 9, 11, 12]. Manifestations of CRS include fever, hypotension, hypoxia, target organ dysfunction, cytopenias, compromised hemostasis, and hemophagocytic lymphohistiocytosis. Neurological toxicities are diverse and include encephalopathy, cognitive defects, dysphasia, seizures, and cerebral edema. Long-term side effects such as prolonged cytopenias and toxicity to other organs (heart, liver, kidney) have been proposed, but because it is a new therapy, the spectrum of these is not yet fully clear [8–11, 13, 14]. Finally, there have been reports of secondary T-cell lymphomas appearing after CAR-T immunotherapy, which is considered a serious risk and is under investigation [15–17].
There are different strategies in the construction of CARs, classified from the first to fifth generation, according to differences in the number and composition of intracellular domains [6]. These domains play a critical regulatory role that determine function and enhance efficacy. First-generation CARs have only the CD3ζ signaling domain. Second-generation CARs include CD3ζ and CD28 or CD137 (4-1BB) (4-1BB signaling can directly suppress Treg or decrease sensitivity to Treg suppression). The third generation combines both co-stimulatory domains, such as CD27, CD28, ICOS, 4-1BB (CD137) or OX40 (CD134), in addition to CD3ζ. The fourth and fifth generation CARs, in addition to the co-stimulatory signals, are also armed with a “nuclear-activated T-cell response expression factor” element for an induced transgene product, such as IL-12 or other cytokines, or signaling domains that activate multiple T cell pathways involved in activation, proliferation, and survival (Figure 2) [6, 18].
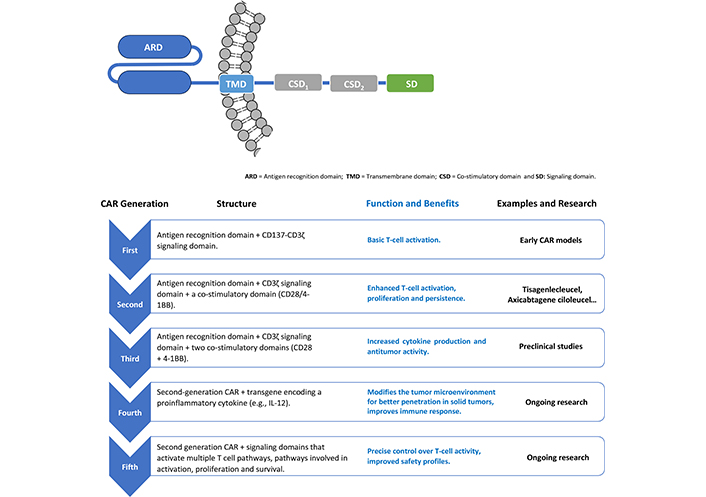
Different strategies and evolution in the construction of CAR molecules. CAR: chimeric antigen receptor
The design of any CAR-T therapy starts with an identifiable antigen to target, and this in and on itself makes the process of creating widespread therapeutics difficult [7]. CAR-T therapies have been successful against different types of “soluble” hematological malignancies, which has led several groups to attempt to apply this strategy against solid tumors [13]. In fact, one of the main limitations in expanding the application of CAR-T to other malignancies is its low penetration and effectiveness against solid tumors [19] showing a risk for on-target, off-tumor (OTOT) toxicity. In solid tumors, the absence of tumor-specific antigens and the heterogeneity of tumor cells complicate the identification of suitable targets, leading to the risk of OTOT toxicity [20]. OTOT occurs when CAR-T attack healthy tissues that express the target antigen, resulting in severe side effects. To mitigate this, researchers are exploring strategies such as bispecific CARs, which target multiple antigens to enhance specificity, nanomaterials, and modulation of the tumor microenvironment to improve CAR-T infiltration and persistence [20]. With the evolution of biotechnology, the prototypical design of CAR genes and vectors has undergone several improvements, such as the addition of co-stimulatory domains for improved intracellular signaling (fourth and fifth-generation receptors, Figure 2), bispecific receptors with more than one target antigen (mentioned above), endogenous production of immune checkpoint antagonist interleukins to enhance T-cell activity and, more recently, the engineering of other immune cells, such as NK cells, macrophages and γδ T cells to express the CAR molecule [5, 13, 18, 21, 22].
Given the characteristics that limit the expansion of CAR applications, alternatives have been sought for effector cells to which the CAR receptor is inserted, such as the use of NK cells and macrophages, which are part of the innate immune system and use an MHC-independent identification system [23, 24]. In turn, these cells can be extracted from various allogeneic sources, their activation is not MHC-dependent, and they have demonstrated antitumor activity in several different models [23]. In addition, Schlegel et al. [19] recently proposed a kind of universal CAR 2.0, which uses a kind of adapter CAR capable of binding to MoAb antibodies containing the connector modules for the CAR receptor. The benefits include the possibility of using different antibodies against different antigens (flexibility), as well as cytolytic cells from different sources, such as NK, MSC-derived and hematopoietic cells, always presenting the universal CAR adapter. The possibility of acquiring ready-to-use CAR cells associated with the flexibility of target antigens (multiple antibodies), results in considerable advances in the popularization and expansion of CAR technology, since its costs and complexity of protocol execution decrease [19].
Due to its flexibility, it is logical to transition this kind of technology from treating hematological malignancies through immunological modulation to treating AID, especially when the incidence of AIDs worldwide has consistently been increasing, without any clear reasons [3]. Among these, type 1 diabetes mellitus (DM-T1), multiple sclerosis (MS), systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), and Crohn’s disease are the most common types of AIDs, which pose enormous health challenges [18, 21, 25]. Historically, anticancer drugs have been strategically repurposed for the management of hematological disorders, a notable example being rituximab—a chimeric MoAb originally engineered for the treatment of B-cell non-Hodgkin lymphomas. Its mechanism of action, which involves targeted depletion of CD20-positive B lymphocytes, has subsequently proven efficacious in a range of AIDs where B cells play a central pathogenic role, thereby exemplifying the translational potential of oncology therapeutics in immunological contexts. Many AIDs have B lymphocytes as a major player in their pathophysiology. Approved CAR therapies for hematologic malignancies (Figure 1) target B-cell receptors (BCR) (CD19 and BCMA), thereby eliminating these cells. Recent studies point to the use of autologous immunotherapy with anti-CD19/BCMA CAR-T to treat AIDs, describing this technique as hopeful for people suffering from these diseases and who do not respond to conventional treatments with immunosuppressants and immunobiological MoAbs [4, 26].
The aim of this paper is to discuss the prospects in the application of autologous immunotherapies based on CAR-T and its variants to eliminate pathogenic AAb by eliminating antibody-producing cells: B lymphocytes and plasma cells.
CAR-T-based therapies targeting specific cell populations in AIDs
B lymphocytes play an important role in AIDs, either by production of AAb or by dysfunction in cytokine production, antigen presentation to T cells and failure of regulatory B lymphocytes. Classical treatments for AIDs, after general immunosuppression medications, focus on various MoAb that bind to antigens present on lymphocytes, such as rituximab (anti-CD20) or inebilizumab (anti-CD19) [27]. These monoclonals act as checkpoint inhibitors, delaying cell activation, or bind to surface receptors of certain populations and recruit the effector part of the antibody-induced immune system, such as the complement system.
The anti-CD19 CAR consists of an extracellular domain derived from an antibody that specifically binds to the CD19 receptor of B lymphocytes [28]. This CAR construct includes a single-chain variable fragment (scFv) of the anti-CD19 antibody and intracellular signaling domains (Figure 2) [21, 29, 30]. Upon encountering CD19-expressing cells, the scFv binds to the CD19 antigen, activating T cells and initiating a cytotoxic response to eliminate B cells. This strategy is particularly effective because it targets CD19, a surface molecule widely expressed on B cells and some plasma cells. This allows B-cell depletion without the need to identify specific pathogenic antigens.
CAR-T-CD19 cells present a significant advantage by allowing the elimination of a large number of B cells with different epitopes. This is especially useful in AIDs with several autoreactive clones or when specific information on the antigens involved is lacking. An example of this is anti-synthetase syndrome (ASS), a rare disease characterized by chronic inflammation causing idiopathic myopathy, in which B cells attack mainly muscle and joints. In a recent study, treatment with CAR-T-CD19 cells in a patient with ASS showed promising results [31]. Before treatment, the patient had anti-Jo-1 antibody levels of 331 U/L. After CAR-T treatment, the levels of these antibodies decreased significantly to 5 U/L (cut-off point is 25 U/L). This dramatic reduction in anti-Jo-1 antibodies correlated with a marked improvement in the patient’s physical function and muscle strength, observed at 180 days post-treatment [31]. These findings suggest a significant therapeutic potential of CAR-T in the management of ASS and similar diseases.
Jin et al. [32] demonstrated the efficacy of CAR-T-CD19 cell application in MRL-lpr mice, a model of SLE. The treatment not only prevented SLE pathogenesis before the onset of symptoms, but also showed therapeutic benefits at later stages of disease progression. Mice treated with CAR-T/4-1BB (second-generation CAR, Figure 2) showed improved life expectancy, regression of splenomegaly, and decreased levels of anti-dsDNA (double-stranded DNA) and antinuclear AAb, biomarkers in SLE. In addition, reduced proteinuria, reduced lymphocytic infiltration in the kidneys and reduced granular immune complexes were observed, along with normalization of CD4/CD8 levels [32]. These results underscore the potential of CAR-T-CD19 therapy for the treatment of SLE.
Successful results have been described with CAR-T-CD19 cells in various AIDs. These results range from in vitro research stages, preclinical stages, various case reports and clinical trials conducted by leading groups worldwide (Table 1). Some examples of AIDs where CAR-T-CD19 has been tested include: SLE, ASS, MS, RA, systemic sclerosis (SS), ANCA (anti-neutrophil cytoplasmic antibody) associated vasculitis, Myositis, Idiopathic inflammatory myopathy and myasthenia gravis (MG) [33–41], listed in Table 1. Taking the case of SLE as an example, CAR-T-CD19 can cause significant B cell depletion, eliminating autoimmune B cell clones and anti-dsDNA antibodies. Importantly, although anti-dsDNA is specific to this disease, about 180 AAbs have been described in SLE [42]. After treatment, the reappearing B-cell population is mainly composed of immature cells expressing IgD and IgM, indicating an immature B-cell state without class change. This profound depletion decreases AAb production, which helps explain the absence of detectable anti-dsDNA antibodies after B-cell repopulation [30, 34, 43].
Another target is BCMA, which is expressed on long-lived plasma cells, sources of circulating antibodies. A phase 1 clinical trial with 12 patients showed that anti-BCMA CAR-T reduced antibody production in patients with neuromyelitis optica spectrum disorder (NMOSD) [44]. This is an AID in which antibodies are produced against aquaporin-4 (AQP-4), a water channel present in astrocytes, causing severe inflammation of the central nervous system. Long-lived plasma cells are the main source of the pathogenic AAbs in this disease. The use of anti-BCMA CAR-T as a proof-of-concept resulted in improved neurological disabilities and quality of life in most participants [44]. In addition, there are ongoing clinical trials with application of CAR-T-BCMA in other AIDs, such as MG, various neuroimmunological diseases, scleroderma, Sjögren’s syndrome and SLE, in some cases including a combination of CAR-T-BCMA together with CAR-T-CD19 [45]. Initial publications on these trials demonstrate improvements in the clinical characteristics of patients [37, 38].
CAR-based therapies project promising results in a broad spectrum of AIDs. For example, in MS, a chronic AID that affects the central nervous system causing inflammation and myelin damage, CD20-targeted CAR-T (CAR-T-CD20) have been shown to be effective in both animal models and patients. These therapies have been successful in reducing disease severity and progression by depleting pathogenic B cells [30, 46].
CD25+ regulatory T lymphocytes (Treg) are crucial for maintaining immune system tolerance and preventing autoimmune processes, since part of their role is to suppress and eliminate autoreactive T lymphocytes [25, 47]. CARs on Treg cells (CAR-Treg) recognize the antigen that is expressed on the target cell (in this case cells involved in the autoimmune or inflammatory process are chosen) and, upon binding to it, exert the suppressive effect. This effect can be by secretion of anti-inflammatory cytokines, by direct effects of cell contact, such as inhibition of cell activation or proliferation processes, or by modulation of antigen-presenting cells [47]. In DM-T1, an AID in which autoreactive T cells destroy pancreatic beta cells, CAR-Treg cells have been engineered to suppress autoreactive T cells and promote immune tolerance [36]. CAR-Tregs have shown potential in preclinical models of DM-T1, MS and ulcerative colitis [36, 48–50]. In non-obese type 1 diabetic mouse models, the application of a specific CAR-T system containing the extracellular part of the receptor the variable region of a MoAb that binds to an epitope of the insulin B-chain (MoAb-287), termed CAR-T-287, has successfully identified and targeted diabetogenic and antigen-presenting T cells, minimizing insulitis and delaying the onset of DM-T1, although the protection has been temporary [51]. Preclinical studies have shown the potential of CAR-T to prevent beta-cell destruction and control DM-T1 [30, 52]. These examples highlight the broad applicability of CAR-based therapies to modulate the immune system and provide relief from various autoimmune conditions.
CAR-T-CD19/CD20/BCMA (and others) cells have the advantage of combining antigen recognition and effector function in a single entity, eliminating the need for other cells or molecules for target cell (B lymphocytes and plasma cells) depletion. Furthermore, they can migrate to any organ, including the brain and bone marrow, achieving robust and widespread depletion of long-lived B cells and plasma cells. Taken together, recent studies demonstrate that cytolytic CAR-T-based therapies directed at B-cell family target antigens offer significant therapeutic potential for AIDs refractory to conventional treatments. Examples such as SLE, ES, ASS, MS, and MG, among others (Table 1), demonstrate the reduction of respective AAbs and thus remarkable improvements in the clinical outcome of patients after CAR-T treatment. The ability of these cells to migrate to various organs and effectively eliminate autoimmune B cells suggests that CAR-T therapies could revolutionize the management of complex AIDs, providing new hope for patients with limited therapeutic options.
Examples of AIDs with research evaluating potential application of CAR technology as a therapeutic option
| Disease | CAR target antigens | Research phase/Clinical phase | Examples of products(Name of drugs) | References |
|---|---|---|---|---|
| Systemic lupus erythematosus (SLE) | CD19, BCMA | Clinical phase | Descartes-08; SC291; SCRI-CAR19v3; YTB323; CABA-201; CC-97540; KYV-101; MB-CART19.1; BRL-301; ATHENA CAR-T; RJMty19; CD19 Universal CAR-γδ T; Relma-cel; RD06-04 Cells; CNCT19 cell; ATA3219; ADI-001; PRG-1801; FKC288; IMPT-514; LMY-920; PRG-2311; GC012F injection; UTAA09; C-CAR168 | [53–67] |
| Anti-synthetase syndrome (ASS) | CD19 | Case reports | CABA-201 | [31, 40, 68–71] |
| Multiple sclerosis (MS) | CD19, CD20 | Preclinical phase; clinical phase | KYV-101; CC-97540; CT103A; C-CAR168 | [71–74] |
| Rheumatoid arthritis (RA) | CD19, CD20 | Preclinical phase | KYV101; BRL-301 | [55, 71, 75] |
| Systemic sclerosis (SSc) and scleroderma | CD19#, CD20, BCMA | Case reports; preclinical/clinical phase | CC-97540; MB-CART19.1; BRL-301; KYV-101; CABA-201; Relma-cel; RD06-04 Cells; UTAA09 | [41, 55, 61–63, 71, 75] |
| ANCA-associated vasculitis | CD19 | Case reports | SC291; BRL-301; RD06-04 Cells; PRG-1801 | [54, 61, 76] |
| Myositis (dermatomyositis) | CD19# | Clinical phase | MB-CART19.1; CABA-201; KYV-101 | [40, 41, 71, 75] |
| Idiopathic inflammatory myopathy | CD19 | Clinical phase | CABA-201; CC-97540; MB-CART19.1; BRL-301; RD06-04 Cells; C-CAR168 | [41, 61–63, 77] |
| Myasthenia gravis (MG) | BCMA, CD19, Musk-CAAR | Clinical phase | Descartes-08; CABA-201; CT103A; C-CAR168; MuSK-CAART | [60, 71, 73, 78–80] |
| Neuromyelitis optica spectrum disorder (NMOSD) | BCMA, CD19, CD20 | Clinical phase | CT103A; C-CAR168; anti-CD19/CD20 CAR-T | [44, 59, 73] |
| Autoimmune hemolytic anemia (AHAI) | CD19 | Clinical phase | CNCT19 CAR-T; ThisCART19A | [75, 81] |
| Diabetes mellitus type 1 (DM-T1) | CAR-Treg | Preclinical phase | GNTI-122 | [71, 82, 83] |
| Inflammatory bowel disease (Crohn’s disease) | CAR-Treg | Preclinical phase | IL23R-Specific CAR Tregs; FliC-CAR Tregs | [84–86] |
| Sjögren’s syndrome | BCMA, CD19 | Preclinical phase; clinical phase | BRL-301; RD06-04 Cells; UTAA09 | [55, 61, 71, 75] |
| Pemphigus vulgaris (PV) | DSG3-CAAR*, DSG3-CAAR | Clinical phase | DSG3-CAART; CABA-201 | [71, 87–89] |
| Anti-NMDAR autoimmune encephalitis | NMDAR-CAAR*, NMDAR-CAAR | Preclinical phase | NMDAR-CAAR | [89–91] |
* CAAR-T, depletion of antigen-specific B lymphocytes. # Including tests with T lymphocytes from an allogeneic (not autologous) source [41]. AIDs: autoimmune diseases; BCMA: B-cell maturation antigen; CAAR: chimeric autoantibody receptor; CAR: chimeric antigen receptor; Treg: regulatory T cell
Antigen-specific B-lymphocyte suppression using cytolitic T-cells
Although CAR-T CD19/CD20 cells have the advantage of eliminating B cells with different epitopes, which is especially beneficial in SLE where there is a large number of autoreactive clones against many autoantigens [42], the main side effect and risk for the patient is the deletion of healthy B cells followed by opportunistic infections, a common problem in immunosuppressive therapies. In diseases where there is a well-described pathogenic AAb, such as pemphigus vulgaris (PV), anti-N-methyl-D-aspartate receptor (NMDAR) autoimmune encephalitis, and MG, if immunosuppressive therapies could be tailored to eliminate only B lymphocytes presenting BCR with specific reactivity to the given autoantigen (resulting in a consequent decrease in autoreactive plasma cells), the treatment would present great potential. This is because it would minimize side effects by preserving the patient’s immune potential to eliminate invading microbes. In this context, a strategy based on chimeric autoantibody receptor (CAAR) has been expanding in the last 5 years. In this strategy, instead of the variable region of an antibody directed against a cell receptor, for example, the CAR-T-CD19 is a CAR with the antigen recognition domain against CD19 (Figure 2), the antigen itself is used in the extracellular part of the chimeric receptor, as detailed bellow for different AIDs, in this case attracting and attacking B lymphocytes with the specific autoreactive BCR. CAAR is also transduced into autologous cytolytic T or NK cells, thus being referred to as CAAR-T or CAAR-NK.
PV is an AID that affects the skin and mucous membranes forming painful lesions in areas such as the mouth, throat, genitalia and nostrils. It is considered a potentially fatal disease and although its etiology is unknown it is associated with two specific and well characterized AAbs against desmoglein 1 and 3 proteins (DSG1, DSG3). Anti-DSG AAbs target intercellular junction proteins of the epidermis [88, 92]. Its diagnosis is frequently based on biopsies where the characteristic lesions of this disease are evidenced, such as loss of intercellular cohesion with formation of supra basal vesicles, presence of acantholytic cells and mixed inflammation with presence of eosinophils and neutrophils [92]. There are other complementary analyses to histopathology that consist of the detection of circulating AAbs by different methods such as indirect immunofluorescence and others [93].
Ellebrecht et al. [94] developed a CAAR containing the DSG3 antigen fused to CD137-CD3ζ signaling domains (DSG3-CAAR-T), with the aim of targeting cytolytic T lymphocytes to attack B lymphocytes with autoreactive anti-DSG3 BCR, resulting in a decrease in cells producing pathogenic anti-DSG3 antibodies in vitro. Lee et al. [88] demonstrated the efficacy of the DSG3-CAAR-T system to lyse target B lymphocytes without suppressing the rest of the immune system in PV patients, leading to a reduction of pathogenic AAbs, both in serum and at the tissue level (evidenced in preclinical models). This reduction generated an improvement in symptoms that reduced patients’ quality of life. Their toxicological screening showed no “off-target” cytotoxic interactions. These therapies are already under evaluation in human clinical trials to assess methods of administration, safety, and efficacy (Table 1) [88, 92, 95].
MG is an AID in which discomfort and lesions are generated at the level of the neuromuscular synaptic junction, clinically evidenced as muscle fatigue. It is caused by AAbs against receptors presented by muscle cells at the neuromuscular synapse. The AAbs in MG mainly bind to the acetylcholine receptor (AChR), with anti-AChR appearing in about 80% of patients. However, up to 50% of patients are negative for anti-AChR, the pathogenic AAbs that bind to muscle tyrosine kinase (MuSK) and generate weakness at the muscle level where AAbs interfere with neuromuscular signaling. Oh et al. [79] created MuSK-CAAR-T cells with CD137-CD3ζ signaling domains and the MuSK antigen with the aim of targeting and depleting anti-MuSK B cells, thereby reducing the pathological effects of AAbs without the side effect of widespread immunosuppression of more conventional therapies. The preclinical trials conducted were effective, depleted anti-MuSK B cells, reduced anti-MuSK IgG levels and even maintained their activity in the presence of anti-MuSK antibodies in solution. In turn, no “off-target” effects were seen in in vivo experiments, primary screens in human cells or high-throughput human membrane proteome arrays. This specificity and efficacy promotes further research for new pharmacological applications of CAAR-T in MG patients seropositive for anti-MuSK antibodies [79, 96, 97].
In autoimmune encephalitis with anti-NMDAR AAbs, symptoms such as dyskinesia, seizures, agitation, hallucinations, or catatonia, among others, are common. The NMDAR is a cation channel and has two NR1 units (GluN1), where glycine binds, and two units which can be NR2 (GluN2) or a combination of NR2 and NR3, where the neurotransmitter glutamate binds. The AAbs act against GluN1 and disrupt glycine binding. As the binding of two glycins and two glutamates is necessary for the channel to open, the presence of anti-NMDR AAbs profoundly affects neuronal communication and brain function [98]. Reincke et al. [91] developed a chimeric CAAR receptor containing a complex composed of extracellular domains of GluN1 and GluN2B, subunits of NMDAR, fused to the intracellular signaling domains 4-1BB-CD3ζ, called NMDAR-CAAR. NMDAR-CAAR transfected into cytolytic T cells (NMDAR-CAAR-T) was able to specifically eliminate anti-NMDAR-secreting clones without being “off-target”, both in vitro and in vivo, in a passive transfer mouse model. These results are promising for treatments for autoimmune encephalitis [90, 91, 99].
In primary immune thrombocytopenia, the AAbs are against platelets, accelerating platelet destruction, affecting megakaryocytes, resulting in a dangerous and potentially fatal bleeding disorder. Several glycoprotein (GP) complexes, composed of different subunits, are formed on the surface of platelets, but AAbs in autoimmune thrombocytopenia are mostly against GP IIb/IIIa and/or GPIb-IX-V complexes [100]. Zhou et al. [101] constructed a chimeric receptor containing in the extracellular part the GPIbα subunit fused to a second-generation structure (Figure 2), which contains the intracellular domains 4-1BB and CD3ζ. The chimeric receptor, named GPIbα-CAAR, after transfection into cytolytic CD8 T cells (GPIbα-CAAR-T), was evaluated both in vitro and in vivo, demonstrating its potential to specifically eliminate autoreactive B lymphocytes against GPIbα (anti-GPIbα), preserving healthy B-cell clones. The initial study by Zhou et al. [101] demonstrated that the application of GPIbα-CAAR-T is promising for the treatment of patients with refractory primary immune thrombocytopenia.
In Graves’ disease, a thyroid-specific AID, patients present AAbs against the thyrotropin receptor, also called thyroid stimulating hormone receptor (TSHR). These AAbs are stimulatory, i.e., when they bind to TSHR, they trigger exaggerated secretion of thyroid hormones T3 and T4, resulting in hyperthyroidism. Duan et al. [102] constructed a chimeric receptor containing the extracellular part of TSHR and the intracellular part of CAR (4-1BB and CD3ζ). In the study, the authors called the receptor TSHR-CAR, but for ease of reading in this text, we will call the receptor TSHR-CAAR because of its receptor for AAbs. Human T lymphocytes transfected with the receptor (TSHR-CAAR-T) were able to eliminate anti-TSHR autoreactive B lymphocytes both in vitro and in vivo in an animal model that received an anti-TSHR-secreting hybridoma, i.e., a Graves’ disease model [102]. This study demonstrated that the TSHR-CAAR-T system is a specific and promising treatment for pathogenic AAb-mediated diseases, such as Graves’ disease.
Considering the difficulty of transfecting primary lymphocytes [103] in a patient-specific autologous immunotherapy setting, it makes more sense to use CD8 T cells as cytolytic cells, due to their abundance in the blood. However, this makes the process of CAR-T generation more expensive and complicated, therefore, as mentioned above, some have proposed the use of “ready-to-use” NK cells as cytolytic cells [23, 104]. One of the autoimmune renal diseases is membranous nephropathy (also called, membranous glomerulonephritis), in which pathogenic AAbs against proteins expressed by glomerular podocytes are found, directly affecting the renal function of patients. The main autoantigens in membranous nephropathy are phospholipase A2 receptor (PLA2R1) and thrombospondin type 1 domain-containing protein 7A (THSD7A). Seifert et al. [105] constructed a chimeric receptor containing in its extracellular part the immunodominant regions (preferred target of AAbs) for PLA2R1 and for THSD7A, and in the intracellular part of CAR, 4-1BB and CD3ζ, named PLA2R1/THSD7A-CAAR. However, in their study, Seifert et al. [105] used an NK cell line as cytolytic cells, called NK-92. PLA2R1/THSD7A-CAAR-NK cytolytic cells (as well as T lymphocytes that were also evaluated in the study) were able to adequately lyse anti-PLA2R1 and anti-THSD7A hybridomas in vitro. Subsequent studies should evaluate the potential for specific elimination of autoreactive cells by PLA2R1/THSD7A-CAAR-NK/Ts in membranous nephropathy [104–106].
The myelin sheath damage observed in MS also has a strong involvement of AAbs, such as those against myelin basic protein (MBP). Sahlolbei et al. [107] generated a receptor with the extracellular portion containing a peptide from the extracellular part of MBP (amino acids 83-99), coupled to CD137 and CD3ζ as intracellular domains, the constructed receptor being named MBP-CAAR. The receptor was transfected into T lymphocytes (MBP-CAAR-T) and its potential to eliminate anti-MBP autoreactive B cells was evaluated and confirmed in vitro [107]. For this study, the next step is in vivo evaluation.
The suppression and elimination of antigen-specific reactive B lymphocytes by using cytolytic cells with CAAR represents a promising approach for the treatment of various AIDs, primarily those where pathogenic AAbs are encountered. This strategy offers the potential to precisely and effectively eliminate autoreactive B cells without compromising the functionality of the overall immune system, thus minimizing the side effects common in general immunosuppressive therapies, including immunobiologics (MoAbs) and CAR-T. The already mentioned limitations associated with CAR-T also apply to CAAR-T; however, an additional feature complicates the development of this technology even further. AAbs are polyclonal and undergo epitope spreading, meaning a given patient can present multiple AAbs against associated antigenic clusters. In addition, maintaining the natural structure of the antigen in the CAAR presents additional challenges, especially in AIDs where the pathogenic AAb targets a complex structure. For example, in MG, AAbs against the AChR are among the most difficult to measure in clinical diagnostics because the AChR is a five-subunit channel, making solid-phase assays perform poorly [108]. Developing a CAAR molecule capable of binding AChR and thereby eliminating B cells with anti-AChR BCRs will require extensive research.
Overall, in vitro and preclinical studies have demonstrated the efficacy and specificity of both CAR-T and CAAR-T therapies in various AIDs. However, lengthy human clinical trials will be crucial to evaluate their safety and applicability in the clinical setting.
Conclusions
B cells are central to the pathogenesis of AIDs due to their role in AAb production, cytokine dysfunction, antigen presentation, and failure of regulatory B cell function, in diseases such as DM-T1, PV, and MS they play the crucial role in generating pathogenic AAbs. About 10% of patients with AIDs do not respond favorably to conventional immunosuppressive therapies. Autologous immunotherapies based on CAR-T and its variants, targeting specific cell populations (CD19/CD20/BCMA, etc.), represent a transformative advance in the treatment for these patients, because they appear as a therapeutic option when all else fails. The complexity and expertise required to engineer CARs and expand cytolytic T cells in the laboratory before reinjecting them into the patient [27] is reflected in the treatment cost and in the associated risks and side effects. However, as protocols become more widespread, costs are expected to decrease, making this therapy more accessible to a greater number of patients.
Within this last decade, the possibility of directly boosting the cellular effector system against malignant cells, known as autologous cellular immunotherapy, has been developed. This approach has been particularly successful in hematological cancers with the use of CAR-T. In the near future, the application of CAR-T is envisaged in the treatment of AIDs, especially those where there is no specific and well-defined autoantigen, or where there are multiple AAbs, with no clear pathogenic AAb, examples of such AIDs include SLE and RA. In these refractory conditions, it is hoped that CAR-T can be used for the ablation of specific cell populations, as in the case of B lymphocytes with CAR-T-CD19/CD20 and plasma cells with CAR-T-BCMA, offering new hope for patients who do not respond to conventional therapies.
Probably within the next decade we foresee a future where we have the ability to suppress the immune system against specific antigens [109]. This is the main challenge we currently have for the treatment of AIDs, especially those mediated by known pathogenic AAbs. Here we discussed the potential of CAAR-T cell therapy, and how it combines precise antigen recognition with robust effector functions. CAAR-T cell therapy offers a targeted approach to antigen-specific B-cell depletion, eliminating only autoreactive cells. Although lengthy phase 3 clinical trials are still required to assess its applicability in the clinical setting, preclinical and phase 1 results are promising. Other techniques have been proposed for eliminating autoreactive B cells in an antigen-specific manner and without affecting healthy cells. One of them is affinity matrix [110] or knockout (modification by deletion or inactivation) by CRISPR-Cas9 targeting V(D)J recombinants that generate AAbs, as previously published by our group [111]. These innovative approaches aim to specifically eliminate cells responsible for the production of pathogenic AAbs. A comprehensive review of these techniques can be found in Stensland et al. [112]. It is likely that a combination of technologies will be applied specifically to individual diseases and patients, allowing for personalized treatments for patients and the application of precision medicine to the clinical practice [109].
Abbreviations
| AAbs: | autoantibodies |
| AChR: | acetylcholine receptor |
| AIDs: | autoimmune diseases |
| ASS: | anti-synthetase syndrome |
| BCMA: | B-cell maturation antigen |
| BCR: | B-cell receptor |
| CAAR: | chimeric autoantibody receptor |
| CAR: | chimeric antigen receptor |
| CAR-T: | chimeric antigen receptor T cells |
| CRS: | cytokine release syndrome |
| DM-T1: | type 1 diabetes mellitus |
| dsDNA: | double-stranded DNA |
| MBP: | myelin basic protein |
| MG: | myasthenia gravis |
| MoAb: | monoclonal antibodies |
| MS: | multiple sclerosis |
| MSCs: | mesenchymal stem cells |
| MuSK: | muscle tyrosine kinase |
| NK: | natural killer |
| NMDAR: | N-methyl-D-aspartate receptor |
| OTOT: | on-target, off-tumor |
| PLA2R1: | phospholipase A2 receptor 1 |
| PV: | pemphigus vulgaris |
| RA: | rheumatoid arthritis |
| scFv: | single-chain variable fragment |
| SLE: | systemic lupus erythematosus |
| THSD7A: | thrombospondin type 1 domain-containing protein 7A |
| Treg: | regulatory T cell |
| TSHR: | thyroid stimulating hormone receptor |
Declarations
Acknowledgments
The manuscript was originally written in Spanish, the mother language of the authors, and translated to English using online tools such as ChatGTP (by OpenAI) and Gemini (by Google/Alphabet). The final text underwent proof by an English language expert, therefore, after using the online tools, authors reviewed and edited the content and take full responsibility for the content of the publication.
Author contributions
MFS and DL: Conceptualization, Investigation, Writing—original draft, Writing—review & editing. YD and RCJ: Data curation, Validation, Writing—review & editing. CAFO: Data curation, Validation, Writing—review & editing. GDK: Conceptualization, Supervision, Project administration, Writing—review & editing. All authors reviewed, discussed, and agreed to their individual contributions before submission. All authors read and approved the final version of the manuscript.
Conflicts of interest
The authors declare that they have no conflicts of interest.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Availability of data and materials
Not applicable.
Funding
GDK was supported by Sao Paulo State Research Foundation, FAPESP (Fundação de Amparo à Pesquisa do Estado de São Paulo - Brasil) during development of this work. MFS was supported by Agencia Nacional de Investigación y Desarrollo de Chile (ANID), Estudios de postgrado financiados por ANID, ANID-Subdirección de Capital Humano/Doctorado Nacional [2024-n.21241121]. ANID also support this work with project No. [86220018]. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© The Author(s) 2025.
Publisher’s note
Open Exploration maintains a neutral stance on jurisdictional claims in published institutional affiliations and maps. All opinions expressed in this article are the personal views of the author(s) and do not represent the stance of the editorial team or the publisher.